

Abstract
Lenalidomide (LEN) treatment in multiple myeloma (MM) results in superior outcome. However, there is concern for increased myelodysplastic syndromes/acute myeloid leukemias (MDS/AML) associated with LEN. Thus, bone marrow morphology and cytogenetic studies from 40 patients were evaluated for early signs of MDS prior to therapy, during therapy and at follow-up. Newly diagnosed MM patients treated with LEN and dexamethasone (LD) alone or followed by ASCT (LD/ASCT), or relapsed/refractory MM patients treated with LEN, bendamustine and dexamethasone (BLD) were included. One patient developed MDS. Baseline prevalence of mild morphologic myelodysplasia was highest in pretreated MM patients (BLD, 71%), but was also seen in newly diagnosed patients (LD and LD/ASCT, 17%). Prevalence of myelodysplasia did not increase over time. Thus, this study did not reveal rapidly emerging MDS in 39 of 40 MM patients treated with LEN. Development of MDS in one patient suggests that longer follow up is needed for all.
Keywords: Myeloma, Lenalidomide, Myelodysplastic Syndrome
INTRODUCTION
Lenalidomide (LEN) induces high response rates in multiple myeloma (MM). Recently, two randomized phase 3 trials (IFM 2005-02, CALGB 100104) have shown that LEN maintenance given after autologous stem cell transplant (ASCT) in newly diagnosed MM patients resulted in significant prolongation of the progression-free survival [1,2] and, in one of the trials, a survival benefit [2]. Unfortunately, an increased rate of second cancer with concern for the development of myelodysplastic syndrome/acute myeloid leukemia (MDS/AML) was observed in both trials [2,3], resulting in an FDA safety alert: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm250606.htm. The MM-015 trial, combining LEN, melphalan and prednisone with LEN maintenance in transplant-ineligible patients ≥65 years, also showed excellent response data, but found an increased incidence of MDS/AML compared to melphalan and prednisone alone [4]. In contrast, a recent pooled analysis of LEN-based therapy for relapsed/refractory MM did not reveal an increased incidence of invasive solid tumor or hematologic malignancies, including MDS/AML [5]. Overall, the benefit of LEN maintenance therapy likely outweighs the risk for MDS/AML for the majority of MM patients [6]. However, the possible role of LEN in the development of MDS/AML in some MM patients requires further investigation [7]. In this study, longitudinal bone marrow and corresponding cytogenetic data of newly diagnosed MM patients treated with LEN and dexamethasone (LD) alone or in combination with ASCT or relapsed/refractory MM patients treated with bendamustine were re-evaluated to analyze the impact of LEN alone, in combination with ASCT, prior myeloma treatment, and concomitant alkylator treatment on development of early signs of MDS.
PATIENTS AND METHODS
Patients and Treatments
The study was approved by the University of Pittsburgh Institutional Review Board and patients gave written informed consent in accordance with the Declaration of Helsinki. The diagnosis of myeloma was established according to the latest international myeloma working group criteria [8]. The study included patients from three different treatment groups:
Newly diagnosed MM: LD alone 6–9 cycles (LD alone)
Newly diagnosed MM: LD 4 cycles followed by ASCT with high dose melphalan (LD/ASCT) [9]
Relapsed/refractory MM: (median 3 prior treatments; range 1–6): bendamustine, lenalidomide, dexamethasone 4–8 cycles (BLD) [10]
Newly diagnosed MM patients were randomized within a clinical trial at the University of Pittsburgh Cancer Institute (UPCI) to either LD alone or LD/ASCT (UPCI# 07–134; ClinicalTrials.gov Identifier NCT00777881). The patients underwent stem cell mobilization with up to 4 g/m2 cyclophosphamide regardless of which treatment group they were assigned (LD alone or LD/ASCT), but only those assigned to the LD/ASCT group received high dose melphalan. Patients with relapsed/refractory MM were treated with BLD within a separate clinical trial (UPCI# 07–089; ClinicalTrials.gov Identifier NCT01042704).
The time points for morphologic and cytogenetic review are specified in Figure 1. Patients were included if they had at least two adequate specimens available for morphologic review corresponding to the specified evaluation time points. Diagnostic criteria for MDS was in accordance with the 2008 WHO Classification [11].
Figure 1.
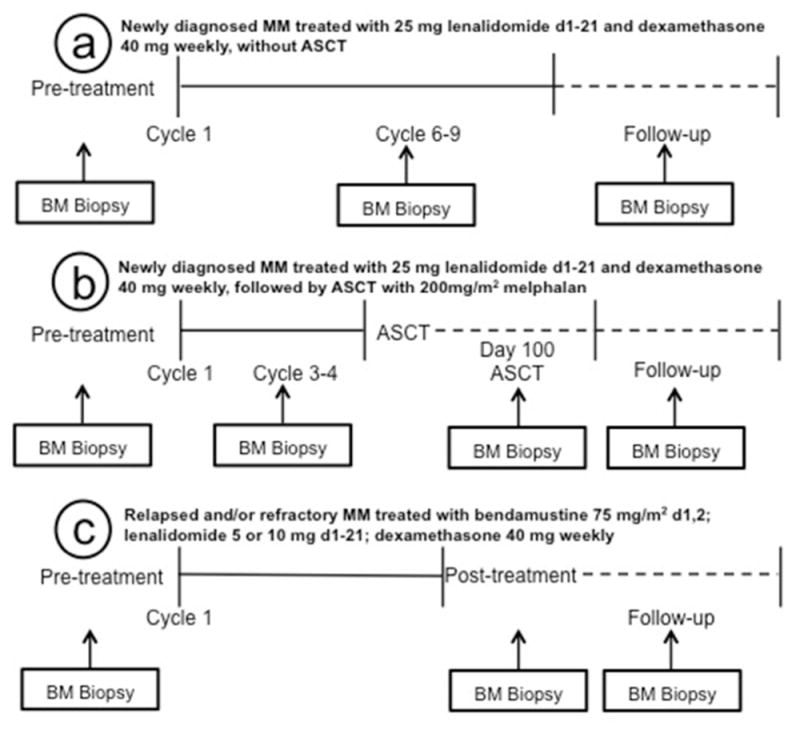
Bone marrow biopsy timeline in different groups. (a) Specimens evaluated among newly diagnosed MM patients assigned to the LD alone group included a pretreatment baseline specimen, after 6–9 cycles and at last follow-up. (b) Specimens evaluated among newly diagnosed MM patients assigned to the LD/ASCT group included a pretreatment specimen, after 3–4 cycles of LD, 3 months after ASCT and at last follow-up. (c) Specimens evaluated among relapsed/refractory MM patients treated with BLD included a pretreatment specimen, at the end of treatment (median 6 cycles, range 4–8 cycles) and at last follow-up.
Peripheral Blood and Bone Marrow Evaluations
Peripheral blood films, bone marrow aspirate smears and biopsies were systematically reassessed by one hematopathologist (S.A.M.) blinded as to treatment group. Manual differential counts were performed on aspirate smears. Dysplastic forms were enumerated among 100 erythroid precursors, 100 maturing neutrophils, and 50 (minimum 20) megakaryocytes when available. Dysplasia in a cell line was deemed present when dysplastic forms, defined according to the 2008 WHO Classification, accounted for ≥10% of the cells [11]. In addition, morphology was assessed for forms likely to be more specific for MDS (pseudo Pelger-Huët anomaly, agranular neutrophils & precursors, micromegakaryocytes, mature megakaryocytes with completely round non-lobated nuclei, megakaryocytes with widely separated nuclei, and ring sideroblasts) according to standard practice and precedence in the literature [12–16]; these findings were designated as “severe dysplasia” when such forms accounted for ≥10% of cells.
Images were captured using an Olympus BX51 microscope, an Olympus DP26 digital camera and cellSens Entry 1.6 digital imaging software (Olympus America Inc., Center Valley, PA).
Classical and Molecular Cytogenetic Studies
Results of classical cytogenetic studies and interphase fluorescence in situ hybridization (FISH) panel studies for myeloma were reviewed to evaluate for abnormalities not likely attributable to MM. For classical chromosome analysis, trypsin–Giemsa banded metaphase cells were analyzed from 24-hour harvests of unstimulated and 72-hour harvest of PHA-stimulated bone marrow aspirate cell cultures. The FISH panel used Abbott Molecular probes (Des Plaines, IL) to detect abnormalities involving CCND1 (11q13), ATM (11q22.3), D13S319/LAMP1 (13q14/13q34), IGH@ (14q32.3) and TP53 (17p13.1).
FISH assays were carried out to assess for NUP98 gene arrangement using archived bone marrow aspirate smears from two patients with evidence of 11p15 rearrangement detected by classical cytogenetic analysis. A dual-color breakapart probe for detection of NUP98 (11p15) rearrangements was designed using two BAC clones RP11-258P13 (SpectrumGreen) and RP11-120E20 (SpectrumOrange) [17]. Fusion signals indicate an intact NUP98 gene and signal separation supports NUP98 gene rearrangement. A normal concurrent blood control slide showed two fusion signals in 99% (100/101) of the cells. Images were captured using an Olympus BX61 epifluorescence microscope (Olympus America Inc.) and Genus software platform on the Cytovision System (Leica Microsystems, San Jose, CA).
All classical chromosome analyses and FISH assays were performed on specimens without selection procedures for plasma cells or CD34 positive progenitor cells.
Statistical Analysis
Fisher’s exact test was used to evaluate relationships between categorical variables. A generalized random effects model [18] was used to evaluate the effect of time since treatment initiation on the prevalence of morphological dysplasia. SAS v9.2 (SAS Institute, Cary, NC) was used for statistical analyses.
RESULTS
We retrospectively analyzed 113 bone marrow biopsy samples of patients prior, during and after LEN-based treatment (Table I). The study included newly diagnosed MM patients randomized to either LD alone (14 patients, average age ± 2SD: 61 ± 13 years, M:F = 7:7) or LD/ASCT (18 patients, average age ± 2SD: 60 ± 14 years, M:F = 9:9) and relapsed/refractory MM patients treated with BLD (8 patients, average age ± 2SD: 62 ± 11 years, M:F = 6:2).
Table I.
Morphologic and Classical Cytogenetic Findings in Three Different Groups Treated with Lenalidomide for Myeloma
| Evaluation Time Point | Newly diagnosed MM patients
|
Relapsed/refractory MM patients
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre-treatment LD+LD/ASCT groups |
LD Alone Treatment Group (14 patients)
|
LD/ASCT Treatment Group (18 patients)
|
BLD Treatment Group (8 patients)
|
||||||
| Post 6–9 LD cycles | Follow-up* (median 14 mos.; range 12–29 mos.) | Post 3–4 LD cycles | 3 months post ASCT | Follow-up* (median 18 mos.; range 12–27 mos.) | Pre-treatment | End of treatment post 4–8 BLD cycles | Follow-up* (median 15 mos.; range 13–23 mos.) | ||
| Mean marrow blast % ± 2SD | 0.5 ± 0.8 | 0.4 ± 1.0 | 0.1 ± 0.8 | 0.3 ± 0.8 | 0.3 ± 0.6 | 0.5 ± 1.5 | 0.4 ± 0.5 | 0.3 ± 0.3 | 0.3 ± 0.6 |
| Severe dysplasia, n/specimens (%) | 0/29 | 0/13 | 1/8 (12)† | 0/18 | 0/15 | 0/10 | 0/7 | 0/8 | 0/5 |
| Non-severe dysplasia, n/specimens (%) | 5/29 (17) | 6/13 (46) | 4/8 (50) | 2/18 (11) | 3/15 (20) | 0/10 (0) | 5/7 (71) | 7/8 (88) | 3/5 (60) |
| Erythroid, n | 2 | 3 | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| Granulocytes, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Megakaryocytes, n | 3 | 4 | 3 | 2 | 3 | 0 | 5 | 7 | 3 |
| Classical cytogenetic studies, n/specimens (%) | |||||||||
| Normal karyotype | 23/27 (85) | 12/13 (92) | 6/8 (75) | 17/17 (100) | 14/15 (93) | 8/10 (80) | 6/7 (86) | 7/8 (88) | 3/4 (75) |
| Clonal changes related to MM | 3/27 (11) | 1/13 (8) | 0 | 0 | 0 | 1/10 (10) | 1/7 (14) | 1/8 (12) | 1/4 (25) |
| Clonal changes not likely related to MM | 1/127 (4)‡ | 0 | 2/8 (25)† | 0 | 1/15 (7)§ | 1/10 (10)§ | 0 | 0 | 0 |
LD, lenalidomide & dexamethasone; ASCT, autologous stem cell transplant; BLD, bendamustine, lenalidomide and dexamethasone.
One patient from LD alone had three follow-up evaluations, one patient from LD/ASCT had two follow-up evaluations and one patient from BLD had two follow-up evaluations; the median and range for follow-up are determined from the last follow-up evaluation.
Severe dyserythropoiesis and dysmegakaryopoiesis was seen in the specimen diagnostic for MDS in 1 patient from the LD alone group 13 mos. after initiation of LD alone therapy (i.e., also 3 months after initiation of Velcade/dexamethasone salvage therapy); a clonal cytogenetic abnormality was detected at this time and persisted on follow-up 18 mos. after initiation of LD alone therapy.
A clonal cytogenetic abnormality was detected in 1 patient from the LD/ASCT therapy group at 3 mos. post ASCT and on follow-up at 10 mos. post ASCT, but was not detected on a final follow-up specimen (details described under results)
Development of MDS in one patient
One newly diagnosed MM patient developed severe dysplasia and clonal cytogenetic abnormalities, consistent with emergence of MDS during follow-up after 7 cycles of LD alone treatment and subsequent salvage therapy. The pretreatment evaluation of this 63 year old female revealed 30% plasma cells in the bone marrow. Mild dyserythropoiesis (i.e., nuclear budding, nuclear irregularities and megaloblastic changes) was present. Classical cytogenetic analysis revealed a normal karyotype, 46,XX[20], and the FISH panel studies for MM were negative. The bone marrow after 7 cycles of therapy was insufficient for morphologic evaluation, but did yield a normal karyotype. The patient achieved a very good partial response, but a follow-up bone marrow 10 months after initiation of therapy demonstrated recurrent MM with occasional ring sideroblasts (<10%) and a normal karyotype. She was treated with salvage Velcade/dexamethasone. A bone marrow evaluation 3 months after initiation of salvage therapy (i.e., 13 months after initiation of LD alone) revealed severe megakaryocytic dysplasia and ring sideroblasts (≥10%) without evidence of myeloma [Figure 2(a-c)]. The classical cytogenetic analysis revealed an abnormal clone consistent with MDS: 46,XX,t(4;11)(q31;p15)[3]/46,XX[18]. She underwent ASCT for relapsed MM and a follow-up bone marrow evaluation 3 months after ASCT (i.e., 18 months after initiation of the LD alone) revealed persistent ring sideroblasts (<10%) and the same chromosomal abnormality in one metaphase cell: 46,XX,t(4;11)(q31;p15)[1]/46,XX[19].
Figure 2.

Bone marrow morphology and FISH analysis in MM patients treated with LEN. (a) The biopsy from time of diagnosis of MDS in one patient from the LD alone group, 13 months after initiation of therapy, was hypercellular (60%) with loose clusters of megakaryocytes (Hematoxylin & Eosin, original magnification X200). (b) Severe dysmegakaryopoiesis, including forms with widely separated nuclei and rare micromegakaryocytes (arrows), were seen at diagnosis of MDS in the same patient as in panel (a) (Wright-Giemsa, original magnification X500). (c) Ring sideroblasts (arrow) were easily seen at diagnosis of MDS in the same patient as in panels (a and b) (Prussian blue, original magnification X1000). (d) FISH analysis performed on a bone marrow aspirate smear at diagnosis of MDS in the same patient as in panels (a–c), whose concurrent classical cytogenetic analysis revealed 46,XX,t(4;11)(q31;p15)[3]/46,XX[18]. There are two normal cells with two fusion signals and a third cell with one fusion signal and one separate orange and green signal each (arrows), which suggest a NUP98 gene rearrangement. (e) Mild dysmegakaryopoiesis, including this small form with a hypolobated nucleus (arrow), was seen in a patient from the LD alone group after 9 cycles of therapy (Wright-Giemsa, original magnification X500). (f) Mild dyserythropoiesis, including this normoblast with nuclear irregularity, budding and megaloblastoid changes (arrow), was seen in a pretreatment specimen from a patient assigned to the LD alone group (Wright-Giemsa, original magnification X500).
Mild Morphologic Myelodysplasia in Patients without MDS
Severe dysplasia affecting ≥ 10% of any cell line, strongly suggesting MDS, was not identified in any other patient. However, mild dysplasia predominantly affecting megakaryocytes was present in 5 (71%) of 7 specimens from relapsed/refractory patients even prior to beginning therapy in the BLD group (Table I, Figure 3). The baseline prevalence in untreated newly diagnosed MM patients (LD alone and LD/ASCT groups) was significantly lower, being detected in 5 (17%) of 29 pretreatment evaluations. The proportion of specimens with dysplasia from the newly diagnosed MM patients did slightly increase with exposure to lenalidomide, but there was no evidence for a significantly increased prevalence over time within any of the three treatment groups (generalized random effects model, p=0.27).
Figure 3.

Proportion of specimens with dysplasia according to treatment group [lenalidomide and dexamethasone (LD) alone, LD followed by autologous stem cell transplant (LD/ASCT) and bendamustine, lenalidomide and dexamethasone (BLD)] and time point. “n” represents the number of specimens that were evaluated at each time point. One specimen (*) with MDS at follow-up in the LD alone group had severe dyserythropoiesis and dysmegakaryopoiesis, but the dyserythropoiesis and dysmegakaryopoiesis in all other specimens was mild.
Dysmegakaryopoiesis
Among patients with newly diagnosed MM assigned to either LD or LD/ASCT treatment group, dysmegakaryopoiesis was seen in 3 (10%) of 29 of patients prior to the start of treatment (Table I, Figure 3). Among these patients, the incidence of dysmegakaryopoiesis involving bone marrow specimens immediately following therapy or upon longer follow-up ranged from 0% to 50%. Dysmegakaryopoiesis was significantly higher among relapsed/refractory and heavily pretreated MM patients treated with BLD therapy and was seen in 5 (71%) of 7 pretreatment specimens with a similar prevalence observed at the end of therapy (88%) and during follow-up (60%).
Except for the specimen from the patient who developed MDS, all 30 remaining specimens with dysmegakaryopoiesis displayed a predominance of mild dyspoietic forms with nuclear hypolobation [Figure 2(e)] and only occasional severe dysplastic forms (<10% of the cells) without any micromegakaryocytes.
Dysmegakaryopoiesis among patients in the three treatment groups was transient in 7 (37%) of 19 patients and appeared persistent in 7 (37%) patients. Persistence could not be evaluated in 5 (26%) patients because the dysmegakaryopoiesis was detected only once on their final specimens.
Dyserythropoiesis
Among patients with newly diagnosed MM assigned to either LD or LD/ASCT treatment group, dyerythropoiesis was detected in 2 (7%) of 29 patients prior to start of treatment (Table I, Figure 3). Among patients treated with LD alone, dyserythropoiesis was seen in 23% patients after 6–9 cycles of LD therapy and in 25% of specimens during follow-up. No dyserythropoiesis was detected during therapy or follow-up from patients assigned to the LD/ASCT group. Dyserythropoiesis was seen in the BLD group in 1 (14%) of 7 patients at the pretreatment evaluation, but was not detected during therapy or follow-up.
Severe dysplasia (i.e., ring sideroblasts) was only detected in specimens from the patient diagnosed with MDS, subsequent to completion of LD alone therapy. The dyserythropoiesis in the other 7 specimens was mild [Figure 2(f)].
Dyserythropoiesis was transient in 3 (50%) of 6 patients and appeared persistent in 1 (17%) patient in the LD alone treatment group. Persistence could not be evaluated in 2 (33%) patients because the dyserythropoiesis was detected only once on their final specimens.
Dysgranulopoiesis
No specimen had dysgranulopoiesis involving ≥10% of the cells. A trend was found for a shift to more immature granulocytic precursors after 3–4 cycles of LD in the LD+ASCT treatment group when compared to their pretreatment specimens, based on mean percentage of promyelocytes and myelocytes (25% vs. 22%), but this did not reach statistical significance (p = 0.09).
Classical and Molecular Cytogenetic Studies
Classical chromosome analysis was performed in 109 of the 113 bone marrow samples. The analysis was performed on ≥ 20 consecutive metaphases (range: 9–26 metaphases) in 108 of the 109 samples.
In addition to the clonal abnormality already described in the patient who developed MDS, a transient clonal cytogenetic abnormality not likely related to MM was detected in a 53 year old male during LD/ASCT treatment. His bone marrow after 4 cycles of LD therapy revealed MM, accounting for 15% of the cellularity, and a normal karyotype, 46,XY[20]. Three months after ASCT (9 months after therapy initiation), his bone marrow was hypocellular without MM, but classical cytogenetic analysis revealed a clonal abnormality in two cells: 46,XY,t(2;17)(p23;q11.2)[2]/46,XY[17]. In addition, one nonclonal cell had a 46,XY,t(4;11)(q12;p15) chromosome pattern at this time, an interesting finding given that rearrangement of 11p15 with a different partner was the clonal abnormality seen in the patient who developed MDS. A follow-up bone marrow 16 months after therapy initiation revealed persistence of t(2;17), but the t(4;11) was not detected: 46,XY,t(2;17)(p23;q11.2)[1]/46,XY[19]; there was no definitive morphologic evidence of MM at this time The karyotype returned to normal with no cytogenetic abnormalities noted at 18 months after therapy initiation, but a low level of MM involving the marrow emerged. Morphologic dysplasia was not detected at any time point in this patient. Transient loss of the Y-chromosome was also detected in a pretreatment bone marrow specimen without morphologic myelodysplasia from a 66 year old male going on to the LD alone treatment group: 45,X,-Y[13]/46,XY[7]; however, the abnormality was no longer present on subsequent evaluations.
FISH assays were carried out on bone marrow aspirate smears from the two patients with evidence for 11p15 rearrangement detected by classical cytogenetic analysis: in one patient at the time of diagnosis of MDS associated with clonal t(4;11)(q31;p15) at 13 months after initiation of LD alone therapy and, in the other patient, at the time of detection of one cell with t(4;11)(q12;p15) that was associated with transient clonal t(2;17)(p23;q11.2). Two fusion signals were detected in 317 (99%) of 320 interphase nuclei in the MDS patient; three (0.9%) of 320 interphase nuclei displayed a pattern that may represent a low level of NUP98 gene rearrangement with one fusion signal and one pair of separate green and orange signals each [Figure 2(d)]. The aspirate smear available from the MDS patient was hemodilute and this could have precluded the detection of evidence for NUP98 gene rearrangement in a greater number of nuclei. The slide from the second patient did not hybridize uniformly and the findings are not reported.
DISCUSSION
Treatment with melphalan is associated with an increased incidence of MDS/AML in MM patients [19–23]. It has been difficult to pinpoint factors other than melphalan-based therapy that contribute to the increased risk for MDS/AML among MM patients. Conventional chemotherapy preceding ASCT appears to play a larger role in contributing to MDS/AML than does the high-dose melphalan-based myeloablative therapy used for ASCT in MM patients [22,24], but one study did support the role of factors other than chemotherapy preceding ASCT [25]. Indeed, there is evidence that some patients with MM and related plasma cell disorders may inherently be at a higher risk for MDS/AML. An 8-fold increased risk of MDS/AML in patients with IgG/IgA MGUS has been reported [7,22]. Furthermore, an increased prevalence of a single-nucleotide polymorphism for the erythropoietin gene promoter, associated with MDS in general, has been found among MM patients who developed MDS [26]. As LEN has become part of the therapeutic armamentarium for MM in the last decade, it has become important to consider what contribution, if any, LEN may make toward increasing this risk.
In two randomized phase 3 trials (IFM 2005-02, CALGB 100104), maintenance treatment with LEN improved progression-free survival compared to placebo after first line ASCT for MM [1,2]. The CALGB 100104 trial demonstrated a survival advantage among the patients receiving maintenance therapy with LEN despite 80% of the patients having crossed over from the placebo arm [2]. However, both trials have reported evidence that maintenance therapy with LEN is associated with an increase in second malignancies, including a concern for a low-level increase in the incidence of MDS/AML. In addition, the phase 3 MM-015 trial reported an increased incidence of MDS/AML among transplant-ineligible MM patients ≥65 years treated with either melphalan-prednisone-LEN (MPR) or MPR followed by LEN maintenance (MPR-R) when compared to those treated with melphalan-prednisone [4]. Notably, the benefit of superior progression-free survival observed with MPR-R was upheld even when second primary malignancies were considered. In contrast, an increased incidence of hematologic malignancies and solid tumors was not found upon retrospective comparison of pooled data from multiple clinical trials of LEN-based therapy for relapsed/refractory multiple myeloma when compared to the general population of the United States or upon comparison between a subset of patients randomized to either LEN/dexamethasone or placebo/dexamethasone [5]. However, adverse-event data were not mandated for the follow-up period after discontinuation of LEN therapy for that study. An increase in solid tissue or hematologic second malignancies was also not found upon comparison of patients from four trial components of the Total Therapy 2 and 3 trials, which differed in the use of thalidomide or LEN with Velcade and dexamethasone as maintenance therapy for MM patients [27]. Thus, more investigation is required to determine whether or not LEN plays any role in the development of MDS/AML in MM patients.
A detailed, longitudinal evaluation of bone marrows for morphologic dysplasia among MM patients treated with LEN has not been previously reported. Our study is the first to focus on early signs of MDS in association with LEN treatment. Our evaluation of bone marrow specimens and corresponding cytogenetic studies from MM patients treated with LEN alone, in combination with ASCT or bendamustine did not reveal clear-cut evidence for early or overt MDS related to LEN therapy in 39 (98%) of 40 patients. Only one patient developed severe dysplasia and a clonal cytogenetic abnormality diagnostic of MDS. This patient developed MDS 13 months after initiation of LD alone therapy, but was also 3 months status post initiation of salvage therapy with Velcade/dexamethasone. The short latency between LEN initiation and MDS in this patient contrasts with a median of 45.3 months between the diagnosis of MM and MDS/AML reported recently in patients receiving primarily melphalan-based therapy [22]. Although her pretreatment morphologic and cytogenetic evaluations would not have distinguished her from other patients in this study, the short latency suggests that this patient may have had a higher predisposition to develop MDS regardless of therapy for MM. Thus, the role LEN had in the development of MDS in a single patient in this study is unclear.
Although severe dysplasia was not detected in specimens from any patient other than the one who developed MDS, mild morphologic dysplasia was common in MM patients in this study and, in some, was transient. As might be expected, dysplasia was seen most often in heavily pretreated MM patients assigned to the BLD treatment group. However, mild dysplasia was also seen in some of the pretreatment specimens from newly diagnosed MM patients. Mild dysmegakaryopoiesis was most common while mild dyserythropoiesis was only occasionally observed. Dysgranulopoiesis was not found, but a trend was noted for an increased proportion of promyelocytes and myelocytes after 3–4 cycles of LD. This trend is consistent with our previous study, which demonstrated that LEN therapy causes downregulation of the transcription factor PU.1 in maturing granulocytes and leads to a block in maturation [28]. Interestingly, LEN and the related immunomodulatory drug pomalidomide have also recently been found to inhibit maturation of megakaryocytes [29], which may, in part, be contributing to the mild dysmegakaryopoiesis observed in this study. It is worth acknowledging that consistent assessment for morphologic dysplasia is difficult. In a prior study using the same methods for morphologic assessment of myelodysplasia [30], we found that discrepancies between two hematopathologists that required consensus review ranged from 10–30% depending on the cell lineage (data not shown). Although the highest rate of discrepancy was for megakaryocytes, the majority (77%) was between “no dysplasia” and “mild dysplasia”. Recognizing this difficulty and knowing that mild dyspoiesis is not uncommon in MM patients treated with LEN are both important so that undue significance is not assigned to mild morphologic myelodysplasia in these patients.
Cytogenetic abnormalities not attributable to myeloma were found at a low frequency in this study. In addition to the clonal abnormality detected in the specimen diagnostic for MDS, an unusual balanced translocation was detected in one patient from the LD/ASCT treatment group when there was no detectable evidence for MM. The clonal t(2;17)(p23;q11.2) persisted on one subsequent analysis, but was not associated with morphologic dysplasia and was ultimately transient. To our knowledge, this particular translocation pattern has not been previously reported in MDS or other neoplasms. Translocations involving 2p23 (ALK gene rearrangements) are well recognized in lymphoma and inflammatory myofibroblastic tumors [31–34], but breakpoints at 2p23 have only rarely been described in myeloid neoplasms [35]. Rearrangement of 17q11 has occasionally been reported in myeloid neoplasms [36,37] and is a chromosomal band in which several cancer-related genes map, including the NF1 gene. Notably, some cytogenetic abnormalities not directly attributable to MM or related plasma cell disorders that emerge in these patients may be less likely to herald MDS/AML than others. Among melphalan-treated MM patients without clinical signs of MDS, 14–17% of them harbored 5q- when evaluated by FISH studies whereas none had 7q- [38,39]. Nilsson et al. [40] found isolated 20q- in six (5%) MM patients and in two patients (8%) with untreated MGUS/smoldering MM, all of whom lacked clinical or morphologic signs of MDS/AML. Notably, myelodysplasia-type abnormalities after melphalan-based ASCT for MM were reported with an overall frequency of 6% at 10 years and, even though the majority of abnormalities were transient, they were associated with significantly shorter survival [41]. Therefore, there could be longer term clinical significance of the transient clonal abnormality detected in one patient in this study, but there is no feasible way to predict this for an individual patient.
In addition to the clonal t(2;17)(p23;q11.2) that developed in the one patient without MDS, a single metaphase cell from the same specimen expressed a nonclonal t(4;11)(q12;p15). Notably, rearrangements of the NUP98 gene at 11p15 with various partner genes are associated with myeloid malignancies, including therapy-related MDS/AML [17,42–44]. It is important to confirm NUP98 rearrangements by FISH because they are detected in only a subset of patients with translocations involving 11p15. In our study, FISH analysis offered some support for a low level of NUP98 gene rearrangement in a hemodilute aspirate smear from the patient with MDS and clonal t(4;11)(q31;p15), but was not successful in the other patient. Thus, further studies to investigate the role of NUP98 gene rearrangements in MM patients may be of interest.
Of note, our FISH panel for abnormalities associated with MDS [−5/del(5)(q31), −7/del(7)(q31),+8, del(20q)] was not carried out in these patients, as it was not indicated clinically or feasible financially. Most studies have shown that FISH panel studies for targeted MDS-associated abnormalities do not yield a significant rate of abnormalities in the setting of a normal karyotype when ≥20 consecutive well-banded, well-spread metaphases are evaluated by classical cytogenetics [45–50]. However, some investigators have found an increased rate of abnormalities detected by FISH in higher-risk MDS or in association with a higher blast count [51–54]. Thus, the role of FISH in screening for early or emerging MDS currently does not seem sufficiently supported for routine practice. However, no study has systematically investigated this question in myeloma patients. In addition, there are techniques [e.g., array comparative genomic hybridization (aCGH) comprised of combined, targeted and well-distributed oligonucleotides and single nucleotide polymorphisms (SNP) to detect zygosity, deep sequencing analysis, and profiling to detect epigenetic abnormalities) that might detect MDS-associated abnormalities in cases that are normal by classical cytogenetic analysis. However, like other molecular genetic methods that analyze pooled DNA, these techniques may not be as sensitive as the cell-by-cell analysis of FISH studies. A recent study of MDS-related myeloid malignancies showed that, among three methods (classical cytogenetics, FISH for MDS-associated abnormalities, and SNP aCGH), no single method detected defects in all cases that had abnormalities and that together, they increased the detection rate by about 5% [55]. However, the application and feasibility of newer techniques for detecting an early or emerging MDS has not been evaluated thoroughly and was beyond the scope of the present study.
Limitations of this study are the small number of patients in each of the three different treatment groups and short follow-up. The exposure to LEN therapy that patients received was also short. It is reassuring that an overt increase in rapidly emerging MDS was not detected. However, greater numbers of patients on carefully designed clinical trials with longer follow-up are needed. The timing of LEN therapy within a therapeutic sequence and/or a specific combination of therapy with LEN may be important factors because, to date, an increased rate of MDS has been reported in MM patients receiving maintenance LEN after ASCT [2,3] or in newly diagnosed MM patients treated with LEN in combination with melphalan [4]. Newly diagnosed MM patients receiving frontline LEN may not be the ones with an increased risk for MDS/AML [56]. Therefore, it is possible that an increase in MDS was not suggested by our study also because the majority of patients were newly diagnosed and treated upfront with LEN and dexamethasone.
In summary, this longitudinal study provides a detailed description of morphology of hematopoietic precursors and reveals rare cytogenetic findings not attributable to MM among 40 patients treated with LEN for MM. Mild myelodysplasia was more frequent in heavily pretreated MM patients, but was also seen in pretreatment specimens from newly diagnosed MM patients. The low incidence of cytogenetic abnormalities not directly attributable to MM in this study does not raise alarming concern for rapidly emerging MDS in patients treated with LEN. Mild forms of dysplasia and even cytogenetic abnormalities not directly related to myeloma that develop during and after LEN therapy may not necessarily herald MDS in MM patients. However, larger studies with longer follow-up are still needed. Using newer molecular genetic assays, future studies may also be able to identify which MM patients are particularly prone to develop MDS when being treated with specific therapies, possibly including LEN, and may provide information to better tailor therapy for individual patients.
Acknowledgments
The authors are grateful to Mr. Dale Lewis for outstanding technical assistance with the molecular cytogenetic assays, carried out in the University of Pittsburgh Cell Culture and Cytogenetics Facility. This work was supported, in part, by the Multiple Myeloma Research Foundation, the Leukemia & Lymphoma Society, the Pennsylvania Department of Health, and the National Cancer Institute (P30CA047904); funding sources had no role in study design, collection, analysis and interpretation of data, in writing the report or in the decision to submit the paper for publication.
Footnotes
Trials were registered at www.clinicaltrials.gov as #NCT00777881 and #NCT01042704.
Potential Conflicts of Interest
During the period that this study was conducted, Dr. Lentzsch received research funding from Celgene Corporation; Celgene Corporation had no role in study design, collection, analysis and interpretation of data, in writing the report or in the decision to submit the paper for publication. Drs. Monaghan, Dai, Normolle, Gollin, and Mapara have nothing to disclose. The authors declare no conflict of interest.
References
- 1.Attal M, Lauwers-Cances V, Marit G, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366:1782–1791. doi: 10.1056/NEJMoa1114138. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366:1770–1781. doi: 10.1056/NEJMoa1114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attal M, Oliver P, Lauwers-Cances V, et al. Maintenance treatment with lenalidmide after transplantation for myeloma: analysis of secondary malignancies within the IFM 2005–02 trial [abstract] Haematologica. 2011;96(Suppl 1):S23. [Google Scholar]
- 4.Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med. 2012;366:1759–1769. doi: 10.1056/NEJMoa1112704. [DOI] [PubMed] [Google Scholar]
- 5.Dimopoulos MA, Richardson PG, Brandenburg N, et al. A review of second primary malignancy in patients with relapsed or refractory multiple myeloma treated with lenalidomide. Blood. 2012;119:2764–2767. doi: 10.1182/blood-2011-08-373514. [DOI] [PubMed] [Google Scholar]
- 6.Landgren O, Thomas A, Mailankody S. Myeloma and second primary cancers. N Engl J Med. 2011;365:2241–2242. doi: 10.1056/NEJMc1111010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas A, Mailankody S, Korde N, Kristinsson SY, Turesson I, Landgren O. Second malignancies after multiple myeloma: from 1960s to 2010s. Blood. 2012;119:2731–2737. doi: 10.1182/blood-2011-12-381426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117:4701–4705. doi: 10.1182/blood-2010-10-299529. [DOI] [PubMed] [Google Scholar]
- 9.Dai L, O’Sullivan A, Kennedy R, et al. A randomized clinical trial of lenalidomide and dexamethasone with and without autologous stem cell transplant in patients with newly diagnosed mulitple myeloma: interim study results. Blood (ASH Annual Meeting Abstracts) 2011;118:Abstract 4142. [Google Scholar]
- 10.Lentzsch S, O’Sullivan A, Kennedy RC, et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood. 2012;119:4608–4613. doi: 10.1182/blood-2011-12-395715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunning RD, Orazi A, Germing U, et al. Myelodysplastic syndromes/neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4. Lyon, France: IARC Press; 2008. pp. 87–107. [Google Scholar]
- 12.Bain BJ. The bone marrow aspirate of healthy subjects. Br J Haematol. 1996;94:206–209. doi: 10.1046/j.1365-2141.1996.d01-1786.x. [DOI] [PubMed] [Google Scholar]
- 13.Matsuda A, Jinnai I, Yagasaki F, et al. Refractory anemia with severe dysplasia: clinical significance of morphological features in refractory anemia. Leukemia. 1998;12:482–485. doi: 10.1038/sj.leu.2400966. [DOI] [PubMed] [Google Scholar]
- 14.Ramos F, Fernandez-Ferrero S, Suarez D, et al. Myelodysplastic syndrome: a search for minimal diagnostic criteria. Leuk Res. 1999;23:283–290. doi: 10.1016/s0145-2126(98)00166-0. [DOI] [PubMed] [Google Scholar]
- 15.Vardiman JW. Hematopathological concepts and controversies in the diagnosis and classification of myelodysplastic syndromes. Hematology (Am Soc Hematol Educ Program) 2006:199–204. doi: 10.1182/asheducation-2006.1.199. [DOI] [PubMed] [Google Scholar]
- 16.Verburgh E, Achten R, Louw VJ, et al. A new disease categorization of low-grade myelodysplastic syndromes based on the expression of cytopenia and dysplasia in one versus more than one lineage improves on the WHO classification. Leukemia. 2007;21:668–677. doi: 10.1038/sj.leu.2404564. [DOI] [PubMed] [Google Scholar]
- 17.Romana SP, Radford-Weiss I, Ben Abdelali R, et al. NUP98 rearrangements in hematopoietic malignancies: a study of the Groupe Francophone de Cytogenetique Hematologique. Leukemia. 2006;20:696–706. doi: 10.1038/sj.leu.2404130. [DOI] [PubMed] [Google Scholar]
- 18.Breslow NE, Clayton DG. Approximate inference in generalized linear mixed models. Journal of the American Statistical Association. 1993;88:9–25. [Google Scholar]
- 19.Acute leukaemia and other secondary neoplasms in patients treated with conventional chemotherapy for multiple myeloma: a Finnish Leukaemia Group study. Eur J Haematol. 2000;65:123–127. doi: 10.1034/j.1600-0609.2000.90218.x. [DOI] [PubMed] [Google Scholar]
- 20.Cuzick J, Erskine S, Edelman D, Galton DAG. A comparison of the incidence of the myelodysplastic syndrome and acute myeloid leukaemia following melphalan and cyclophosphamide treatment for myelomatosis. A report to the Medical Research Council’s working party on leukaemia in adults. Br J Cancer. 1987;55:523–529. doi: 10.1038/bjc.1987.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dores GM, Cote TR, Travis LB. New malignancies following Hodgkin lymphoma, non-Hodgkin lymphoma and myeloma. In: Curtis RE, Freedman MD, Ron E, et al., editors. New malignancies among cancer survivors: SEER Cancer Registries, 1973–2000. Bethesda, MD: National Cancer Institute; 2006. pp. 397–434. NIH Publ. No. 05–5302. [Google Scholar]
- 22.Mailankody S, Pfeiffer RM, Kristinsson SY, et al. Risk of acute myeloid leukemia and myelodysplastic syndromes after multiple myeloma and its precursor disease (MGUS) Blood. 2011;118:4086–4092. doi: 10.1182/blood-2011-05-355743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergsagel DE, Bailey AJ, Langley GR, MacDonald RN, White DF, Miller AB. The chemotherapy on plasma-cell myeloma and the incidence of acute leukemia. N Engl J Med. 1979;301:743–748. doi: 10.1056/NEJM197910043011402. [DOI] [PubMed] [Google Scholar]
- 24.Govindarajan R, Jagannath S, Flick JT, et al. Preceding standard therapy is the likely cause of MDS after autotransplants for multiple myeloma. Br J Haematol. 1996;95:349–353. doi: 10.1046/j.1365-2141.1996.d01-1891.x. [DOI] [PubMed] [Google Scholar]
- 25.Przepiorka D, Buadi F, McClune B, Franz G, Walsh W, White F. Myelodysplastic syndrome after autologous peripheral blood stem cell transplantation for multiple myeloma. Bone Marrow Transplant. 2007;40:759–764. doi: 10.1038/sj.bmt.1705814. [DOI] [PubMed] [Google Scholar]
- 26.Landgren O, Ma W, Kyle RA, Rajkumar SV, Korde N, Albitar M. Polymorphism of the erythropoietin gene promotor and the development of myelodysplastic syndromes subsequent to multiple myeloma. Leukemia. 2012;26:844–845. doi: 10.1038/leu.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Usmani SZ, Sexton R, Hoering A, et al. Second malignancies in total therapy 2 and 3 for newly diagnosed multiple myeloma: influence of thalidomide and lenalidomide during maintenance. Blood. 2012;120:1597–1600. doi: 10.1182/blood-2012-04-421883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pal R, Monaghan SA, Hassett AC, et al. Immunomodulatory derivatives induce PU. 1 down-regulation, myeloid maturation arrest, and neutropenia. Blood. 2010;115:605–614. doi: 10.1182/blood-2009-05-221077. [DOI] [PubMed] [Google Scholar]
- 29.Liu A, Donnenberg A, Li S, et al. IMiD(R) Immunomodulatory drugs lenalidomide and pomalidomide inhibit the maturation of megakaryocytes by suppressing the expression of GATA1. Blood (ASH Annual Meeting Abstracts) 2011;118:Abstract 1840. [Google Scholar]
- 30.Monaghan SA, Surti U, Doty K, Craig FE. Altered neutrophil maturation patterns that limit identification of myelodysplastic syndromes. Cytometry B Clin Cytom. 2012;82:217–228. doi: 10.1002/cyto.b.21016. [DOI] [PubMed] [Google Scholar]
- 31.Cools J, Wlodarska I, Somers R, et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2002;34:354–362. doi: 10.1002/gcc.10033. [DOI] [PubMed] [Google Scholar]
- 32.De Paepe P, Baens M, van Krieken H, et al. ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood. 2003;102:2638–2641. doi: 10.1182/blood-2003-04-1050. [DOI] [PubMed] [Google Scholar]
- 33.Gascoyne RD, Lamant L, Martin-Subero JI, et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood. 2003;102:2568–2573. doi: 10.1182/blood-2003-03-0786. [DOI] [PubMed] [Google Scholar]
- 34.Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59:2776–2780. [PubMed] [Google Scholar]
- 35.Melnick A, Fruchtman S, Zelent A, et al. Identification of novel chromosomal rearrangements in acute myelogenous leukemia involving loci on chromosome 2p23, 15q22 and 17q21. Leukemia. 1999;13:1534–1538. doi: 10.1038/sj.leu.2401513. [DOI] [PubMed] [Google Scholar]
- 36.Ahmad F, Dalvi R, Mandava S, Das BR. Acute Myelogeneous Leukemia (M0/M1) with novel chromosomal abnormality of t(14;17) (q32; q11. 2) Am J Hematol. 2007;82:676–678. doi: 10.1002/ajh.20846. [DOI] [PubMed] [Google Scholar]
- 37.Haferlach C, Bacher U, Schnittger S, et al. ETV6 rearrangements are recurrent in myeloid malignancies and are frequently associated with other genetic events. Genes Chromosomes Cancer. 2012;51:328–337. doi: 10.1002/gcc.21918. [DOI] [PubMed] [Google Scholar]
- 38.Amiel A, Fridman K, Elis A, et al. Deletion 5q31 in patients with stable, melphalan-treated multiple myeloma. Cancer Genet Cytogenet. 1999;113:45–48. doi: 10.1016/s0165-4608(98)00279-9. [DOI] [PubMed] [Google Scholar]
- 39.Amiel A, Yukla M, Yogev S, Manor Y, Fejgin MD, Lishner M. Deletion of 5q31 and 7q31 in patients with stable melphalan treated multiple myeloma. Cancer Genet Cytogenet. 2004;152:84–87. doi: 10.1016/j.cancergencyto.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 40.Nilsson T, Nilsson L, Lenhoff S, et al. MDS/AML-associated cytogenetic abnormalities in multiple myeloma and monoclonal gammopathy of undetermined significance: evidence for frequent de novo occurrence and multipotent stem cell involvement of del(20q) Genes Chromosomes Cancer. 2004;41:223–231. doi: 10.1002/gcc.20078. [DOI] [PubMed] [Google Scholar]
- 41.Barlogie B, Tricot G, Haessler J, et al. Cytogenetically defined myelodysplasia after melphalan-based autotransplantation for multiple myeloma linked to poor hematopoietic stem-cell mobilization: the Arkansas experience in more than 3,000 patients treated since 1989. Blood. 2008;111:94–100. doi: 10.1182/blood-2007-06-097444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Block AW, Carroll AJ, Hagemeijer A, et al. Rare recurring balanced chromosome abnormalities in therapy-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes Chromosomes Cancer. 2002;33:401–412. doi: 10.1002/gcc.10044. [DOI] [PubMed] [Google Scholar]
- 43.Gough SM, Slape CI, Aplan PD. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood. 2011;118:6247–6257. doi: 10.1182/blood-2011-07-328880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobzev YN, Martinez-Climent J, Lee S, Chen J, Rowley JD. Analysis of translocations that involve the NUP98 gene in patients with 11p15 chromosomal rearrangements. Genes Chromosomes Cancer. 2004;41:339–352. doi: 10.1002/gcc.20092. [DOI] [PubMed] [Google Scholar]
- 45.Beyer V, Castagne C, Muhlematter D, et al. Systematic screening at diagnosis of −5/del(5)(q31), −7, or chromosome 8 aneuploidy by interphase fluorescence in situ hybridization in 110 acute myelocytic leukemia and high-risk myelodysplastic syndrome patients: concordances and discrepancies with conventional cytogenetics. Cancer Genet Cytogenet. 2004;152:29–41. doi: 10.1016/j.cancergencyto.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Cherry AM, Brockman SR, Paternoster SF, et al. Comparison of interphase FISH and metaphase cytogenetics to study myelodysplastic syndrome: an Eastern Cooperative Oncology Group (ECOG) study. Leuk Res. 2003;27:1085–1090. doi: 10.1016/s0145-2126(03)00104-8. [DOI] [PubMed] [Google Scholar]
- 47.Costa D, Valera S, Carrio A, et al. Do we need to do fluorescence in situ hybridization analysis in myelodysplastic syndromes as often as we do? Leuk Res. 2010;34:1437–1441. doi: 10.1016/j.leukres.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 48.Jiang H, Xue Y, Wang Q, et al. The utility of fluorescence in situ hybridization analysis in diagnosing myelodysplastic syndromes is limited to cases with karyotype failure. Leuk Res. 2012;36:448–452. doi: 10.1016/j.leukres.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Ketterling RP, Wyatt WA, VanWier SA, et al. Primary myelodysplastic syndrome with normal cytogenetics: utility of ‘FISH panel testing’ and M-FISH. Leuk Res. 2002;26:235–240. doi: 10.1016/s0145-2126(01)00117-5. [DOI] [PubMed] [Google Scholar]
- 50.Pitchford CW, Hettinga AC, Reichard KK. Fluorescence in situ hybridization testing for −5/5q, −7/7q, +8, and del(20q) in primary myelodysplastic syndrome correlates with conventional cytogenetics in the setting of an adequate study. Am J Clin Pathol. 2010;133:260–264. doi: 10.1309/AJCPZ4JL5ZMRPFTD. [DOI] [PubMed] [Google Scholar]
- 51.Bernasconi P, Cavigliano PM, Boni M, et al. Is FISH a relevant prognostic tool in myelodysplastic syndromes with a normal chromosome pattern on conventional cytogenetics? A study on 57 patients. Leukemia. 2003;17:2107–2112. doi: 10.1038/sj.leu.2403108. [DOI] [PubMed] [Google Scholar]
- 52.Rigolin GM, Bigoni R, Milani R, et al. Clinical importance of interphase cytogenetics detecting occult chromosome lesions in myelodysplastic syndromes with normal karyotype. Leukemia. 2001;15:1841–1847. doi: 10.1038/sj.leu.2402293. [DOI] [PubMed] [Google Scholar]
- 53.Romeo M, de Chauffaille ML, Silva MR, Bahia DM, Kerbauy J. Comparison of cytogenetics with FISH in 40 myelodysplastic syndrome patients. Leuk Res. 2002;26:993–996. doi: 10.1016/s0145-2126(02)00047-4. [DOI] [PubMed] [Google Scholar]
- 54.Yang W, Stotler B, Sevilla DW, et al. FISH analysis in addition to G-band karyotyping: utility in evaluation of myelodysplastic syndromes? Leuk Res. 2010;34:420–425. doi: 10.1016/j.leukres.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 55.Makishima H, Rataul M, Gondek LP, et al. FISH and SNP-A karyotyping in myelodysplastic syndromes: improving cytogenetic detection of del(5q), monosomy 7, del(7q), trisomy 8 and del(20q) Leuk Res. 2010;34:447–453. doi: 10.1016/j.leukres.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossi AC, Mark TM, Jayabalan D, et al. Incidence of second primary malignancies (SPM) after 6-years follow-up of continuous lenalidomide in first-line treatment of multiple myeloma (MM) J Clin Oncol. 2011;29(Suppl):Abstract 8008. [Google Scholar]