

Much of the clinical disability in Charcot-Marie-Tooth disease 1A results from secondary axon loss. Hantke et al. show that the transcription factor c-Jun is upregulated in Schwann cells in a mouse model of the disorder, and that this protects against axon loss. Targeting this axon-supportive mechanism may have therapeutic potential.
Keywords: axonal degeneration, demyelinating disease, neural repair, neuron-glial interaction, neuropathy
Abstract
Charcot–Marie–Tooth disease type 1A is the most frequent inherited peripheral neuropathy. It is generally due to heterozygous inheritance of a partial chromosomal duplication resulting in over-expression of PMP22. A key feature of Charcot–Marie–Tooth disease type 1A is secondary death of axons. Prevention of axonal loss is therefore an important target of clinical intervention. We have previously identified a signalling mechanism that promotes axon survival and prevents neuron death in mechanically injured peripheral nerves. This work suggested that Schwann cells respond to injury by activating/enhancing trophic support for axons through a mechanism that depends on upregulation of the transcription factor c-Jun in Schwann cells, resulting in the sparing of axons that would otherwise die. As c-Jun orchestrates Schwann cell support for distressed neurons after mechanical injury, we have now asked: do Schwann cells also activate a c-Jun dependent neuron-supportive programme in inherited demyelinating disease? We tested this by using the C3 mouse model of Charcot–Marie–Tooth disease type 1A. In line with our previous findings in humans with Charcot–Marie–Tooth disease type 1A, we found that Schwann cell c-Jun was elevated in (uninjured) nerves of C3 mice. We determined the impact of this c-Jun activation by comparing C3 mice with double mutant mice, namely C3 mice in which c-Jun had been conditionally inactivated in Schwann cells (C3/Schwann cell-c-Jun−/− mice), using sensory-motor tests and electrophysiological measurements, and by counting axons in proximal and distal nerves. The results indicate that c-Jun elevation in the Schwann cells of C3 nerves serves to prevent loss of myelinated sensory axons, particularly in distal nerves, improve behavioural symptoms, and preserve F-wave persistence. This suggests that Schwann cells have two contrasting functions in Charcot–Marie–Tooth disease type 1A: on the one hand they are the genetic source of the disease, on the other, they respond to it by mounting a c-Jun-dependent response that significantly reduces its impact. Because axonal death is a central feature of much nerve pathology it will be important to establish whether an axon-supportive Schwann cell response also takes place in other conditions. Amplification of this axon-supportive mechanism constitutes a novel target for clinical intervention that might be useful in Charcot–Marie–Tooth disease type 1A and other neuropathies that involve axon loss.
Introduction
Inherited neuropathies that affect peripheral nerves are among the most common hereditary neurological diseases. The most frequent genetic form of these conditions is Charcot–Marie–Tooth disease type 1A (CMT1A). It is generally caused by heterozygous inheritance of a duplication within an area of chromosome 17p11.2 that contains a gene encoding the 22 kDa protein PMP22, which is expressed by Schwann cells and serves as a minor myelin component. The duplication results in mild elevation of PMP22 levels in compact myelin and a disease characterized by the loss of Schwann cell myelin, weakness of distal muscles and muscle atrophy, sensory and reflex deficiencies, foot deformities and reduced nerve conduction velocity (Scherer and Wrabetz, 2008; Shy et al., 2008).
It is notable that although overexpression of PMP22 in Schwann cells is thought to be the cause of the disease, a key feature of CMT1A is secondary death of axons (Lewis et al., 2003; Suter and Scherer, 2003). Axon death is also seen in many other conditions where nerves are diseased or traumatized: it is common in alternative forms of inherited neuropathies in addition to CMT1A, it is characteristic of the acute motor axonal neuropathy and acute motor-sensory axonal neuropathy form of acquired neuropathies, and it is an important feature of diabetic neuropathy (Hughes and Cornblath, 2005; Said et al., 2008; Scherer and Wrabetz, 2008). Furthermore, there is strong evidence that clinical disabilities associated with different forms of neuropathy, including CMT1A, are to a significant extent due to axon death rather than demyelination. Accordingly, prevention of axonal loss is considered an important target for clinical intervention (Krajewski et al., 2000; Scherer and Wrabetz, 2008).
It is therefore of interest that endogenous signalling mechanisms that potentially promote axon survival and prevent neuron death have been identified within peripheral nerves (Keswani et al., 2004; Arthur-Farraj et al., 2012). One of these mechanisms depends on the activation of the transcription factor c-Jun in Schwann cells of injured nerves (cut or crush) (Parkinson et al., 2008; Arthur-Farraj et al., 2012). In these experiments c-Jun was found to exert broad control over the Schwann cell response to nerve damage, since it directs the conversion of myelin and non-myelin (Remak) cells to repair (Bungner) Schwann cells that are specialized for supporting growth and guidance of regenerating axons. Most relevant in the present context is the observation that mice with selective inactivation of Schwann cell c-Jun (c-Jun-cKO mice) show not only poor regeneration after nerve injury, but also significant neuronal death (Arthur-Farraj et al., 2012; Fontana et al., 2012). This indicates that in damaged nerves, c-Jun activation prompts the expression of Schwann cell factors that support the survival of distressed neurons. Quantitatively this effect is substantial: the number of small dorsal root ganglion and facial motor neurons that die after sciatic or facial nerve injury is 2–3-fold higher in c-Jun-cKO mice than in wild-type mice. Even the large dorsal root ganglion neurons, which do not show significant death after nerve injury in wild-type control mice were reduced in number by 30–40% in c-Jun-cKO animals.
The observation that c-Jun orchestrates Schwann cell support for distressed neurons after mechanical injury raises a significant question. Do Schwann cells also activate a c-Jun dependent programme to rescue neurons and axons in other types of nerve pathology, including inherited demyelinating disease? This possibility is consistent with our finding that Schwann cell c-Jun is elevated in Schwann cells in nerve biopsies from patients with CMT1A (Hutton et al., 2011), and has recently received strong support from observations on Cx32def mice that model CMT1X neuropathy (Klein et al., 2014). In uninjured nerves of these mice, c-Jun and GDNF are co-upregulated in Schwann cells associated with axons with deficient neurofilament phosphorylation, suggesting activation of Schwann cell trophic support for perturbed axons.
Here this hypothesis was tested using the C3 mouse model of CMT1A. The C3 mouse carries three to four copies of the human PMP22 gene and shows intermediate disease severity (Verhamme et al., 2011). We found that Schwann cell c-Jun was elevated in Schwann cells of (uninjured) nerves of C3 mice, in line with previous findings in humans. We determined the impact of this c-Jun activation by comparing C3 mice with double mutant mice, namely C3 mice in which c-Jun has been conditionally inactivated in Schwann cells (C3/c-Jun-cKO mice). To do this we carried out sensory-motor tests and electrophysiological measurements and counted axons in proximal and distal nerves. The results indicate that c-Jun elevation in the Schwann cells of CMT1A nerves serves to improve CMT1A behavioural symptoms, maintain F-wave persistence and prevent loss of axons, particularly in distal nerves. This suggests that Schwann cells have two contrasting functions in CMT1A: on the one hand they are the genetic source of the disease, on the other, they respond to it by mounting a c-Jun-dependent response that significantly reduces its impact. Amplification of this axon-supportive mechanism constitutes a novel target for clinical intervention that might be useful in CMT1A and other neuropathies that involve axon loss.
Materials and methods
Conditional ablation of Schwann cell c-Jun in C3 mice
Animal experiments were approved by the UK Home Office. The C3/c-Jun-cKO line generated here is of mixed C57BL6J and FVB background.
To generate the C3/c-Jun-cKO line we used the C3 mouse (Verhamme et al., 2011), the Junfl/fl mouse (Behrens et al., 2002) and the P0 Cre mouse (Feltri et al., 1999), which expresses Cre recombinase under the control of the P0 promoter. The C3 mouse was crossed twice with the Junfl/fl mouse to generate Junfl/fl C3 mice. The Junfl/fl P0 Cre+ mouse was generated by crossing the P0 Cre+ mouse twice with the Junfl/fl mouse (Parkinson et al., 2008). In a final step the Junfl/fl C3 mice were crossed with Junfl/fl P0 Cre+ mice to generate the C3 Junfl/fl P0 Cre+ (C3/c-Jun-cKO mouse).Three other genotypes also resulted, namely the C3 Junfl/fl P0 Cre− (called C3 mouse), the Junfl/fl P0 Cre− and the Junfl/fl P0 Cre+ (c-Jun-cKO mouse) (Arthur-Farraj et al., 2012) (Supplementary Fig. 1A). Junfl/fl P0 Cre− and Junfl/fl P0 Cre+ mice showed no difference in sensory-motor function (Supplementary Fig. 2). Therefore they were combined into one group (called control) for all experiments involving C3 and C3/c-Jun-cKO mice.
Genotyping of mice
DNA was extracted from ear or tail samples using the HotSHot method (Truett et al., 2000). Primers used for typing the PMP22 transgene were 5’-CTTCAGGCCCTGCACCTC-3’ and 5’-CATTCCGCAGACTTGGATG-3’, for the P0 Cre transgene 5’-GCTGGCCCAAATGTTGCTGG-3’ and 5’-CCACCACCTCTCCATTGCAC-3’ and for the Jun flox locus were 5’-CCGCTAGCACTCACGTTGGTAGGC-3’ and 5’-CTCATACCAGTTCGCACAGGCGGC-3’.
Behavioural tests
Beam walking
Five and 12 mm beams 1 -m long and 20-cm high were used. A score taking both foot slips and beam falls into account was given in accordance with performance: 0 and 1 foot slip = 1; 2 to 5 foot slips = 2; over 5 foot slips or at least 1 beam slip = 3.
Sciatic functional index
Sciatic functional index was measured as described elsewhere (Klapdor et al., 1997; Inserra et al., 1998; Arthur-Farraj et al., 2012).
Hanging wire test
The mouse was placed on a cage lid, which was slowly turned around. Latency for hind limbs to come off the lid for longer than 10 s was timed with a cut-off set at 2 min.
Rotarod test
Mice were trained for five trials using acceleration from 4 to 20 rpm. On the third day three trials were performed accelerating to 40 rpm.
Grid test
The walkway was 1 -m long, 5-cm wide and 25-cm high. For the equidistant grid test the distance between metal bars was 2 cm and for the random grid test random distances of 1, 2 or 3 cm. Hindpaw slips were counted for each trial and analysed.
Von Frey test
The von Frey test was conducted using a dynamic plantar aesthesiometer (Ugo Basile 37450).
Hotplate test
The hotplate (Ugo Basile 7280) was heated to 50°C. The animal was placed on the plate and the latency to respond with either a hindpaw flick, hindpaw lick, or a jump was recorded. Following a response the mouse was removed from the hotplate. The cut-off value for response was set at 2 min to avoid tissue damage.
Electrophysiological measurements
Nerve conduction studies on isoflurane anaesthetized mice used a Viking Quest EMG machine. Compound action potentials were recorded from small foot muscles with subcutaneous needle electrodes and 10 Hz high-pass and 10 kHz low-pass filters. The recording electrode was inserted midway between the metatarsal-phalangeal joints and the ankle. The reference electrode was inserted into the hallux. The tibial nerve was stimulated at the ankle and the sciatic nerve at the sciatic notch using a pair of monopolar needle electrodes. Nerve conduction velocity, distal and proximal CMAP amplitudes and distal motor latency were determined after supramaximal stimulation and minimal F-wave latency and persistence was determined from 20 consecutive stimuli.
Western blotting
After dissection, nerves were snap frozen in liquid nitrogen, extracted and blotted (Arthur-Farraj et al., 2012). Antibodies: mouse anti-c-Jun Ig (BD Biosciences #610326; 1:2500), mouse anti-GAPDH Ig (Abcam #ab8245; 1:5000) and P0 mouse monoclonal antibody (Astexx; 1:1000).
Immunocytochemistry
Schwann cell cultures
Schwann cell cultures were prepared from sciatic nerves and brachial plexus from 7 day-old mice (Dong et al., 1999). For immunocytochemistry cells were fixed with 4% paraformaldehyde (PFA) in PBS for 20 min, then for 10 min in methanol, −20°C. Application of anti-S100 (DAKO A/S; 1:100), was followed by Alexa Fluor® 488 anti-rabbit Ig (Molecular Probes; 1:400). For double immunolabelling, c-Jun mouse monoclonal antibodies (BD Biosciences #610326; 1:500 in antibody diluting solution (ADS)) were subsequently applied overnight at 4°C, followed by donkey anti-mouse Ig Cy3 (Jackson Research Labs; 1:500) and 4',6-diamidino-2-phenylindole (DAPI).
Teased nerves
Nerves were fixed in 4% PFA for 10 min, washed, teased, dried and post-fixed in acetone at −20°C for 10 min (Klein et al., 2014). They were double immunolabelled with rabbit monoclonal antibodies to c-Jun overnight (Cell Signalling; 1:200), anti-rabbit Ig Alexa Fluor® 488 (1:500; 2.5 h), 324 rat anti-mouse Ig L1 antibodies (1:500; 1.5 h), anti-rat Ig Cy3 (1:500; 1 h) and DAPI as above.
Frozen nerve sections
Ten micrometre sciatic or saphenous nerve sections were fixed in acetone, and double labelled sequentially with goat anti-Sox10 antibodies overnight (R&D AF2864; 1:100) to label Schwann cells, anti-goat Ig Alexa Fluor® 488 (1:1000; 1 h), rabbit c-Jun antibodies overnight, anti-rabbit Ig Cy3 (1 h) and DAPI. For macrophage counts nerve sections were fixed in 4% PFA in PBS (10 min), labelled with monoclonal antibodies F4/80 and anti-rat Ig Alexa Fluor® 488 and DAPI as above.
Morphometry
Mice were perfused with 4% PFA in PBS. Tissues were fixed overnight in 1% PFA, 1% glutaraldehyde in 0.1 M PIPES buffer pH 7.6. Toes were decalcified in 5% EDTA in 0.1 M PIPES buffer pH 7.6. Nerves were osmicated in 1% osmium tetroxide and embedded in Agar resin (Agar Scientific).
Ultrathin 70-nm sections were cut and stained with lead citrate. To calculate g-ratios ×3000 electron micrographs were taken (Gatan software) and analysed in ImageJ. In the saphenous nerve, dorsal root and ventral root the number of myelinated fibres and the number of axons >1.5 µm were counted in every field.
ATPase staining
Soleus muscles were dissected from 10-month-old mice, and myofibrillar actomyosin ATPase histochemistry was performed as described (http://neuromuscular.wustl.edu/pathol/histol/atp.htm, Brooke and Kaiser, 1970).
Statistical analysis
For comparisons between control, C3 and C3/c-Jun-cKO animals, one way analysis of variance (one-way ANOVA) was used with the Bonferroni test as a post test for multiple comparisons (Graphpad Prism5) (with P-values designated as: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Comparisons between the two control groups Junf/f and Junf/f P0 cre and between C3 and control mice were conducted using Student’s t-test. Frequency distribution data were analysed using two-way ANOVA with Bonferroni post-test. Graphical data is expressed as means ± SEM.
Results
c-Jun levels are increased in Schwann cells of the C3 mouse
c-Jun protein levels were increased in uninjured peripheral nerves of C3 mice by western blotting. This increase had already occurred at post-natal Day 13 and continued with age. Both predominantly sensory (saphenous) and motor (quadriceps) nerves were affected (Fig. 1A–C). To confirm that c-Jun was elevated in Schwann cells, we carried out double label immunolabelling on sciatic and saphenous nerve sections with c-Jun antibodies and Sox10 antibodies to identify Schwann cells. In both nerves, the number of Schwann cells with detectable nuclear c-Jun in C3 mice was close to double that seen in control nerves. Similarly, in teased sciatic and saphenous nerves from C3 mice, the number of myelin Schwann cells with nuclear c-Jun was about double that found in teased control nerves. The intensity of Schwann cell c-Jun immunoreactivity was also noticeably higher in C3 nerves compared to controls. The general intensity of c-Jun labelling in uninjured C3 nerves was, however, substantially lower than that previously described in injured nerves (Fig. 1D–G). The number of F4/80+macrophages was not significantly different between saphenous nerves or sciatic nerves in C3 and control mice (saphenous: 1.7 ± 0.40 and 1.5 ± 0.56 macrophages/field; sciatic: 3.2 ± 0.84 and 1.8 ± 1.24 macrophages /field; n = 3 in control and C3 nerves, respectively).
Figure 1.

c-Jun levels are elevated in Schwann cells of the C3 mouse. (A–C) Western blots of (uninjured) peripheral nerves from control and C3 mice showing elevated c-Jun protein levels at 6, 8 and 13 months, and post-natal Day 13. (A) Strongly elevated c-Jun levels are seen in the brachial plexus (BP) and femoral nerve (FN) of C3 mice compared to controls (6-month-old mice). (B) In the sciatic nerve (SN) of C3 mice, c-Jun protein levels increase with age (m = months). (C) In C3 mice, elevated c-Jun is already seen in saphenous (SaN) and quadriceps (QN) nerves at post-natal Day 13. (D) Counts showing that in C3 nerves, the number of c-Jun+ Schwann cell nuclei is about double that in control nerves. The numbers for saphenous and sciatic nerves represent the percentage of SOX10+ Schwann cell nuclei that were also c-Jun+ in double immunolabelled transverse sections. The numbers for teased nerves represent the percentage of DAPI labelled nuclei that were also c-Jun+ in myelin Schwann cells identified by phase contrast in teased sciatic and saphenous nerves immunolabelled with L1 antibodies to identify non-myelin (Remak) cells. (E) Transverse sciatic nerve sections double labelled with SOX10 antibodies to identify Schwann cells, and c-Jun antibodies. The images for c-Jun were captured using identical parameters and illustrate the difference in intensity of c-Jun labelling typically seen between C3 and control nerves. In controls, c-Jun is hard to detect in this comparison, although the signal is unambiguous using other imaging settings. Scale bar = 10 µm.
Ablation of Schwann cell c-Jun in the C3 mouse
To investigate the role of elevated c-Jun in CMT1A Schwann cells, we selectively deleted c-Jun in Schwann cells of the C3 mouse by breeding together C3 mice, Junfl/fl mice (Behrens et al., 2002) and P0 Cre mice (Feltri et al., 1999), (see ‘Materials and methods’ section and Supplementary Fig. 1A). This resulted in the generation of a C3 mouse without c-Jun in Schwann cells (C3 Junfl/fl P0Cre+ mouse), referred to as the C3/c-Jun-cKO mouse, and three other genotypes, which served as controls.
We examined the success of this strategy by immunolabelling c-Jun in Schwann cell cultures prepared from post-natal Day 7 sciatic nerves, using S100 as a Schwann cell marker. Nuclear c-Jun was seen in nearly all Schwann cells from Junfl/fl P0 Cre- mice (controls) or C3 mice as expected, whereas c-Jun was essentially absent in cells from Junfl/fl P0 Cre+ and C3/c-Jun-cKO mice (Supplementary Fig. 1B). In parallel experiments, comparable lack of c-Jun was seen in cells from mice with conditional inactivation of c-Jun in Schwann cells only (Junfl/fl P0Cre+ mice) as reported previously (Arthur-Farraj et al., 2012). In confirmatory experiments, nuclear c-Jun was undetectable in cultured Schwann cells from 5-month-old C3/c-Jun-cKO mice, whereas c-Jun expression was retained in other cell types and in Schwann cells from Junfl/fl P0 Cre− controls (Supplementary Fig. 1C).
C3 mice show impaired performance in sensory-motor tests
As a first step towards determining the function of c-Jun in Schwann cells with a genetic modification akin to the human CMT1A duplication, we used a number of behavioural tests to establish a quantitative profile of sensory- motor performance of the C3 mouse compared to controls. The tests included the accelerating rotarod, the hanging wire test to measure grip strength, grid walking on a horizontal ladder to measure paw misplacements, sciatic functional index measurement and beam walking with two beam widths.
In every test, using 1.5-, 3- and 6-month-old mice, the C3 animals showed a clear trend towards impaired performance compared to controls. This reached statistical significance for the Rotarod, grid walking and sciatic functional index in 3-month old-mice, for the hanging wire test in 6-month-old mice, and for 1.5-month-old mice in the narrow beam walking test (Fig. 2 and Supplementary Fig. 3).
Figure 2.

C3 mice show sensory-motor deficits that are amplified in the absence of Schwann cell c-Jun in C3/c-Jun-cKO mice. (A) Rotarod: Using the accelerating rotarod all mouse lines show some decline in performance with age. C3 mice show a trend towards worse performance than controls which reaches significance in 3-month-old mice. Similarly, C3/c-Jun-cKO mice tend to perform worse than C3 mice, reaching significance in 3-month-old mice. (B) Hanging wire test: C3 mice perform significantly worse than controls at 6 months. C3/c-Jun-cKO mice perform significantly worse than C3 mice at 1.5 and 3 months. (C) Grid walking: C3 mice show a trend towards worse performance than controls, which is significant at 3 months. C3/c-Jun-cKO mice perform significantly worse that C3 mice at 3 and 6 months and show a strong trend towards deteriorating performance with age. (D) Sciatic function index: C3 mice tend to perform worse than controls at all ages and this is significant at 1.5 and 3 months. In 6-month-old mice there is a clear trend towards worse performance by C3/c-Jun-cKO mice compared to C3 but this does not reach significance. (E) Beam walking 1.2 cm beam: There is little difference between C3 and control mice. But C3/c-Jun-cKO mice show a strong decline in performance compared to C3 that is significant at 3 and 6 months. (F) Beam walking 0.5 cm beam: C3 mice perform significantly worse than controls at 1.5 months but the deterioration in C3/c-Jun-cKO mice compared to C3 mice is not significant. *P < 0.05, **P < 0.01, ***P < 0.001.
The C3 mouse also showed reduced heat sensitivity compared to controls, shown by increased response time in a hotplate test (Fig. 3A). The von Frey hair test failed to reveal abnormal touch sensation in C3 mice (Fig. 3B).
Figure 3.

Sensory tests and electrophysiological measurement. (A) Hotplate test: both C3 and C3/c-Jun-cKO mice show a prominent sensory deficit in perception of thermal stimuli, which is significantly different from control animals (B) Automatic von Frey: Applying the von Frey test in an automatic version detected no significant differences between the three mouse lines. (C–H) Electrophysiology. The figures obtained in the C3 mouse reveal several deficits compared to control animals that are typical for CMT1A. One parameter, F wave persistence, is normal in C3 mice but significantly reduced in C3/c-Jun-cKO mice. Nerve conduction studies were performed on 3-month-old control, C3 and C3/c-Jun-cKO animals. (C–F) C3 and C3/c-Jun-cKO mice have significantly decreased motor neuron conduction velocity (MNCV), and proximal and distal amplitude, whereas distal motor latency is increased in both mouse lines in comparison to the control. (G) F wave latency was increased in the C3 and C3/c-Jun-cKO mice. (H) C3/c-Jun-cKO mice showed significantly reduced F wave persistence compared to both C3 and control mice. *P < 0.05, **P < 0.01, ***P < 0.001.
C3 nerves have conduction defects
Nerve conduction studies revealed changes typical for inherited neuropathies. In comparison to values for control animals, motor nerve conduction velocities as well as proximal and distal amplitudes of compound muscle action potentials were significantly reduced, whereas distal motor latency values and minimal F-wave latencies increased in line with the reduced conduction velocity of the hindlimb. F-wave persistence, however, was similar in C3 and control mice (Fig. 3). These findings are in agreement with previous data from rodent models of CMT1A and patients with PMP22 duplication (see ‘Discussion’ section).
C3 nerves show dysmyelination in motor nerves and abnormal muscle fibre patterns
As both motor and sensory nerves showed a similar increase in c-Jun protein we first examined mixed nerves by counting myelin sheaths in light micrographs of toluidine blue stained semi-thin sections. In the tibial branch of the sciatic nerve (at the sciatic notch) we found that myelination appeared mostly normal in C3 nerves, although some myelin sheaths were obviously thin. Counting together thin and apparently normal myelin we found that the total number of myelin sheaths was comparable in C3 and control nerves (reduced by a non-significant 8% in C3) (Fig. 4A, and C). We also examined the distal nerves at the base of toe 1. Here we found a significant reduction in C3 compared to control nerves of ∼30% in the number of myelin sheaths. (Fig. 4B and D). Next we carried out separate evaluations of predominantly sensory nerves (dorsal root and saphenous nerve) and motor nerves (ventral root and quadriceps nerve).
Figure 4.

Light microscopy of mixed nerves. (A) There is a trend towards lower numbers of myelinated axons from control to C3 to C3/c-Jun-cKO mice. (B) In the distally located nerves in toe 1, the number of myelinated axons in nerves from C3 and C3/c-Jun-cKO mice is significantly lower than in controls. Removal of Schwann cell c-Jun (the C3/c-Jun-cKO compared to the C3 mouse) does not increase the number of myelin sheaths. (C and D) General appearance of the tibial and toe nerves from control, C3 and C3/c-Jun-cKO mice, as shown in semithin sections. Scale bar = 20 µm. *P < 0.05, **P < 0.01, ***P < 0.001.
In sensory nerves, light microscopy counts showed a drop of ∼18% in the number of myelin sheaths in dorsal roots of C3 mice compared to controls, whereas no significant reduction was seen in the saphenous nerve (Fig. 5A). Electron microscopy of dorsal roots showed that several axons in C3 mice had too thin or too thick myelin, whereas some myelin-competent (i.e. > 1.5 µm) axons were amyelinated (i.e. had no myelin or one or two myelin wraps) (Fig. 5B). This seemed to be dysmyelination rather than demyelination, as there was no evidence of myelin debris, myelin containing macrophages or other signs of myelin breakdown. These observations were confirmed by scatter plots of G-ratios versus axon diameter which showed that the small diameter axons in the C3 mouse often showed a propensity to a thickened myelin sheath, whereas large diameter axons were more frequently hypomyelinated. There was also a reduction in the proportion of larger myelinated axons (Supplementary Fig. 5A).
Figure 5.

Light microscopy of sensory nerves. (A) Counts of myelinated fibres in light microscopy photographs show that C3 mice have small but significant loss of myelin sheaths in dorsal roots but not in saphenous nerves when compared to control nerves. Both nerves showed significant loss of myelin sheaths in C3/c-Jun-cKO compared to C3 mice. (B) Electron micrographs illustrating amyelinated and thickly and thinly myelinated axons. These myelin abnormalities are less pronounced in the saphenous nerve than in the dorsal root. Scale bar = 5 µm. (C) Analysis of myelinated axon diameters in dorsal root and saphenous nerve in control, C3 and C3/c-Jun-cKO mice. *P < 0.05, **P < 0.01, ***P < 0.001.
Using electron microscopy, we counted the total number of myelin competent axons in sensory nerves, dividing them into two categories (i) axons with normal, thick or thin myelin and (ii) amyelinated axons > 1.5 µm (Table 1). The total numbers of myelin-competent axons [category (i) plus category (ii)] in the dorsal root and saphenous nerves were not significantly different from that found in control nerves (reduced by non-significant 5.2% and 8.0%, respectively) (Fig. 5A and Table 1). The size distribution of the diameters of myelinated axons was also similar in C3 and control mice (Fig. 5C). Consistent with the light microscopic observations, fewer of the myelin-competent axons were myelinated in C3 nerves, the incidence of amyelinated axons rising from 1.2% in control dorsal roots to 7.5% in C3 roots and from 0.1% in control saphenous nerves to 0.5% in C3 nerves (Table 1).
Table 1.
In the C3 mouse, lack of c-Jun in Schwann cells results in loss of sensory but not motor axons
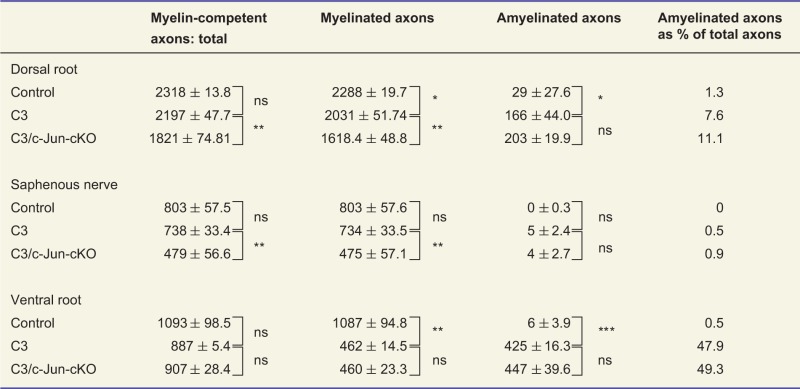 |
Counts of myelinated and amyelinated axons (including axons with less than two layers of myelin) with a diameter >1.5 µm. The counts were carried out on complete transverse sections of the tibial branch of the sciatic nerve from 3-month-old control, C3 and C3/c-Jun-cKO animals. Myelinated and amyelinated axons together make up myelin-competent axons.
*P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
In motor nerves the situation was different, as light microscopic counts of myelin sheaths revealed a substantial reduction in C3 mice compared with controls. This amounted to about 55% and 25%, respectively, in ventral roots and quadriceps nerves (Fig. 6A). Electron microscopy of the ventral root showed that many axons in C3 mice had thin myelin and revealed a large number of amyelinated axons (Fig. 6B). As in sensory nerves and for the same reasons, this is likely to represent dysmyelination. Scatter plots of G-ratios versus axon diameter revealed a similar pattern of change between C3 and control nerves as seen in sensory nerve, namely a reduction in the proportion of larger myelinated axons together with a tendency towards hypermyelination of smaller axons and hypomyelination of larger ones (Supplementary Fig. 5B). Examination of the diameter of myelinated axons in the ventral root also indicated that dysmyelination in C3 mice primarily affected the largest diameter axons (Fig. 6C).
Figure 6.

Light microscopy of motor nerves. (A) Counts of myelinated fibres in light microscopy photographs show that C3 mice have about 50% fewer myelin sheaths in ventral roots and ∼20% fewer sheaths in quadriceps nerves compared to controls. No significant increase in the number of myelin sheaths was seen in C3/c-Jun-cKO nerves compared to C3 nerves. (B) Electron micrographs show the marked dysmyelination in motor nerves of C3 and C3/c-Jun-cKO mice. Scale bar = 5 µm. (C) Analysis of myelinated axon diameters in ventral root and quadriceps nerve in control, C3 and C3/c-Jun-cKO mice. *P < 0.05, **P < 0.01, ***P < 0.001.
We counted the total number of ventral root axons in the electron microscope, using the same two categories as for the sensory nerve counts (Table 1). The total number of myelin-competent axons [category (i) plus category (ii)] in the ventral root was reduced from control by ∼18.7% but this did not reach statistical significance. These counts showed also that as many as 47.9% of axons were amyelinated in the C3 nerves compared to only 0.5% in the controls, in agreement with the counts by light microscopy.
As muscle weakness and wasting are classical symptoms of CMT1A, we looked at muscle pathology in the C3 mouse. Staining of the muscle ATPase at pH 4.3 in soleus muscles revealed the typical checkerboard pattern in control and C3 muscles, although there appeared to be a trend towards greater fibre grouping in C3 muscles (Fig. 7A). Fibre type distribution was also comparable in control and C3 mice (Fig. 7B). There was a trend towards increased average muscle fibre area in C3 muscles (Fig. 7C). This was confirmed by the observation that in C3 muscles there was a significant decrease of fibre numbers in the smaller size category 1000 to 1499 µm2 for type 1 and type 2a/b fibres, whereas larger fibre categories had greater percentages of fibres (Fig. 7D, E and Supplementary Fig. 5).
Figure 7.

Analysis of muscle fibres. (A) Light micrographs of muscles in control, C3 and C3/c-Jun-cKO mice. Both C3 and C3/c-Jun-cKO have immature (2c), angulated (black arrowhead), hypertrophic (black arrow) and atrophied fibres (empty arrow head). Scale bar = 50 µm. (B) Fibre type distribution in control, C3 and C3/c-Jun-cKO animals. (C–E) Area distribution of type 1 and type 2 fibres. *P < 0.05, **P < 0.01, ***P < 0.001.
Together these results document and quantify a set of performance faults and electrophysiological defects in the C3 model of CMT1A. A major contributor to these functional defects is likely to be the extensive dysmyelination of motor axons in the C3 mouse.
Expression of c-Jun in CMT1A Schwann cells improves sensory-motor performance, maintains normal F-wave persistence and prevents loss of sensory fibres in C3 mice
Comparison of C3 mice, in which Schwann cell c-Jun expression is present and elevated, with C3/c-Jun-cKO mice, in which c-Jun is absent, allowed us to determine the impact of c-Jun expression in Schwann cells in this model of CMT1A.
Sensory-motor performance
In sensory-motor tests, we found that C3 mice expressing c-Jun consistently outperformed C3/c-Jun-cKO mice in every test. The difference between the C3 and C3/c-Jun-cKO mice was significant in the rotarod test using 3-month-old mice, in the hanging wire test with 1.5- and 3-month-old mice, in grid walking using 3- and 6-month-old mice, and in the 1.2 cm wide beam walk for 3- and 6-month-old mice (Fig. 2). Moreover, while the difference in the SFI between control and C3 mice was not significant due to large variability, abnormalities of paw placement by C3 mice were clearly visible in the foot prints (Supplementary Fig. 2). No change in heat sensation or von Frey tests was seen in C3 compared to C3/c-Jun-cKO mice (Fig. 3A and B).
Confirming the trend towards impaired sensory-motor function from control to C3 to C3/c-Jun-cKO mice, we found that for mice at every age, and in every test, the difference in performance between control and C3/c-Jun-cKO was statistically significant, with the sole exception of broad beam walking at 1.5 months (Fig. 2).
These results indicate that in CMT1A mice, expression of Schwann cell c-Jun is adaptive, and a significant factor in reducing the sensory-motor deficits caused by PMP22 over-expression.
Maintenance of F-wave persistence
Comparing control and C3 mice we showed that out of the six electrophysiological parameters that were measured, only one, F-wave persistence, remained normal in C3 mice (Fig. 3). We found that this maintenance of normal F-wave persistence depends on Schwann cell c-Jun expression in the C3 mice, as F-wave persistence was strongly reduced by ∼70% in the C3/c-Jun-cKO mouse (Fig. 3F). The other five electrophysiological parameters that were abnormal in C3 mice remained so in the absence of c-Jun (Fig. 3A–E). As in C3 mice, normal F-wave persistence is a feature of human CMT1A.
Prevention of axon loss
Comparison of sensory nerves in control and C3 mice showed that some axons in both C3 dorsal roots and saphenous nerves had too thick or too thin myelin (Fig. 5). Nevertheless, counts in the electron microscope showed that the total number of myelin-competent axons (axons with normal, thick, thin or no myelin but >1.5 µm) was not significantly different in the sensory nerves of C3 mice compared to the same nerves in control mice (Table 1). A different picture was seen in C3/c-Jun-cKO mice. These animals showed substantial loss of myelin-competent axons, which was more extensive in distal nerves (distal axonopathy). Comparing dorsal roots in C3 and C3/c-Jun-cKO mice showed that the number of myelin competent axons was 25.5% (P < 0.01) higher in C3 nerves (that express c-Jun) compared to C3/c-Jun-cKO nerves where c-Jun is absent. And in the more distal saphenous nerve 54.5% (P < 0.01) more axons were found in C3 nerves compared to C3/c-Jun-cKO nerves. Nevertheless, the distribution of the diameters of myelinated axons was similar in the two mouse lines, indicating that axon loss in C3/c-Jun-cKO nerves occurred at all axon diameters (Fig. 5C).
Previously we showed that c-Jun dependent Schwann cell signalling provides survival support to dorsal root ganglion sensory neurons, when nerves are damaged by crushing. Comparable survival support was not found for injured spinal cord motor neurons (Arthur-Farraj et al., 2012). Interestingly, we found the same selectivity, with respect to axon loss, in the C3 mouse, although in this mouse the neurons are distressed by Schwann cell PMP22 over-expression, rather than mechanical damage. This was shown by the fact that the number of spinal cord motor axons in ventral roots, which was normal in C3 mice, remained unchanged in C3/c-Jun-cKO mice, rather than declining. The myelin abnormalities we had seen in C3 ventral roots in comparison with control, including extensive dysmyelination, too thick and too thin myelin sheaths (Fig. 6), did not change significantly in C3/c-Jun-cKO mice. The size distribution of the diameters of myelinated axons was also similar in the two genotypes (Fig. 6C).
These observations indicate that up regulation of Schwann cell c-Jun is a necessary component of an axon-supportive mechanism in this mouse model of CMT1A.
Muscle phenotype
The muscle phenotype was mostly comparable in C3 and C3/c-Jun-cKO mice (Fig. 7). The C3/c-Jun-cKO mouse showed a tendency to increased hypertrophy of muscle fibres of all types (Fig. 7C), which was also manifested in a significant decrease in type 1 fibres in the size group 1500–1999 µm2 whereas larger fibres were over-represented (Fig. 7D).
Discussion
The present experiments show, first, that the transcription factor c-Jun is upregulated in Schwann cells in the C3 mouse model of CMT1A, in line with the presence of Schwann cell c-Jun in human peripheral neuropathy, and Cx32def mice (Hutton et al., 2011; Klein et al., 2014). Second, inactivation of c-Jun results in a substantial worsening of CMT1A-related behavioural deficits and abnormal shortening of F-wave latency, suggesting impairment in the function of spinal cord motor neurons. Third, when Schwann cell c-Jun is inactivated in C3 nerves, there is a striking loss of myelin-competent dorsal root ganglion sensory axons that is more pronounced in distal nerves. From this and our previous work on c-Jun we conclude that activation of Schwann cell c-Jun is an adaptive response to disease that results in axonal protection and the support of sensory-motor performance.
Distal axonopathy is a key feature of human CMT1A and axon death accounts for many of the clinical manifestations (Krajewski et al., 2000). Axon death is also seen in severe mouse models of CMT1A (Sancho et al., 1999) and we found a (non-significant) trend towards reduced axonal number in the C3 mouse. Thus PMP22 over-expression and its consequences in Schwann cells act to distress axons/neurons and adversely affect their function, resulting in axonal death.
Mechanical nerve injury (transection or crush) of wild-type nerves also elevates Schwann cell c-Jun and prevention of c-Jun activation by genetic means, as in the present study, results in substantial axon and neuron loss in injured nerves, malfunctioning Bungner repair cells and regeneration failure. In both C3 mice and cut/crushed nerves, loss of Schwann cell c-Jun affects dorsal root ganglion neurons but not spinal cord motor neurons (Arthur-Farraj et al., 2012). Together these studies suggest that nerve pathology, whether caused by mechanical damage or PMP22 over-expression, activates a common neuron-supportive mechanism. This depends on the activation of Schwann cell c-Jun and results in damage limitation, likely due to trophic signalling from Schwann cells to axons (Arthur-Farraj et al., 2012; Fontana et al., 2012). We suggest that normal axon numbers in the C3 CMT1A nerves are maintained by this c-Jun-dependent axon supportive mechanism. Genetic inactivation of Schwann cell c-Jun removes/reduces trophic Schwann cell support, revealing an underlying distal axonopathy in sensory neurons in the C3 mouse. Conversely, it is possible that the amplification of this mechanism would be of clinical benefit in nerve pathology.
The role of c-Jun in Schwann cells of injured and diseased nerves
Upregulation of c-Jun in Schwann cells of injured nerves is well established (De Felipe and Hunt, 1994; Shy et al., 1996). The functional significance of this started to emerge when it was found that c-Jun suppressed myelin genes, opposed the function of pro-myelin signals such as cyclic AMP elevation and the transcription factor Krox-20 (now known as EGR2), and inhibited myelination in neuron-Schwann cell co-cultures. On this basis, c-Jun was characterized as negative regulator of myelination (Jessen and Mirsky, 2008). Other transcriptional regulators that share some of these functions include NOTCH, SOX2 and PAX3 (Le et al., 2005; Jessen and Mirsky, 2008; Doddrell et al., 2012). More recent work has shown that the ability of c-Jun to suppress myelin genes and drive dedifferentiation of myelin Schwann cells is only one facet of a much broader function. This is to control the genetic reprogramming of Schwann cells in intact nerves to form the repair–supportive Schwann cell phenotype in injured nerves (Arthur-Farraj et al., 2012). This process includes reduction in myelin gene expression and dedifferentiation of myelin cells, but also activation of autophagy for myelin breakdown, recruitment of macrophages, and formation of regeneration tracks (Bands of Bungner). Most importantly for the present work, it also involves the c-Jun mediated upregulation of important trophic factors such as GDNF, artemin and sonic hedgehog and cell surface proteins such as P75NTR, L1, NCAM and N-cadherin. All of these molecules are likely to mediate Schwann cell-axon interactions. In line with this, one of the functions of the Bungner repair cell is to support the survival of injured dorsal root ganglion sensory and facial motor neurons, although not spinal cord motor neurons (Arthur-Farraj et al., 2012; Fontana et al., 2012). c-Jun seems to serve a similar neuron-supportive function in C3 mice, to the extent that removal of Schwann cell c-Jun results in loss of dorsal root ganglion axons in dorsal roots and distal sensory nerves (saphenous), while leaving axon numbers in ventral roots unchanged.
In C3 mice, elevation of Schwann cell c-Jun is not associated with demyelination, as is the case in injured wild-type nerves: dorsal root ganglion axons in C3 mice, with or without c-Jun, largely retain their myelin sheaths. Further, in wild-type mice, a significant number of Schwann cells express low but detectable c-Jun while remaining myelinated, and c-Jun is elevated in myelin Schwann cells of Cx32def mice (present work and Klein et al., 2014). Another disconnection between c-Jun and myelin status is the finding that dysmyelinated axons in C3 mice do not myelinate in C3/c-Jun-cKO mice in which c-Jun has been removed.
The facts that c-Jun can be expressed at detectable levels in cells that do not demyelinate, and that removal of c-Jun does not always promote myelination both argue against a simple view of c-Jun as a negative regulator of myelination. There are a number of plausible explanations for these observations. Perhaps higher c-Jun levels are required to initiate myelin breakdown than are needed to activate expression of neuron-supportive factors. Alternatively, c-Jun-mediated demyelination requires co-activation of other signals that are not present PMP22 over-expressing cells in intact nerves. Dysmyelination in C3 mice may not be due to the action of negative regulators but derive from faults in the drivers of myelination, or negative regulators other than c-Jun may be at play.
In addition to c-Jun dependent support for axons, other mechanisms that are affected by c-Jun could play a part in explaining the differences in axon numbers and sensory-motor performance between C3 and C3/c-Jun-cKO mice. For instance, c-Jun promotes Schwann cell autophagy (unpublished data), and is regulated in the unfolded protein response, processes that might have beneficial effects on Schwann cell and axonal health in C3 mice (Lee and Notterpek, 2013; D`Antonio et al., 2013).
Mouse models of CMT1A
Several rodent models of CMT1A have used random insertion of human or mouse PMP22 transgenes which produce extra PMP22 protein in addition to the endogenously made PMP22. Models with severe disability, such as the C22 mouse (Huxley et al., 1996), the my41 mouse (Robertson et al., 2002) and the TgN 247-249 mice (Magyar et al., 1996) represent the extreme end of the human CMT1A spectrum. In contrast, the intermediate disease severity in the rat model (Sereda et al., 1996), the C61 mouse (Huxley et al., 1998) and the C3 mouse (Verhamme et al., 2011) mirrors human CMT1A more closely.
To control for the effects of removing c-Jun from Schwann cells in the C3 mouse, it was necessary to perform our own characterization of the C3 CMT phenotype. We confirmed and extended the description by Verhamme et al. (2011), including defining a previously undescribed sensory deficit detectable with the hotplate test, and showing that the muscles of the C3 mouse show a tendency towards immature fibre types, a general fibre hypertrophy and characteristic fibre type grouping.
F wave persistence
The electrophysiological defects that we observed in C3 mice are in line with findings in CMT1A rodent models and patients with PMP22 duplication (Kaku et al., 1993; Magyar et al., 1996; Sereda et al., 1996; Birouk et al., 1997; Krajewski et al., 2000; Robaglia-Schlupp et al., 2002; Verhamme et al., 2011).
F-waves provide a functional measure of proximal nerve and anterior horn cells, measuring the ability of motor neurons in the anterior horn to generate and propagate action potentials. F-wave persistence relies on antidromic conduction in the alpha motor neurons and its ‘reflection’ in a small proportion of motor neurons and the orthodromic propagation back to the muscle. It is not observed under physiological conditions, but serves as an electrophysiological diagnostic tool. It has been reported in patients with Guillain-Barré syndrome and in rats with experimental autoimmune neuritis, an animal model of Guillain-Barré syndrome (Kuwabara et al., 2000; Taylor and Pollard, 2003). F-wave persistence is strikingly reduced in C3/c-Jun-cKO mice compared to C3 mice. This indicates reduced conduction or excitability of ventral horn motor neurons. This is not likely to be due to increased demyelination or axonal pathology, as we observed no significant differences in ventral roots of these two lines. Another explanation could be that in C3/c-Jun-cKO mice, death of sensory axons and the resulting loss of sensory input to motor neurons leaves these cells hyperpolarized relative to their counterparts in C3 mice. This would result in reduced excitability likely reflected in reduction in F-wave persistence.
Conclusion
The present paper and previous work suggests that Schwann cells respond to nerve injury or disease by activating/enhancing trophic support for axons through a mechanism that depends on Schwann cell c-Jun (Arthur-Farraj et al., 2012; Fontana et al., 2012). This response results in substantial sparing of axons that would otherwise die. Further work will be needed to test individual steps of this process, including analysing the molecular nature of trophic Schwann cell-axon signalling, learning why PMP22 over-expression in Schwann cells adversely affects axon health, and determining to what extent c-Jun elevation is induced by axonal signals or results from PMP22 over-expression through cell-autonomous mechanisms. Another question is whether the failure to detect c-Jun-dependent Schwann cell support for spinal cord motor axons, in this study and previous work reflects the absence of distress-induced Schwann cell-mediated trophic support for these axons. It is more likely that this mechanism is present, but depends on molecular mechanisms other than c-Jun, in line with other molecular differences between Schwann cells of sensory and motor fibres (Hoke et al., 2006).
Axonal death is a central feature of much nerve pathology, and it will be important to establish whether this adaptive Schwann cell response to promote axon health also takes place in other conditions such as diabetes. Amplification of this response represents a new target for therapy.
Acknowledgements
We thank M.L. Feltri and L. Wrabetz for the gift of P0-Cre mice.
Glossary
Abbreviation
- CMT
Charcot–Marie–Tooth
Funding
This work was funded by Wellcome Trust Programme grants (091119 and 074665) to K.R.J. and R.M. and an MRC project grant (G0600967) to K.R.J. and R.M. Funding was also received from the European Community’s Seventh Framework Program (FP7/2007-2013) under grant agreement No. HEALTH-F2-2008-201535. C3 mice were generated with support of the Netherlands Organization for Scientific Research (NWO) to F.B.
Supplementary material
Supplementary material is available at Brain online.
References
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, et al. c-Jun Reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron. 2012;75:633–47. doi: 10.1016/j.neuron.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, David JP, Mohle-Steinlein U, Tronche F, Schutz G, et al. Impaired postnatal hepatocyte proliferation and liver regeneration in mice lacking c-jun in the liver. EMBO J. 2002;21:1782–90. doi: 10.1093/emboj/21.7.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birouk N, Gouider R, Le Guern E, Gugenheim M, Tardieu S, Maisonobe T, et al. Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain. 1997;120:813–23. doi: 10.1093/brain/120.5.813. [DOI] [PubMed] [Google Scholar]
- Brooke MH, Kaiser KK. Muscle fiber types: how many and what kind? Arch Neurol. 1970;23:369–79. doi: 10.1001/archneur.1970.00480280083010. [DOI] [PubMed] [Google Scholar]
- D'Antonio M, Musner N, Scapin C, Ungaro D, Del Carro U, Ron D, et al. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J Exp Med. 2013;210:821–38. doi: 10.1084/jem.20122005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felipe C, Hunt SP. The differential control of c-jun expression in regenerating sensory neurons and their associated glial cells. J Neurosci. 1994;14:2911–23. doi: 10.1523/JNEUROSCI.14-05-02911.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Sinanan A, Parkinson D, Parmantier E, Mirsky R, Jessen KR. Schwann cell development in embryonic mouse nerves. J Neurosci Res. 1999;56:334–48. doi: 10.1002/(SICI)1097-4547(19990515)56:4<334::AID-JNR2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Doddrell RD, Dun XP, Moate RM, Jessen KR, Mirsky R, Parkinson DB. Regulation of Schwann cell differentiation and proliferation by the Pax-3 transcription factor. Glia. 2012;60:1269–78. doi: 10.1002/glia.22346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999;883:116–23. [PubMed] [Google Scholar]
- Fontana X, Hristova M, Da Costa C, Patodia S, Thei L, Makwana M, et al. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J Cell Biol. 2012;198:127–41. doi: 10.1083/jcb.201205025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoke A, Redett R, Hameed H, Jari R, Li JB, Griffin JW, Brushart TM. Schwann cells express motor and sensory phenotypes that regulate axon regeneration. J Neurosci. 2006;26:9646–9655. doi: 10.1523/JNEUROSCI.1620-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366:1653–66. doi: 10.1016/S0140-6736(05)67665-9. [DOI] [PubMed] [Google Scholar]
- Hutton EJ, Carty L, Laura M, Houlden H, Lunn MP, Brandner S, et al. c-Jun expression in human neuropathies: a pilot study. J Peripher Nerv Syst. 2011;16:295–303. doi: 10.1111/j.1529-8027.2011.00360.x. [DOI] [PubMed] [Google Scholar]
- Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, et al. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet. 1996;5:563–9. doi: 10.1093/hmg/5.5.563. [DOI] [PubMed] [Google Scholar]
- Huxley C, Passage E, Robertson AM, Youl B, Huston S, Manson A, et al. Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum Mol Genet. 1998;7:449–58. doi: 10.1093/hmg/7.3.449. [DOI] [PubMed] [Google Scholar]
- Inserra MM, Bloch DA, Terris DJ. Functional indices for sciatic, peroneal, and posterior tibial nerve lesions in the mouse. Microsurgery. 1998;18:119–24. doi: 10.1002/(sici)1098-2752(1998)18:2<119::aid-micr10>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia. 2008;56:1552–65. doi: 10.1002/glia.20761. [DOI] [PubMed] [Google Scholar]
- Kaku DA, Parry GJ, Malamut R, Lupski JR, Garcia CA. Nerve conduction studies in Charcot-Marie-Tooth polyneuropathy associated with a segmental duplication of chromosome 17. Neurology. 1993;43:1806–8. doi: 10.1212/wnl.43.9.1806. [DOI] [PubMed] [Google Scholar]
- Keswani SC, Buldanlioglu U, Fischer A, Reed N, Polley M, Liang H, et al. A novel endogenous erythropoietin mediated pathway prevents axonal degeneration. Ann Neurol. 2004;56:815–26. doi: 10.1002/ana.20285. [DOI] [PubMed] [Google Scholar]
- Klapdor K, Dulfer BG, Hammann A, Van der Staay FJ. A low-cost method to analyse footprint patterns. J Neurosci Methods. 1997;75:49–54. doi: 10.1016/s0165-0270(97)00042-3. [DOI] [PubMed] [Google Scholar]
- Klein D, Groh J, Wettmarshausen J, Martini R. Nonuniform molecular features of myelinating Schwann cells in models for CMT1: distinct disease patterns are associated with NCAM and c-Jun upregulation. Glia. 2014;62:736–50. doi: 10.1002/glia.22638. [DOI] [PubMed] [Google Scholar]
- Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123(Pt 7):1516–27. doi: 10.1093/brain/123.7.1516. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Ogawara K, Mizobuchi K, Koga M, Mori M, Hattori T, et al. Isolated absence of F waves and proximal axonal dysfunction in Guillain-Barre syndrome with antiganglioside antibodies. J Neurol Neurosurg Psychiatry. 2000;68:191–5. doi: 10.1136/jnnp.68.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci USA. 2005;102:2596–601. doi: 10.1073/pnas.0407836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Notterpek L. Dietary restriction supports peripheral nerve health by enhancing endogenous protein quality control mechanisms. Exp Gerontol. 2013;48:1085–90. doi: 10.1016/j.exger.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RA, Li J, Fuerst DR, Shy ME, Krajewski K. Motor unit number estimate of distal and proximal muscles in Charcot-Marie-Tooth disease. Muscle Nerve. 2003;28:161–7. doi: 10.1002/mus.10419. [DOI] [PubMed] [Google Scholar]
- Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, et al. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J Neurosci. 1996;16:5351–60. doi: 10.1523/JNEUROSCI.16-17-05351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, et al. c-Jun is a negative regulator of myelination. J Cell Biol. 2008;181:625–37. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robaglia-Schlupp A, Pizant J, Norreel JC, Passage E, Saberan-Djoneidi D, Ansaldi JL, et al. PMP22 overexpression causes dysmyelination in mice. Brain. 2002;125:2213–21. doi: 10.1093/brain/awf230. [DOI] [PubMed] [Google Scholar]
- Robertson AM, Perea J, McGuigan A, King RH, Muddle JR, Gabreels-Festen AA, et al. Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J Anat. 2002;200:377–90. doi: 10.1046/j.1469-7580.2002.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said G, Baudoin D, Toyooka K. Sensory loss, pains, motor deficit and axonal regeneration in length-dependent diabetic polyneuropathy. J Neurol. 2008;255:1693–702. doi: 10.1007/s00415-008-0999-z. [DOI] [PubMed] [Google Scholar]
- Sancho S, Magyar JP, Aguzzi A, Suter U. Distal axonopathy in peripheral nerves of PMP22-mutant mice. Brain. 1999;122(Pt 8):1563–77. doi: 10.1093/brain/122.8.1563. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–89. doi: 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sereda M, Griffiths I, Puhlhofer A, Stewart H, Rossner MJ, Zimmerman F, et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–60. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- Shy ME, Chen L, Swan ER, Taube R, Krajewski KM, Herrmann D, et al. Neuropathy progression in Charcot-Marie-Tooth disease type 1A. Neurology. 2008;70:378–83. doi: 10.1212/01.wnl.0000297553.36441.ce. [DOI] [PubMed] [Google Scholar]
- Shy ME, Shi Y, Wrabetz L, Kamholz J, Scherer SS. Axon-Schwann cell interactions regulate the expression of c-jun in Schwann cells. J Neurosci Res. 1996;43:511–25. doi: 10.1002/(SICI)1097-4547(19960301)43:5<511::AID-JNR1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4:714–26. doi: 10.1038/nrn1196. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Pollard JD. Neurophysiological changes in demyelinating and axonal forms of acute experimental autoimmune neuritis in the Lewis rat. Muscle Nerve. 2003;28:344–52. doi: 10.1002/mus.10432. [DOI] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodiumhydroxide and tris (HotSHOT) Biotechniques. 2000;29:52, 54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Verhamme C, King RH, ten Asbroek AL, Muddle JR, Nourallah M, Wolterman R, et al. Myelin and axon pathology in a long-term study of PMP22-overexpressing mice. J Neuropathol Exp Neurol. 2011;70:386–98. doi: 10.1097/NEN.0b013e318217eba0. [DOI] [PubMed] [Google Scholar]