

Leucine-rich, glioma inactivated 1 (LGI1) is a secreted protein linked to seizures of both genetic and autoimmune aetiology. Using conditional mouse knockouts, Boillot et al. show that LGI1 depletion in cortical excitatory neurons, but not inhibitory interneurons, contributes to seizure pathogenesis. LGI1 is essential for proper circuit functioning throughout life.
Keywords: epilepsy, LGI1, ADEAF, genetics, conditional knockout
Abstract
Leucin-rich, glioma inactivated 1 (LGI1) is a secreted protein linked to human seizures of both genetic and autoimmune aetiology. Mutations in the LGI1 gene are responsible for autosomal dominant temporal lobe epilepsy with auditory features, whereas LGI1 autoantibodies are involved in limbic encephalitis, an acquired epileptic disorder associated with cognitive impairment. We and others previously reported that Lgi1-deficient mice have early-onset spontaneous seizures leading to premature death at 2–3 weeks of age. Yet, where and when Lgi1 deficiency causes epilepsy remains unknown. To address these questions, we generated Lgi1 conditional knockout (cKO) mice using a set of universal Cre-driver mouse lines. Selective deletion of Lgi1 was achieved in glutamatergic pyramidal neurons during embryonic (Emx1-Lgi1cKO) or late postnatal (CaMKIIα-Lgi1cKO) developmental stages, or in gamma amino butyric acidergic (GABAergic) parvalbumin interneurons (PV-Lgi1cKO). Emx1-Lgi1cKO mice displayed early-onset and lethal seizures, whereas CaMKIIα-Lgi1cKO mice presented late-onset occasional seizures associated with variable reduced lifespan. In contrast, neither spontaneous seizures nor increased seizure susceptibility to convulsant were observed when Lgi1 was deleted in parvalbumin interneurons. Together, these data showed that LGI1 depletion restricted to pyramidal cells is sufficient to generate seizures, whereas seizure thresholds were unchanged after depletion in gamma amino butyric acidergic parvalbumin interneurons. We suggest that LGI1 secreted from excitatory neurons, but not parvalbumin inhibitory neurons, makes a major contribution to the pathogenesis of LGI1-related epilepsies. Our data further indicate that LGI1 is required from embryogenesis to adulthood to achieve proper circuit functioning.
Introduction
Most genes mutated in monogenic epilepsies encode ion channel subunits, suggesting that familial epilepsies are effectively classed as channelopathies (Reid et al., 2009). One exception is the leucine-rich glioma-inactivated 1 (LGI1) gene, encoding a secreted protein. Mutations in LGI1 have been reported in families with autosomal dominant epilepsy with auditory features (ADEAF) (Kalachikov et al., 2002; Morante-Redolat et al., 2002). ADEAF is a well-defined inherited condition consisting of adolescence/early adulthood-onset lateral temporal seizures. The clinical distinctive feature of this syndrome is the presence of prominent auditory component, either as an aura or as a triggering factor of seizures (Michelucci et al., 2009). One-third of the 39 published ADEAF-causing mutations result in a premature stop codon, which causes haploinsufficiency. Numerous missense mutations prevent LGI1 secretion from transfected cells (Senechal et al., 2005; Sirerol-Piquer et al., 2006; Chabrol et al., 2007; Striano et al., 2008; de Bellescize et al., 2009; Nobile et al., 2009; Di Bonaventura et al., 2011; Leonardi et al., 2011; Fanciulli et al., 2012; Sadleir et al., 2013; Lee et al., 2014). Other pathogenic mutations may impair LGI1 interactions with protein partners rather than suppressing protein secretion (Striano et al., 2011). Overexpression of two different truncating LGI1 mutant proteins in mammalian cells (Schulte et al., 2006) and in transgenic mice (Zhou et al., 2009) point to dominant negative effects. In addition to its role in inherited epilepsies, LGI1 is involved in a subset of patients with acquired autoimmune limbic encephalitis, a neurological disorder of adulthood (Irani et al., 2010; Lai et al., 2010). Patients with autoantibodies directed against LGI1 protein suffer from psychiatric symptoms, including memory loss and confusion, and from epilepsy. Seizures are described as: (i) characteristic faciobrachial dystonic seizures with brief, repeated unilateral motor manifestation; or (ii) mesial temporal lobe seizures in the case of limbic involvement (Irani et al., 2013).
Mechanisms of LGI1-related epilepsies remain unclear (Kegel et al., 2013). Four main functions have been proposed for LGI1 in the CNS: (i) inhibition of the inactivation of the presynaptic voltage-gated potassium channel Kv1.1 (Schulte et al., 2006); (ii) potentiation of AMPA receptor-mediated synaptic transmission in the hippocampus through interaction with ADAM22/23 (‘a disintegrin and metalloprotease’ domain) transmembrane proteins (Fukata et al., 2006, 2010; Ohkawa et al., 2013); (iii) enhancement of neuronal growth on myelin-based inhibitory substrates by interactions with Nogo receptor 1 (Thomas et al., 2010); and (iv) postnatal developmental maturation of glutamatergic transmission, in hippocampal dentate gyrus through pre- and postsynaptic functional maturation, pruning of spines and dendritic branches, and in thalamus through axonal pruning (Zhou et al., 2009, 2012). Remarkably, all animal models of Lgi1-deficiency display spontaneous seizures: Lgi1−/− knockout (KO) mice (Chabrol et al., 2010; Fukata et al., 2010; Yu et al., 2010), Lgi1-mutant rats (Baulac et al., 2012) and lgi1a zebrafish knockdown (Teng et al., 2010). Homozygous Lgi1−/− mice have spontaneous seizures with onset at postnatal Day 10 and all pups die at 2–3 weeks of age, while heterozygous Lgi1+/− mice are more susceptible to sound-induced (Chabrol et al., 2010) or pentylenetetrazole-induced seizures (Fukata et al., 2010). Two studies based on records of hippocampal miniature excitatory postsynaptic currents (mEPSCs) in Lgi1−/− brain slices suggested effects of LGI1 on glutamatergic synapses, though with opposite conclusions: Yu et al. (2010) showed an increased frequency of mEPSCs in Lgi1−/− mice, whereas Fukata et al. (2010) found a reduced amplitude of mEPSCs mediated by AMPA receptors. A specific role for LGI1 in glutamatergic circuits is intriguing as Lgi1 mRNA and protein are expressed in both glutamatergic and gamma amino butyric acidergic (GABAergic) neurons, as well as glial cells, of multiple brain regions (Kalachikov et al., 2002; Senechal et al., 2005; Head et al., 2007; Silva et al., 2011; Ohkawa et al., 2013).
This study was based on conditional knockout (cKO) mice permitting a brain region-, cell type-, and time-restricted deletion of Lgi1. In this way, we first aimed to determine whether spontaneous seizures of Lgi1−/− mice could be reproduced by a selective deficiency of Lgi1 in cortical glutamatergic neurons. Secondly, we aimed to ask whether loss of Lgi1, beyond the early neurodevelopmental period, can also trigger seizures later in life. Independent Lgi1 cKO mice strains were generated using a set of universal Cre-driver mouse lines (Emx1-Cre and CaMKIIα-Cre) targeting cortical neuronal populations: (i) Emx1-Cre targets forebrain neural progenitor cells, the precursors of neocortical and hippocampal glutamatergic neurons at embryonic stage E10.5 (Gorski et al., 2002); and (ii) CaMKIIα-Cre targets neocortical and hippocampal glutamatergic neurons at late-onset postnatal stage (from 5 weeks) (Zeng et al., 2001). We also investigated whether GABAergic cortical interneurons might be involved in seizure generation using PV-Cre mice, which targets parvalbumin (PV) interneurons from the embryonic stage (Hippenmeyer et al., 2005).
Materials and methods
Generation of Lgi1 conditional knockout mice
Three universal Cre-driver mouse lines were selected to excise the floxed allele of Lgi1: Emx1-Cre (JAX # 005628), allowing embryonic glutamatergic forebrain-specific deletion of Lgi1 (Gorski et al., 2002); CaMKIIα-Cre (also reported as CW2-Cre, generated in Tonegawa's laboratory, RIKEN-MIT Neuroscience Research Center, Massachusetts Institute of Technology, Cambridge, MA, USA), allowing postnatal glutamatergic forebrain-specific deletion of Lgi1 (Zeng et al., 2001; Anderson et al., 2005) and PV-Cre (JAX #008069), allowing embryonic deletion of Lgi1 in parvalbumin interneurons (Hippenmeyer et al., 2005). Lgi1 floxed (Lgi1fl/fl) mice and heterozygous KO mice (Lgi1+/−) were previously generated (Chabrol et al., 2010). All mouse lines were maintained on the C57BL/6J genetic background. Mice were housed in groups of four to six littermates/cage with food and water ad libitum and kept in a 21 ± 1°C and 12-h light/dark cycle under specific pathogen-free conditions at the ICM animal core facility and at the Nouvelle Animalerie Commune (Pitié-Salpêtrière, Paris). The Animal Ethics Committee approved all experiments. Animals were treated according to the guidelines of the European Community (authorization number 75-1622). All efforts were made to minimize the number of animals used in the study and their suffering.
Genotyping
All mice were genotyped by PCR at the genotyping and sequencing ICM platform. Genomic DNA was extracted from tail biopsies of mice. Lgi1 wild-type, floxed and null alleles were analysed by PCR amplification using the following primer pairs: 5′-ACATTTCCTTAGTGCCCCTGTTT-3′/5′-CCTCTTAGCCACTGAGGCATCT-3′ (wild-type and floxed alleles) and 5′-ATTTCCTTAGTGCCCCTGTTTTTA-3′/5′-TGTCTGGATTCAATGCTGTCTTAGA-3′ (null allele). A 120-bp band was observed for the wild-type allele, a 160-bp band for the floxed allele, and a 200-bp band for the null allele. Genotyping regularly detected germinal knockout mice in the offspring of Emx1-Cre breedings due to expression of Cre recombinase in germline cells. Germinal knockout mice were systematically excluded from the study. Primers and PCR conditions recommended by the Jackson Laboratory were used to screen for the presence of each Cre transgene. Age-matched littermates of the genotypes of interest were used for all subsequent analyses.
Western blot
Conditional knockout mice and their littermate controls were decapitated and their whole brains were rapidly removed. Cortex, hippocampus and cerebellum were dissected and lysed in 2.5 M urea, 2.5% SDS, 50 mM Tris, 30 mM NaCl buffer. Total protein concentrations were determined by the BCA Protein Assay Kit (Pierce). Twenty micrograms of protein for each sample was separated on 10% Bis–Tris polyacrylamide gels (Novex, Invitrogen). Western blot analyses were performed using the following antibodies: rabbit polyclonal anti-LGI1 antibody (1/500, ab30868, Abcam) and rabbit polyclonal anti-α-actin antibody (1/1000, A2066, Sigma-Aldrich). Quantification of LGI1 expression was done using MultiGauge densitometry software and normalized with actin. Data are reported as mean ± standard error of the mean (SEM).
Histochemistry
Conditional knockout mice and their littermate controls were intraperitoneally injected with a lethal dose of sodium pentobarbital, and then perfused with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Brains were removed, post-fixed in the same fixative for 24 h at 4°C and then paraffin embedded. Paraffin sections of 7-µm thickness were cut. For all experiments, at least two littermate mice of each genotype were processed simultaneously. Antibodies used were rabbit polyclonal anti-LGI1 antibodies (ab30868, ab69491, ab67319; Abcam) and goat polyclonal anti-LGI1 antibodies (sc-9581, sc-9583; Santa Cruz). Nissl staining was used to reveal neuronal cytoarchitecture.
Mouse lifespan
Mice were housed in groups of four to six same-sex littermates under specific pathogen-free conditions. They were observed and weighed daily, but were otherwise left undisturbed until they died. Survival was assessed in male and female mice. Kaplan–Meier survival curves were made using known birth and death dates. Data are reported as mean ± SEM. Mice that died spontaneously were not used for other experiments.
Seizure analysis by SeizureScan
SeizureScan software is designed to automatically identify seizures of rodents in a home cage, providing a non-invasive tool to screen for epileptic phenotype (CleverSys Inc). Mice were placed individually in their home cage and habituated for 30 min before recording. Recurrent video recordings of Emx1-Lgi1cKO (n = 4), CaMKIIα-Lgi1cKO (n = 5) and PV-Lgi1cKO (n = 6) mice and their control littermates (n = 11) were analysed by SeizureScan software. In collaboration with CleverSys, settings fitted to juvenile or adult animals were developed and used for automatic detection of behavioural seizures. A systematic manual control of the results was performed. Data are reported as mean ± SEM.
Animal surgical procedure
This work was performed at Rennes University and followed the European Community Council Directive of 24 November 1986 (86/609/EEC). Conditional knockout mice and their littermate controls were anaesthetized by an intraperitoneal injection of a chloral hydrate solution in NaCl (4%, 10 ml/kg), and then placed in a stereotaxic frame in a flat skull position. Bipolar depth electrodes (homemade polyester insulated stainless steel electrodes) were implanted in both hippocampi (coordinates from bregma: anteroposterior = −2.0 mm, mediolateral = ±1.5 mm, dorsoventral = −1.9 mm) and monopolar surface electrodes over both motor cortex (M1). A reference electrode was placed over the cerebellum.
Intracranial video-EEG recordings
EEG recordings were obtained from Emx1-Lgi1cKO (n = 2), CaMKIIα-Lgi1cKO (n = 7), PV-Lgi1cKO (n = 2) and littermate controls (n = 12) mice using a video-EEG system (Deltamed, 2048 Hz). Recording sessions always started at the same time of the day to minimize diurnal variation. Mice were placed in a transparent cage, positioned in a Faraday cage, first habituated for 30 min and then recorded. EEG from Emx1-Lgi1cKO mice were recorded immediately after complete recovery from surgery, and EEG records were obtained for 4–6 h/day over 2 days, until animal death. EEG from CaMKIIα-Lgi1cKO mice were recorded during >9 months on a weekly basis. EEG from PV-Lgi1cKO mice were recorded over a period of up to 2.5 months on a weekly basis. To verify location of depth electrodes after the experiment, brains were removed and sliced (20-µm thickness) with a cryostat. Data are reported as mean ± SEM.
Pentylenetetrazole administration
Three groups of 2-month-old mice were injected with a 53 mg/kg dose of pentylenetetrazole (Sigma-Aldrich): PV-Lgi1cKO mice (n = 11), PV-Lgi1+/− mice (n = 18) and PV-Lgi1+/+ (n = 20). Pentylenetetrazole was dissolved in saline solution. Behavioural response to pentylenetetrazole was followed 30 min after the injection to assess severity of seizures visually scored to four stages as previously reported (Harai et al., 2012). Mice were observed by two investigators blinded to their genotype. Distribution of maximal score of seizure severity was assessed using the χ2 test. Latency between pentylenetetrazole injection and the occurrence of the first generalized seizure was assessed using the t-test. At the end of the experiments, all animals were euthanized. Data are reported as mean ± SEM.
Results
Conditional deletion of Lgi1 in forebrain pyramidal neurons
We examined the effects of conditional Lgi1 deletion in forebrain glutamatergic pyramidal cells at two different developmental stages by using two standard Cre transgenic mouse lines. We used previously generated homozygous Lgi1-floxed (Lgi1fl/fl) and heterozygous Lgi1 knockout mice (Lgi1+/−) (Chabrol et al., 2010). Lgi1fl/fl and Lgi1+/− mice are fertile, viable and display neither gross structural or histological brain abnormalities nor spontaneous seizures (data not shown). Figure 1 shows the breeding cascade performed to generate distinct mouse lines on a Lgi1fl/− heterozygous knockout background: Lgi1fl/−; Emx1-Cre+/− designated as Emx1-Lgi1cKO, and Lgi1fl/−; CaMKIIα-Cre+/− designated as CaMKIIα-Lgi1cKO (Fig. 1). Littermates carrying the alternative genotypes (Lgi1fl/+; Cre+/− designated as Lgi1c/+), (Lgi1fl/−; Cre−/− designated as Lgi1+/−) and (Lgi1fl/+; Cre−/− designated as Lgi1+/+) were used as controls. In Emx1-Cre (males and females) derived mouse strain, expected genotypes were not recovered in Mendelian ratios due to recurrent ectopic recombination of the floxed Lgi1 allele. At birth, all conditional knockout and littermate control mice were viable with no gross anatomical abnormalities.
Figure 1.

Breeding schemes of the generation of Lgi1cKO mice. Two sequential crosses were made: 1/ breeding Emx1-Cre, CaMKIIα-Cre or PV-Cre mice with Lgi1+/− mice resulted in Lgi1+/−, Emx1-Cre+/−; Lgi1+/−, CaMKIIα-Cre+/− and Lgi1+/−, PV-Cre+/− animals, 2/ these mice were then crossed with Lgi1fl/fl mice to generate experimental genotypes. Results representative of PCR genotyping from Lgi1+/+ and Lgi1c/+ mice (wild-type Lgi1: 120-bp band, floxed Lgi1: 160-bp band), Lgi1+/− and Lgi1cKO mice (floxed Lgi1: 160-bp band, null Lgi1: 200-bp band) are shown.
LGI1 protein expression from conditional knockout mice and their respective littermate controls was assessed by Western blot of hippocampus, neocortex (data not shown) and cerebellum lysates. Immunoblots showed the intensity of the LGI1 64 kDa band in the hippocampus of Emx1-Lgi1cKO mice (n = 2) was greatly reduced compared to controls, whereas it was still expressed in the cerebellum (Fig. 2A). In adult CaMKIIα-Lgi1cKO mice, immunoblots of hippocampus lysates revealed LGI1 expression was reduced to 47 ± 3% at 9 weeks (n = 2) and 37 ± 12% at 11 months (n = 2) after birth compared to CaMKIIα-Lgi1+/− mice (Fig. 2B and C). However, we noted that the expression of LGI1 varied between animals of the same age, which may reflect a variable efficiency of the deletion. As expected, LGI1 expression was similar in the cerebellum of CaMKIIα-Lgi1cKO and CaMKIIα-Lgi1+/− mice up to 11 months of age (Fig. 2B and C). These data demonstrate a successful recombination of the floxed Lgi1 allele in the hippocampus of Emx1-Lgi1cKO and CaMKIIα-Lgi1cKO mice resulting in a specific deletion. Because five commercial LGI1 antibodies (ab30868, ab69491, ab67319, sc-9581 and sc-9583) gave non-specific staining by immunohistochemistry of brain tissue from germinal Lgi1−/−, we could not determine the neuronal populations of the Cre-mediated deletion (Supplementary Fig. 1). Nevertheless, Emx1-Cre and CaMKIIα-Cre mouse strains have been extensively used and proven to be cell-type specific (Tsien et al., 1996; Madisen et al., 2010; Liang et al., 2012) (Allen Brain Institute, mouse brain connectivity section: http://connectivity.brain-map.org).
Figure 2.

Specific deletion of Lgi1 in forebrain glutamatergic neurons. Representative Western blot cropped around LGI1 and actin bands showing (A) a severely reduced LGI1 expression in the hippocampus but not cerebellum of two Emx1-Lgi1cKO mice aged postnatal Day 18, (B) a reduction of LGI1 expression in the hippocampus of two CaMKIIα-Lgi1cKO mice aged 9 weeks and 11 months. LGI1 level was not reduced in the cerebellum of CaMKIIα-Lgi1cKO mice. (C) Quantification of LGI1 protein levels by densitometry of Western blots and normalized with actin for loading. Bar graph with error bars indicating SEM shows mean values of LGI1 band intensity normalized to actin.
Embryonic loss of LGI1 in forebrain glutamatergic neurons is sufficient to generate epileptic seizures
First, we examined gross brain morphology by Nissl staining of coronal and sagittal brain sections of Emx1-Lgi1cKO mice at post-natal Day 19. No major abnormalities of cortical and hippocampal organization and lamination were detected, suggesting that Lgi1 deletion in embryonic forebrain cells does not affect neuronal migration (Supplementary Fig. 2A–D).
We assessed seizure activity by using two complementary methods: (i) a non-invasive video-based approach that automatically recognizes seizure behavioural manifestations (SeizureScan software) which was used to detect the occurrence of seizures and their frequency; and (ii) intracranial video-EEG monitoring to assess electrographic features of the seizures. First, we performed video monitoring coupled to SeizureScan analysis in Emx1-Lgi1cKO mice (n = 4) from the age of postnatal Day 13 up to a maximum of postnatal Day 22, until animals death. We detected recurrent spontaneous behavioural seizures in 100% of Emx1-Lgi1cKO mice. Age at first seizure ranged from postnatal Days 16 to 21 (Table 1). Typically, seizures consisted of stereotyped behavioural sequences of repeated forelimb clonic jerks and a hypertonic neck and tail, generally followed by loss of postural equilibrium and clonic jerks of the head and all limbs (Supplementary Video 1). In some cases, seizures continued with myoclonic jerks and hyperkinetic running or hypertonic posture of the trunk, limbs and tail. During the postictal period, mice were immobile for several seconds. Secondly, video-EEG recordings were monitored to characterize the electrographic epileptic activity. Multiple depth electrodes were implanted in two Emx1-Lgi1cKO mice (at postnatal Days 21 and 26). EEG recordings were limited to 4–6 h/day as a maximum as pups were not weaned, and were obtained during 2 days, until animal death. During spontaneous seizures, ictal electrographic activities were apparent in both hippocampal and cortical electrodes (Fig. 3A). Typically, a high-amplitude spike and wave (5–6 mV/1–2 Hz) generally initiated a seizure in the hippocampus, followed by low voltage fast activities (70–100 Hz), as illustrated in the time-frequency representation (Fig. 3A). Spike discharges and bursts of polyspikes of increasing amplitude in hippocampus and cortex finally ended the seizures, which were followed by a flattening of the EEG activity for 43.3 ± 15 s. The majority of seizures were recorded earlier in the hippocampus than in the cortex (Fig. 3). However, in one seizure, epileptic activities were detected in the cortex before the hippocampus (Supplementary Fig. 3).
Table 1.
Summary of SeizureScan and video-EEG findings and characteristics of spontaneous seizures
| Genotype | Number of mice with seizures (total n) |
% of mice with seizures* | Number of seizures recorded* | Age range of recordings (days)* | Age range at seizure onset (days) | Frequency range of seizures (/h)* | Mean seizure duration (s)** | |
|---|---|---|---|---|---|---|---|---|
| EEG | SeizureScan | |||||||
| Emx1-Lgi1cKO | 2 (2) | 4 (4) | 100 | 18 | 13–27 | 16–21*** | 0.34–1.79 | 31.7 ± 2.9 |
| CaMKIIα-Lgi1cKO | 4 (7) | 3 (5) | 58 | 41 | 46–330 | 94–221* | 0.07–1.4 | 67.7 ± 8.3 |
| PV-Lgi1cKO | 0 (2) | 0 (6) | 0 | 0 | 30–160 | NA | NA | NA |
| Controls | 0 (12) | 0 (11) | 0 | 0 | 13–248 | NA | NA | NA |
*Data combined from SeizureScan and video-EEG.
**Data from video-EEG.
***Data from SeizureScan.
Figure 3.

Spontaneous epileptic seizures and premature death in Emx1-Lgi1cKO mice. (A) Top: Cortical and hippocampal EEG records show a spontaneous seizure from an Emx1-Lgi1cKO mouse aged postnatal Day 27. Monopolar electrodes were implanted in both cortices (EEG traces: Cx left and right) and bipolar electrodes were implanted in both hippocampi (EEG traces: Hip left 1/2, Hip right 1/2). The seizure started with a high-amplitude spike and wave (arrow indicates onset of seizure) and terminated abruptly with pronounced postictal flattening (arrow indicates end of seizure). Behavioural modifications were correlated with EEG changes: 1 = behavioural arrest; 2 = repeated clonic jerks of all limbs and hypertonic neck; 3 = postictal immobility. Enlarged panel: Expanded hippocampal EEG trace showing low voltage fast activities, corresponding to the frequencies with the highest powers in the spectrogram. The time frequency representation was obtained with a wavelet transform of the EEG signal. (B) Epileptic activity was not generated by age-matched controls (n = 4). Representative EEG traces from one cortical and one hippocampal electrode in age-matched control mouse. (C) Kaplan-Meier survival curves of Emx1-Lgi1cKO mice (n = 17) and littermate controls (n = 18) from birth until postnatal Day 32 showing early premature death. Half of Emx1-Lgi1cKO mice died before postnatal Day 20.
Six Emx1-Lgi1cKO mice were investigated for the epileptic phenotype with 18 total seizures recorded. SeizureScan and EEG records provided concordant data that are summarized in Table 1. All mice had seizures, indicating a complete penetrance of the epileptic phenotype. They had frequent, recurrent and severe spontaneous seizures with an age at onset comprised between postnatal Days 16 and 21. The mean seizure frequency was 0.76 ± 0.23/h and the mean duration was 31.7 ± 2.9 s. Emx1-Lgi1cKO mice died within 3 days after seizure onset, presumably due to cardiac or respiratory arrest during the postictal period accompanied by a flat EEG. We also found several mice (n = 4) dead in their cages, in a hypertonic posture. Age-matched controls never showed spontaneous behavioural or electrographic seizures (n = 7 by SeizureScan, n = 4 by EEG) (Fig. 3B).
Lifespan study revealed that all Emx1-Lgi1cKO mice died prematurely, within the first month of age. The majority of mice (15/17) died before postnatal Day 23 but two mice survived up to the age of postnatal Days 28 and 32. The average lifetime was 20 ± 0.9 days according to Kaplan-Meier survival curves (Fig. 3C). Control littermates always survived beyond this period.
Postnatal loss of LGI1 in forebrain glutamatergic neurons leads to occasional epileptic seizures
We examined the consequences of LGI1 depletion induced postnatally in forebrain pyramidal neurons by using the CaMKIIα-Cre transgenic mouse line. CaMKIIα-Lgi1cKO mice were viable with no overt physiological or behavioural phenotypes. No gross morphological abnormalities were evident in Nissl stained whole-brain sections from 9-month-old CaMKIIα-Lgi1cKO mice (Supplementary Fig. 2E–H).
We asked whether spontaneous seizures emerged in CaMKIIα-Lgi1cKO mice using non-invasive video records of behaviour coupled to SeizureScan analysis. Video monitoring was performed twice a week (up to 24 continuous hours) from adult CaMKIIα-Lgi1cKO (n = 5) over a period of 5.5 months (2.5- to 8-month-old mice). We detected spontaneous behavioural seizures in three of five CaMKIIα-Lgi1cKO mice with an age at onset of 3.5 to 5 months (postnatal Days 110-150). Seizures occurred both during sleep (12 seizures) or waking (10 seizures) periods. Typically, behavioural manifestations of seizures started with a hypertonia of the neck and tail, followed by repeated clonic jerks, restricted to forelimbs, sometimes unilaterally, and accompanied by loss of posture (Supplementary Video 2). A brief immobility was observed during the postictal period. In rare seizures, the behavioural manifestations were subtle, consisting only of hypertonia of the neck and brief forelimb and head clonic jerks. Epileptic activity was confirmed from intracranial video-EEG recordings. Intracranial electrodes were stereotaxically implanted in adult CaMKIIα-Lgi1cKO mice (n = 7). Simultaneous video-EEG records of 4 h/week duration were acquired during a period of up to 9 months (mice aged 1.5- to 11-month-old). Occasional behavioural and electrographic seizures (19 seizures total) were recorded from the hippocampus and cortex of both hemispheres of four of seven CaMKIIα-Lgi1cKO mice with an age at onset ranging from 3 to 7 months (postnatal Days 94–221) (Fig. 4A). Typically, seizure onset in the hippocampus was marked by high-amplitude hippocampal spikes or polyspikes (4–7 mV, 8–15 Hz), resembling interictal activity and followed by low voltage fast activities (50–70 Hz). Then, a rhythmic activity of progressively increasing amplitude appeared, which secondarily spread to involve the cortex (Fig. 4A). Electrographic seizures terminated with a decrease in spike frequency and amplitude, and then a postictal EEG flattening period of 7.5 ± 3.2 s. EEG epileptic activity was never detected in three littermate controls (Fig. 4B). In addition to seizures, hippocampal interictal spikes were common in records from epileptic CaMKIIα-Lgi1cKO mice, but never detected in control animals. The number of spikes ranged from ∼100 to ∼1200/h during ictal periods whereas it was ∼ 50/h in records without seizures. We also detected interictal-like spikes in video-EEG records from one CaMKIIα-Lgi1cKO mouse where no seizure was recorded (∼200 spikes/h at age 7 months) (Fig. 4C).
Figure 4.


Late-onset epileptic phenotype and reduced lifespan in CaMKIIα-Lgi1cKO mice. (A) Top: Cortical and hippocampal EEG records of a spontaneous seizure of a 4-month-old CaMKIIα-Lgi1cKO mouse. Monopolar electrodes were implanted in both cortices (EEG traces: Cx left and right) and bipolar electrodes were implanted in both hippocampi (EEG traces: Hip left 1/2, Hip right 1/2). The seizure started with hippocampal polyspikes (Hip right 2, arrow indicates onset) and terminated with brief postictal flattening. Behavioural modifications were correlated with EEG changes: 1 = behavioural arrest; 2 = agitation, gyration; 3 = repeated clonic jerks of head and forelimbs with a hypertonic neck and tail; 4 = postictal immobility. Enlarged panel: Expanded EEG traces of the seizure onset show low voltage fast activities in the right hippocampus, followed by spike discharges of increasing amplitude, which begin earlier in the hippocampus than in the cortex. (B) Epileptic activities were never seen in age-matched controls (n = 3), as illustrated by the representative EEG traces from a CaMKIIα-Lgi1+/− littermate control mouse. (C) Hippocampal ‘interictal-like’ spikes recorded in a CaMKIIα-Lgi1cKO mouse. The mean frequency of spikes recorded at this age (postnatal Day 221) was ∼200/h. Spikes are boxed. (D) Kaplan-Meier survival curves of CaMKIIα-Lgi1cKO mice (n = 25) and littermate control mice (n = 25) from postnatal Day 0 until Day 220. 60% of CaMKIIα-Lgi1cKO mice died in the first seven postnatal months.
Twelve CaMKIIα-Lgi1cKO mice were investigated by both SeizureScan and video-EEG over 2000 hours, and seizures (total of 41) detected in half of them (Table 1). While electrographic manifestations of seizures and their duration (mean 67.7 ± 8.3 s) were similar between animals, their frequency (mean = 0.36 ± 0.1/h, range: 0.07–1.4/h), age at onset (3 to 7 months) and timing of occurrence (from 3 to 7–8 months), assessed by both EEG and SeizureScan varied greatly, possibly due to a variable timing of Lgi1 deletion mediated by CaMKIIα-Cre mouse. Overall, seizures of both Emx1-Lgi1cKO and CaMKIIα-Lgi1cKO mice began with a similar behavioural sequence but were clearly milder in CaMKIIα-Lgi1cKO mice in which hyperkinetic running or hypertonic postures were never observed and postictal flat EEG period was shorter.
Beside the epileptic phenotype, CaMKIIα-Lgi1cKO mice had a shorter lifespan than littermate controls: during the time of the experiment (up to 7 months), 60% of CaMKIIα-Lgi1cKO mice died at various ages (mean lifetime of 193 ± 13.8 days) (Fig. 4D). Death may have been caused by respiratory or cardiac arrest occurring after a seizure during the postictal period of EEG flattening. However, in contrast to Emx1-Lgi1cKO mice, some CaMKIIα-Lgi1cKO mice survived up to 90 days after seizures onset. All littermate controls survived throughout the period.
Parvalbumin interneurons have no major role in the generation of seizures
The expression of LGI1 protein in inhibitory interneurons has recently been confirmed using human serum containing LGI1 autoantibodies, which overcome the poor specificity of commercial antibodies (Ohkawa et al., 2013). Moreover, LGI1 has been suggested to play an important role in the development of cortical interneurons (Friocourt and Parnavelas, 2011). To determine a possible involvement of inhibitory neurons in the epileptic phenotype of Lgi1-deficient mice, we made a genetic deletion of Lgi1 from parvalbumin GABAergic interneurons using a PV-Cre mouse line (Hippenmeyer et al., 2005), which has been shown to be specific to parvalbumin interneurons using a reporter mouse strain (Yi et al., 2014). Animals were generated from two sequential crosses to produce PV-Lgi1cKO mice (Lgi1fl/−; PV-Cre+/−) and littermate controls (Fig. 1). There was no difference in body weight or gross brain morphology (Supplementary Fig. 2I–L) between PV-Lgi1cKO mice and their littermate controls. Moreover, over 8 months of experiments, lifespan was preserved in PV-Lgi1cKO mice.
We next sought for spontaneous seizures in PV-Lgi1cKO mice by SeizureScan and video-EEG monitoring (Table 1). SeizureScan recordings were acquired for 16 h/week (light and dark monitoring) from PV-Lgi1cKO mice aged 1 to 5 months (n = 6). Video-EEG recordings were collected over 4 h/week in two PV-Lgi1cKO mice aged 2 to 4.5 months. Neither observation, nor SeizureScan and EEG data revealed spontaneous seizures or interictal spikes in PV-Lgi1cKO mice over a total period of >1000 h of recordings.
Next, we searched for a more subtle hyperexcitable phenotype by investigating the susceptibility of PV-Lgi1cKO mice to GABAA receptor antagonist pentylenetetrazole-induced seizures. We compared the severity of seizures induced by a 53 mg/kg intraperitoneal injection of pentylenetetrazole in 2-month-old PV-Lgi1cKO, PV-Lgi1+/− and PV-Lgi1+/+ mice. As expected with this pentylenetetrazole dose, all mice exhibited at least myoclonic body jerks and forelimb clonic jerks, corresponding to score 1 according to Harai et al. (2012). There were no significant differences in the global distribution of seizure severity scores between PV-Lgi1cKO and PV-Lgi1+/− mice (PV-Lgi1+/−: score 1: 17.6%, score 2: 70.6%, score 3: 0%, score 4: 11.8%; PV-Lgi1cKO: score 1: 9.1%, score 2: 81.8%, score 3: 0%, score 4: 9.1%; χ2 test P-value = 0.78; Fig. 5). We did not observe significant differences in the latency between pentylenetetrazole injection and the occurrence of the first generalized seizure between PV-Lgi1cKO and PV-Lgi1+/− mice (PV-Lgi1+/−: 246.3 ± 34.3 s; PV-Lgi1cKO: 267.6 ± 70.4 s; t-test P-value = 0.77). However, in contrast to previous work (Fukata et al., 2010), no significant difference in the severity of response to pentylenetetrazole-induced seizures was detected between Lgi1+/+ and Lgi1+/− mice. Possibly the result depended on different experimental conditions (pentylenetetrazole dose, age of mice, or genetic background).
Figure 5.
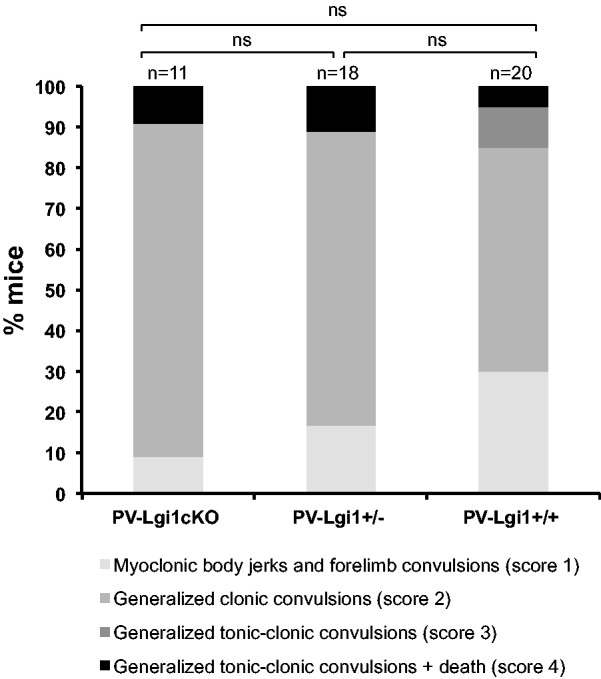
Loss of LGI1 in parvalbumin interneurons does not trigger seizures. Pentylenetetrazole-injection induced seizures of similar severity in PV-Lgi1cKO mice compared to PV-Lgi1+/− mice. Quantification of maximal reaction to pentylenetetrazole injection (53 mg/kg) in 2-month-old PV-Lgi1cKO (n = 11), PV-Lgi1+/− (n = 18) and PV-Lgi1+/+ (n = 20) mice: score 1, myoclonic body jerks and forelimb convulsions; score 2, generalized clonic convulsions; score 3, generalized tonic-clonic convulsions; score 4, generalized tonic-clonic convulsions followed by death. ns = non-significant.
Discussion
LGI1 is a key protein for CNS function as its loss, either due to genetic haploinsufficiency or acquired antibody-mediated depletion, causes epilepsy. However, mechanisms of abnormal brain excitability are not clear. Here we used conditional knockout mice with restricted spatiotemporal and cell-type deletion of Lgi1. These animals let us define contributions of glutamatergic pyramidal neurons and GABAergic interneurons to seizure generation, and ask whether the role of LGI1 in seizure generation is purely neurodevelopmental or whether depletion in adult can also trigger seizures.
Glutamatergic neuron-targeted loss of Lgi1 causes a severe epileptic phenotype
Several studies suggested that LGI1 may regulate glutamatergic transmission (Fukata et al., 2006, 2010; Zhou et al., 2009, 2012; Yu et al., 2010). We attempted to validate this hypothesis in vivo, using two conditional knockout mouse lines (Emx1-Lgi1cKO and CaMKIIα-Lgi1cKO) to achieve a selective deletion of Lgi1 in glutamatergic pyramidal neurons of the neocortex and hippocampus. Our results revealed that both conditional knockout mice presented spontaneous seizures. The epileptic phenotype of Emx1-Lgi1cKO mice, consisting of early-onset, frequent seizures associated with premature death, was reminiscent of that of germinal Lgi1−/− mice (Chabrol et al., 2010). Behavioural manifestations and electrographic pattern of seizures of germinal Lgi1−/− and Emx1-Lgi1cKO mice were very similar, suggesting an identical sequential involvement of different brain areas. We noticed that ictal epileptic activities in the hippocampus of Emx1-Lgi1cKO mice preceded discharges from the motor cortex in most seizures, suggesting seizures in mice originate in mesial temporal structures. Although some patients with ADEAF describe psychic (‘déjà-vu’) and autonomous (epigastric sensations) symptoms, characteristic of mesial temporal lobe auras (Morante-Redolat et al., 2002; Winawer et al., 2002; Ottman et al., 2004), the human syndrome rather implicates lateral temporal lobe. We envisage two explanations for the discrepancy: (i) seizures might originate in lateral temporal structures, but have passed undetected with no electrodes in these areas; (ii) different structures might be involved in mice and in humans. In contrast, restricting deletion of Lgi1 to GABAergic parvalbumin interneurons did not trigger spontaneous seizures or confer increased susceptibility to pentylenetetrazole-induced seizures. Parvalbumin-positive cells account for about half of cortical interneurons (Wonders and Anderson, 2006). They innervate perisomatic regions on pyramidal cells of hippocampus and neocortex and are therefore suited to control the output of these principal cells (Freund and Katona, 2007). Recent evidence points to a direct role for parvalbumin interneurons in the pathogenesis of some genetic epilepsies, in particular Dravet syndrome, a severe epileptic encephalopathy caused by de novo mutations in the SCN1A gene encoding the voltage-gated sodium alpha 1 subunit channel (Ogiwara et al., 2007; Dutton et al., 2012; Rossignol et al., 2013). Although these results do not exclude a contribution of GABAergic interneurons to the genesis of seizures in Lgi1-deficient mice, they do suggest it may be relatively weak. Instead, similarities between the epileptic phenotype of Emx1-Lgi1cKO mice and that of the germinal Lgi1−/−, and the lack of seizures in PV-Lgi1cKO mice suggest that glutamatergic neurons are the main contributors to the pathogenesis of LGI1-related epilepsy. This study also demonstrates that deletion of Lgi1 in the forebrain alone suffices to generate spontaneous seizures in mice.
LGI1 displays an essential role in brain during the whole life
Postnatal deletion of Lgi1 in CaMKIIα-Lgi1cKO mice caused a milder phenotype, with infrequent and late-onset electroclinical spontaneous seizures associated with a variable reduction in lifespan. Long-term EEG recordings will help better characterize the dynamics of the seizure emergence in these mice. Nevertheless, our data imply that LGI1 depletion induces seizures after main synaptic and neuronal developmental processes have matured properly. Our findings are reminiscent of the human acquired autoimmune encephalitis with adult onset, supporting a direct link between LGI1 loss of function due to LGI1 autoantibodies and limbic encephalitis (Ohkawa et al., 2013). We therefore conclude that LGI1 is critical from perinatal through late postnatal development. However, our results clearly indicate that the phenotype of Emx1-Lgi1cKO mice is more severe than that of CaMKIIα-Lgi1cKO mice in terms of behavioural manifestations, seizure frequency (0.76 versus 0.36 seizures/h), duration of postictal period and EEG flattening (43.3 versus 7.5 s), and latency between seizure onset and death (3 versus 90 days). This may result from an enhanced vulnerability of immature brain to seizures (Ben-Ari and Holmes, 2006). Yet, the more severe phenotype associated with the earlier LGI1 deletion is also consistent with the postnatal neurodevelopmental role of LGI1 in transgenic mice expressing a truncating mutant form of LGI1 that was shown to act as a dominant-negative (Zhou et al., 2009, 2012). However, in this conditional loss-of-function mouse model, seizures also arose during adult life, indicating LGI1 function is not limited to early postnatal development processes. LGI1, like reelin, a secreted protein of the extracellular matrix, could serve different functions during brain development and adulthood. During embryonic development, reelin contributes to a correct lamination of cortical and hippocampal regions, while at postnatal and adult stages, it modulates synaptic plasticity and dendritic growth (Campo et al., 2009). The importance of LGI1 in adulthood is also consistent with a role of LGI1 helping circuits respond to seizures by recruiting Kv4.2 potassium voltage-gated ion channel to the membrane as a seizure dampening mechanism (Smith et al., 2012).
Novel insight into the function of LGI1
Various intracellular and extracellular functions have been attributed to LGI1. It remains unclear whether secreted and/or cytoplasmic LGI1 protein is involved in the circuit hyperexcitability that triggers seizures. The identity of neurons that synthesize and secrete LGI1 and the binding sites for secreted LGI1 also remain to be fully characterized. Single-CA1 neuron genetic invalidation by stereotaxic injection of siRNA against Lgi1 could help clarify potential cell-autonomous effects. Our results show that a lack of LGI1 synthesis by glutamatergic neurons can trigger seizures even while secretion from interneurons and glial cells was presumably maintained. This lack of compensation from different cell types might support a cytoplasmic function for LGI1, or possibly suggests a local, rather paracrine action. These findings may limit the search for the effective pro-epileptic site of mutant LGI1 to pre- or extra-synaptic regions at glutamatergic synapses.
Our data emphasize that cortical excitatory neurons, rather than inhibitory interneurons, contribute to the pathogenesis of LGI1-related epilepsy. We also show that LGI1 is an essential protein to maintain normal brain function throughout life. Loss of LGI1 during late postnatal period leads to seizures pointing to a role in adult regulation of neuronal excitability as well as a function in early developmental processes.
Acknowledgements
The authors thank Magali Dumont and Doriane Foret for their technical support at the ICM animal facility and rodent behaviour core facility, Christelle Enond for technical support at the Nouvelle Animalerie Commune, Alberto Bacci for sharing the PV-Cre mice, Vikrant N. Kobla for technical support with SeizureScan, Khalid Hamid El Hachimi for expertise in neuropathology and Eric Noé and Mélanie Morin-Brureau for helpful comments on the manuscript.
Glossary
Abbreviations
- ADEAF
autosomal dominant epilepsy with auditory features
- cKO
conditional knockout
Funding
The ICM animal facility is supported by the ‘Fondation pour la Recherche Médicale’, ‘Institut du Cerveau et de la Moelle épinière’ and ‘Institut Hospitalo-Universitaire’. This study was funded by the Agence Nationale de la Recherche (ANR R10193DD, ANR R11174DD to S.B.), the program ‘Investissements d’avenir’ ANR-10-IAIHU-06, Institut Carnot, Fondation pour la Recherche sur le Cerveau to S.B., National Institute of Health (R01NS057444, R01NS081916, and R21 MH100868 to M.P.A.), the Nancy Lurie Marks Family Foundation (M.P.A.), Autism Speaks/NAAR (M.P.A.), and advanced ERC (322721 to R.M.). While completing this work, S.B. received funding from Fondation Française pour la Recherche sur l'Epilepsie.
Supplementary material
Supplementary material is available at Brain online.
References
- Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, et al. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci USA. 2005;102:1743–8. doi: 10.1073/pnas.0409644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S, Ishida S, Mashimo T, Boillot M, Fumoto N, Kuwamura M, et al. A rat model for LGI1-related epilepsies. Hum Mol Genet. 2012;21:3546–57. doi: 10.1093/hmg/dds184. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055–63. doi: 10.1016/S1474-4422(06)70626-3. [DOI] [PubMed] [Google Scholar]
- Campo CG, Sinagra M, Verrier D, Manzoni OJ, Chavis P. Reelin secreted by GABAergic neurons regulates glutamate receptor homeostasis. PLoS One. 2009;4:e5505. doi: 10.1371/journal.pone.0005505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrol E, Navarro V, Provenzano G, Cohen I, Dinocourt C, Rivaud-Pechoux S, et al. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain. 2010;133:2749–62. doi: 10.1093/brain/awq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrol E, Popescu C, Gourfinkel-An I, Trouillard O, Depienne C, Senechal K, et al. Two novel epilepsy-linked mutations leading to a loss of function of LGI1. Arch Neurol. 2007;64:217–22. doi: 10.1001/archneur.64.2.217. [DOI] [PubMed] [Google Scholar]
- de Bellescize J, Boutry N, Chabrol E, Andre-Obadia N, Arzimanoglou A, Leguern E, et al. A novel three base-pair LGI1 deletion leading to loss of function in a family with autosomal dominant lateral temporal epilepsy and migraine-like episodes. Epilepsy Res. 2009;85:118–22. doi: 10.1016/j.eplepsyres.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Di Bonaventura C, Operto FF, Busolin G, Egeo G, D'Aniello A, Vitello L, et al. Low penetrance and effect on protein secretion of LGI1 mutations causing autosomal dominant lateral temporal epilepsy. Epilepsia. 2011;52:1258–64. doi: 10.1111/j.1528-1167.2011.03071.x. [DOI] [PubMed] [Google Scholar]
- Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, et al. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. 2012;49C:211–20. doi: 10.1016/j.nbd.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanciulli M, Santulli L, Errichiello L, Barozzi C, Tomasi L, Rigon L, et al. LGI1 microdeletion in autosomal dominant lateral temporal epilepsy. Neurology. 2012;78:1299–303. doi: 10.1212/WNL.0b013e3182518328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Parnavelas JG. Identification of Arx targets unveils new candidates for controlling cortical interneuron migration and differentiation. Front Cell Neurosci. 2011;5:28. doi: 10.3389/fncel.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–5. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci USA. 2010;107:3799–804. doi: 10.1073/pnas.0914537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–14. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harai T, Inoue R, Fujita Y, Tanaka A, Horio M, Hashimoto K, et al. Decreased susceptibility to seizures induced by pentylenetetrazole in serine racemase knockout mice. Epilepsy Res. 2012;102:180–7. doi: 10.1016/j.eplepsyres.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Head K, Gong S, Joseph S, Wang C, Burkhardt T, Rossi MR, et al. Defining the expression pattern of the LGI1 gene in BAC transgenic mice. Mamm Genome. 2007;18:328–37. doi: 10.1007/s00335-007-9024-6. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, et al. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain. 2010;133:2734–48. doi: 10.1093/brain/awq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani SR, Stagg CJ, Schott JM, Rosenthal CR, Schneider SA, Pettingill P, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain. 2013;136:3151–62. doi: 10.1093/brain/awt212. [DOI] [PubMed] [Google Scholar]
- Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–41. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel L, Aunin E, Meijer D, Bermingham JR. LGI proteins in the nervous system. ASN Neurol. 2013;5:167–81. doi: 10.1042/AN20120095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci R, Pasini E, Nobile C. Lateral temporal lobe epilepsies: clinical and genetic features. Epilepsia. 2009;50(Suppl 5):52–4. doi: 10.1111/j.1528-1167.2009.02122.x. [DOI] [PubMed] [Google Scholar]
- Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, Saenz A, Poza JJ, Galan J, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119–28. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]
- Lai M, Huijbers MGM, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9:776–85. doi: 10.1016/S1474-4422(10)70137-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Kim SW, Lee JH, Cho YJ, Kim DE, Lee BI, et al. A newly discovered LGI1 mutation in Korean family with autosomal dominant lateral temporal lobe epilepsy. Seizure. 2014;23:69–73. doi: 10.1016/j.seizure.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Leonardi E, Andreazza S, Vanin S, Busolin G, Nobile C, Tosatto SCE. A computational model of the LGI1 protein suggests a common binding site for ADAM proteins. PLoS One. 2011;6:e18142. doi: 10.1371/journal.pone.0018142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Hippenmeyer S, Ghashghaei HT. A Nestin-cre transgenic mouse is insufficient for recombination in early embryonic neural progenitors. Biol Open. 2012;1:1200–3. doi: 10.1242/bio.20122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile C, Michelucci R, Andreazza S, Pasini E, Tosatto SC, Striano P. LGI1 mutations in autosomal dominant and sporadic lateral temporal epilepsy. Hum Mutat. 2009;30:530–6. doi: 10.1002/humu.20925. [DOI] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. 2013;33:18161–74. doi: 10.1523/JNEUROSCI.3506-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman R, Winawer MR, Kalachikov S, Barker-Cummings C, Gilliam TC, Pedley TA, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology. 2004;62:1120–6. doi: 10.1212/01.wnl.0000120098.39231.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87:41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Rossignol E, Kruglikov I, van den Maagdenberg AM, Rudy B, Fishell G. CaV 2.1 ablation in cortical interneurons selectively impairs fast-spiking basket cells and causes generalized seizures. Ann Neurol. 2013;74:209–22. doi: 10.1002/ana.23913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadleir LG, Agher D, Chabrol E, Elkouby L, Leguern E, Paterson SJ, et al. Seizure semiology in autosomal dominant epilepsy with auditory features, due to novel LGI1 mutations. Epilepsy Res. 2013;107:311–7. doi: 10.1016/j.eplepsyres.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klocker N, Sailer CA, Bildl W, Biniossek M, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–20. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- Silva J, Wang G, Cowell JK. The temporal and spatial expression pattern of the LGI1 epilepsy predisposition gene during mouse embryonic cranial development. BMC Neurosci. 2011;12:43. doi: 10.1186/1471-2202-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirerol-Piquer MS, Ayerdi-Izquierdo A, Morante-Redolat JM, Herranz-Perez V, Favell K, Barker PA, et al. The epilepsy gene LGI1 encodes a secreted glycoprotein that binds to the cell surface. Hum Mol Genet. 2006;15:3436–45. doi: 10.1093/hmg/ddl421. [DOI] [PubMed] [Google Scholar]
- Smith SE, Xu L, Kasten MR, Anderson MP. Mutant LGI1 inhibits seizure-induced trafficking of Kv4.2 potassium channels. J Neurochem. 2012;120:611–21. doi: 10.1111/j.1471-4159.2011.07605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striano P, Busolin G, Santulli L, Leonardi E, Coppola A, Vitiello L, et al. Familial temporal lobe epilepsy with psychic auras associated with a novel LGI1 mutation. Neurology. 2011;76:1173–6. doi: 10.1212/WNL.0b013e318212ab2e. [DOI] [PubMed] [Google Scholar]
- Striano P, de Falco A, Diani E, Bovo G, Furlan S, Vitiello L, et al. A novel loss-of-function LGI1 mutation linked to autosomal dominant lateral temporal epilepsy. Arch Neurol. 2008;65:939–42. doi: 10.1001/archneur.65.7.939. [DOI] [PubMed] [Google Scholar]
- Teng Y, Xie X, Walker S, Rempala G, Kozlowski DJ, Mumm JS, et al. Knockdown of zebrafish Lgi1a results in abnormal development, brain defects and a seizure-like behavioral phenotype. Hum Mol Genet. 2010;19:4409–20. doi: 10.1093/hmg/ddq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Favell K, Morante-Redolat J, Pool M, Kent C, Wright M, et al. LGI1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci. 2010;30:6607–12. doi: 10.1523/JNEUROSCI.5147-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, et al. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–26. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Winawer MR, Martinelli Boneschi F, Barker-Cummings C, Lee JH, Liu J, Mekios C, et al. Four new families with autosomal dominant partial epilepsy with auditory features: clinical description and linkage to chromosome 10q24. Epilepsia. 2002;43:60–7. doi: 10.1046/j.1528-1157.2002.45001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7:687–96. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Yi F, Ball J, Stoll KE, Satpute VC, Mitchell SM, Pauli JL, et al. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: roles in cellular excitability, inhibitory transmission and cognition. J Physiol. 2014;592:3463–94. doi: 10.1113/jphysiol.2014.275453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YE, Wen L, Silva J, Li Z, Head K, Sossey-Alaoui K, et al. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet. 2010;19:1702–11. doi: 10.1093/hmg/ddq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, et al. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–29. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med. 2009;15:1208–14. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YD, Zhang D, Ozkaynak E, Wang X, Kasper EM, Leguern E, et al. Epilepsy gene LGI1 regulates postnatal developmental remodeling of retinogeniculate synapses. J Neurosci. 2012;32:903–10. doi: 10.1523/JNEUROSCI.5191-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]