

Abstract
Advances in genetic engineering have made it possible to generate human T-cell products that carry desired functionalities, such as the ability to recognize cancer cells. The currently used strategies for the generation of gene-modified T-cell products lead to highly differentiated cells within the infusion product, and on the basis of data obtained in preclinical models, this is likely to impact the efficacy of these products. We set out to develop a good manufacturing practice (GMP) protocol that yields T-cell receptor (TCR) gene-modified T-cells with more favorable properties for clinical application. Here, we show the robust clinical-scale production of human peripheral blood T-cells with an early memory phenotype that express a MART-1-specific TCR. By combining selection and stimulation using anti-CD3/CD28 beads for retroviral transduction, followed by expansion in the presence of IL-7 and IL-15, production of a well-defined clinical-scale TCR gene-modified T-cell product could be achieved. A major fraction of the T-cells generated in this fashion were shown to coexpress CD62L and CD45RA, and express CD27 and CD28, indicating a central memory or memory stemlike phenotype. Furthermore, these cells produced IFNγ, TNFα, and IL-2 and displayed cytolytic activity against target cells expressing the relevant antigen. The T-cell products manufactured by this robust and validated GMP production process are now undergoing testing in a phase I/IIa clinical trial in HLA-A*02:01 MART-1-positive advanced stage melanoma patients. To our knowledge, this is the first clinical trial protocol in which the combination of IL-7 and IL-15 has been applied for the generation of gene-modified T-cell products.
Introduction
Ex vivo modification of T-cells with genes encoding T-cell receptors (TCR) has proven an attractive strategy for the induction of tumor-specific immune responses against defined antigens. Following early proof-of-concept studies in preclinical mouse models (Morris et al., 2005; de Witte et al., 2006), several groups have now shown the feasibility and clinical activity of TCR gene therapy in clinical trials. In the first of these trials, targeting the melanoma antigen MART-1, the clinical response rate was relatively low (∼13%) (Morgan et al., 2006). However, response rates have been considerably higher in subsequent trials, in particular with the use of a high-affinity NY-ESO-1-specific TCR (Robbins et al., 2011). In addition, T-cell products modified with chimeric antigen receptors (CARs) have been utilized to target cell surface antigens on human cancers. To date, clinical trials targeting CAIX, CD19, CD20, and GD2 have been performed (Jensen et al., 2010; Kochenderfer et al., 2010; Lamers et al., 2011; Louis et al., 2011), and recent data using CD19 CAR-modified T-cells have been particularly promising, showing strong clinical activity in patients with chronic lymphocytic leukemia and acute lymphocytic leukemia (Porter et al., 2011; Brentjens et al., 2013; Grupp et al., 2013).
Over the past years, there has been increasing evidence to suggest that long-term engraftment of infused T-cells is a prerequisite for successful adoptive cell therapy (ACT). Several clinical studies have demonstrated a clear correlation between tumor regression and persistence of tumor-specific cells in blood for weeks or more after infusion (Yee et al., 2002; Robbins et al., 2004). In addition, infusion of less differentiated cells has shown to result in an increased capacity for long-term maintenance and increased antitumor activity in preclinical models, and has been associated with longer-term in vivo persistence in the clinic. Such cells could be isolated from the small T memory stem cell (TSCM) compartment that has recently been postulated (Gattinoni et al., 2011), or from the naïve T-cell (TN) pool (Hinrichs et al., 2009; Cieri et al., 2013). Alternatively, elegant work in nonhuman primates (Berger et al., 2008) and in murine models (Stemberger et al., 2014) employing single-cell transfer indicates that selection of central memory CD8+ T-cells (TCM) may form a very attractive strategy to create T-cell products with a capacity for long-term maintenance. A high frequency of T-cells with a TCM phenotype (defined as CD45RO+CD62L+) within the infusion product has been correlated with clinical response in neuroblastoma patients treated with CAR-modified T-cells (Louis et al., 2011).
The above data demonstrate that the generation of T-cell products with a less differentiated phenotype forms an appealing strategy for ACT. Such cell products could possibly be obtained by selection of cells before adoptive transfer on the basis of their phenotypic markers. As an alternative strategy, it would be attractive to generate T-cell products that are enriched for the desired cell population by adjustment of the culture conditions used. Conceivably, this would require less handling of the product and lead to a higher initial yield.
Most protocols used for the expansion of T-cells for ACT are based on the addition of the cytokine interleukin (IL)-2 that promotes T-cell proliferation. In vitro, this cytokine is known to drive the differentiation of naïve CD8+ T-cells into effector and effector memory cells, and to promote the cytolytic function of these effector cells (Hinrichs et al., 2008; Cha et al., 2009; Yang et al., 2013). On the other hand, the homeostatic cytokines IL-7 and IL-15, which play a key role in the development and maintenance of central memory T-cells in vivo (Schluns et al., 2000; Prlic et al., 2002; Tan, 2002; Melchionda et al., 2005; Wang, 2005), have shown to allow the generation of central memory T-cells in culture systems (Klebanoff et al., 2005; Cha et al., 2009). Furthermore, work from the Bonini group that explored the capacity of human T-cells to induce graft versus host disease (GVHD) in a humanized mouse model has demonstrated that unselected gene-modified T-cells cultured in the presence of IL-7 and IL-15 were markedly more potent than T-cells cultured in the presence of IL-2 (Kaneko et al., 2009). To our knowledge, the combination of IL-7 and IL-15 has thus far not been applied in clinical trials of adoptive T-cell therapy.
Here, we describe the development and validation of a good manufacturing practice (GMP) production protocol that allows the generation of large quantities of TCR-modified cells by using IL-7 and IL-15 expansion in combination with anti-CD3/CD28 bead selection and activation. This protocol results in the production of large quantities of gene-modified T-cells with a phenotype comparable to that of central memory cells or memory stem cells, suggesting that these cell products may allow the long-term induction of a tumor-reactive T-cell compartment.
Materials and Methods
TCR gene optimization and cloning
The MART-1(26–35)-specific 1D3 TCR, selected on the basis of its high affinity, has been described previously (Dietrich et al., 2003; Jorritsma et al., 2007). In order to induce preferential pairing of the 1D3 TCR alpha and beta chains, and thereby reduce mispairing with endogenous chains, the human constant domains within the TCR alpha and beta chains were replaced by their murine counterparts (Cohen et al., 2006). In addition, an additional cysteine pair was introduced in order to allow the formation of a second interchain disulfide bridge (Cohen et al., 2007; Kuball et al., 2007). Modified TCR genes were gene-optimized and produced by GeneArt, and the TCR alpha and beta chains were subsequently cloned into the retroviral vector MP71 vector containing a self-cleaving P2A linker (Szymczak et al., 2004; Leisegang et al., 2008).
Virus production
The retrovirus used in this study to transduce peripheral T-cells with the 1D3HMcys TCR was produced by Eufets AG. Production of retroviral supernatant was started by establishment of a primary seed clone, consisting of PG13 cells stably transduced with vector-containing supernatant from 293T-cells. This primary seed bank was then expanded into a master cell bank (MCB), which was subsequently used for the large-scale production of retroviral supernatant. At several points in the production process, quality controls were performed, including tests on replication-competent retrovirus, sterility, and adventitious virus. Sequence integrity of the viral gene was confirmed by sequence analysis of the MCB.
Gene modification and T-cell culture
TCR-transduced T-cell products were generated under GMP conditions following standard operating procedures of the AmBTU (Amsterdam BioTherapeutics Unit). Cell products were generated in a class B clean room containing a class A biosafety cabinet for cell handling. All clean rooms were subjected to a monitoring program for viable and nonviable particles at operating and at resting state. All personnel performed media fills on a regular basis for validation of aseptic handling of the ACT products.
For the manufacture of TCR-transduced T-cells, all critical media components, viruses, and pharmaceutical excipients were of GMP grade and provided with a certificate of analysis and certificate of origin (when containing animal components) by the supplier.
Apheresis material was collected from patients within the central clinical laboratory of the Netherlands Cancer Institute-Antoni van Leeuwenhoek (NKI-AVL), after signing informed consent. Material was frozen and stored at the Dutch blood bank (Sanquin). The culture medium consisted of 50% AIM-V MED CTS (Life Technologies) and 50% RPMI 1640 containing gentamicin (50 μg/ml) and UltraGlutamine (223 μg/ml) (Lonza), and was mixed by Lonza. Upon thawing, apheresis material was diluted in the culture medium containing 20% human serum (HS; heat-inactivated pool of five AB male donors; Sanquin). Cells were then centrifuged and resuspended in phosphate buffered saline (PBS) containing 1% HS albumin (HSA; Albuman; Sanquin). Subsequently, cells were incubated with anti-CD3/CD28 CTS beads (Life Technologies) at a bead-to-cell ratio of 3:1 for 30 min at room temperature, followed by magnetic selection of cells.
Recovered cells were resuspended in the culture medium containing 5% HS and the cytokines IL-7 and IL-15 at 5 ng/ml each (CellGenix) and incubated for 48 hr at 37°C and 5% CO2. Thawing and culturing was performed in a closed system using transfer bags (Fresenius) and LifeCell bags (Baxter). Two days before retroviral transduction, non-tissue-culture–treated Falcon 24-well plates (Becton Dickinson) were coated with RetroNectin (Takara) and kept at +2–8°C until the day of transduction. At the day of transduction, the RetroNectin-coated plates underwent a blocking step using 2% HSA in PBS to prevent nonspecific binding. Plates were subsequently coated with GMP-grade retroviral supernatant (Eufets), by spinning for 90 min at 439 g and were kept at +2–8°C until further use.
At the day of transduction, the anti-CD3/CD28-activated cells were harvested and resuspended at a concentration of 5×105/ml in the medium. Retroviral supernatant was then removed from the virus-coated plates and 1 ml cell suspension per well was added to the plates. Plates were incubated overnight at 37°C and 5% CO2, and the transduction procedure was repeated the following day using new virus-coated plates. After the second transduction and incubation for minimally 5 hr, cells were collected and transferred to a 1-liter LifeCell culture bag (Baxter). A fresh medium containing IL-7, IL-15 (5 ng/ml each), and 5% HS was added to the cells and cells were cultured at 37°C and 5% CO2. Every 2–4 days, cells were counted and a fresh medium was added such that the concentration was ∼0.25×106 cells/ml. After an 11-day posttransduction expansion phase, TCR-transduced cells were concentrated by volume reduction on a Cytomate (Baxter) followed by magnetic removal of beads (MPCMagnet; Dynal). Cells were then washed twice and resuspended in 0.9% sodium chloride (NaCl) containing 2.5% HSA plus low-dose recombinant IL-2 (200 IU/ml, Proleukin; Novartis).
Melanoma cell lines
Melanoma cell lines Mel526, Mel624 (HLA-A2+, MART1+), and Mel938 (HLA-A2−, MART1+) were described previously (Topalian et al., 1989). Cell lines were maintained in RPMI (Gibco–Invitrogen) in the presence of 5% fetal calf serum (FCS; Sigma Chemical), penicillin (100 U/ml), and streptomycin (100 μg/ml; Roche).
Residual bead count
Aliquots of 5×106 cells were harvested in triplicate and centrifuged at 16,000 g in an Eppendorf tube. The resulting cell pellet was resuspended in distilled water and placed on coated Shandon cytospin slides with marked circles for the specimen (Thermo Scientific). To ensure that the entire pellet was collected, Eppendorf tubes were washed once with water and the collected material was added to the same slide. Slides were dried >30 min on a hot plate and were subsequently embedded in Xyleen and Pertex and covered with a coverslip. The total number of beads was counted on a microscope using a 200× magnification and dark field condenser. The final number of beads in the cell product was calculated by multiplying the number of beads observed by the ratio between the number of cells in the entire harvest and the number of cells per sample analyzed.
Residual compounds
To determine the amount of residual compounds within the infusion product, two different approaches were used. The first approach was based on the reduction of gentamicin levels in the ultimate cell product compared with the starting medium. Gentamicin was measured with a Siemens Viva E using the Emit 2000 gentamicin plus assay according to manufacturer's protocol. The second approach was based on an enzyme-linked immunosorbent assay (Quantikine HS ELISA IL-7; R&D Systems) to evaluate the reduction in IL-7 levels in the final cell product as compared with the starting medium, according to the manufacturer's instruction.
Residual viral particles
To calculate the number of remaining viral particles in the infusion product, the following formula was used: (V/20W×22.4T), where V is the starting titer of the virus, W is the number of wash steps, and T is the number of days of cell culture following retroviral transduction. This formula is derived from the Guideline for Handling of Lentiviral Vector-Transduced Eukaryotic Cells by the Netherlands Commission on Genetic Modification (COGEM, CGM051215-01). The COGEM requires that there will be a maximum of 0.01 viral particles left in the final infusion product.
Endotoxin measurements
Endotoxin levels within the supernatant of the final infusion product stored at −20°C were measured with a Pyrochrome limulus amoebocyte lysate (LAL) assay (Cape Cod Associates) according to manufacturer's instructions and European Pharmacopea guidelines (method A).
Flow cytometric analysis
Surface expression of the 1D3HMcys TCR-transduced T-cells was measured using APC-labeled HLA-A*02:01 MART-1(26–35 A>L) tetramers generated through ultraviolet-induced peptide exchange (Rodenko et al., 2006; Toebes et al., 2006) in combination with anti-CD3-PE- and anti-CD8-FITC-labeled antibodies (BD Biosciences) or in combination with anti-TCR Vb14-PE (CAS1.1.3) (Beckman Coulter). For phenotypic analyses, antibodies directed against CD3 (SK7), CD4 (SK3), CD8 (SK1), CD27 (L128), CD28 (CD28.2), CD45RA (HI100), and CD62L (DREG-56), conjugated with FITC, PE, PerCP Cy5.5, APC, and PE-Cy7, were used in combination with HLA-A*02:01 MART-1 tetramers. For analysis of intracellular cytokine levels, transduced cells were incubated with or without the indicated melanoma cell lines at a ratio of 1:1 (2×105 cells each) for 4–5 hr, in the presence of Golgiplug (Brefeldin A) and Golgistop (Monensin) (BD Biosciences) at 37°C and 5% CO2. Subsequent staining was performed with anti-CD8-PerCP, anti-IFNγ-APC (B27), anti-IL-2-PE (MQ1-17H12), and anti-TNFα-FITC (Mab11) (BD Biosciences). Cells were analyzed using a FACS Calibur (Becton Dickinson) and Flowjo software (Three Star).
Chromium release assay
Target cells were labeled for 1 hr at 37°C with 100 μCi (3.7 MBq) 51Cr (Amersham) and washed three times with RPMI 1640 medium containing 5% FCS. Labeled target cells were incubated with effector cells at the indicated ratios for 4 hr at 37°C and 5% CO2 in 200 μl medium. Maximal and spontaneous release of target cells was determined by the addition of 1% Triton X-100 (Sigma) or addition of medium alone, respectively. 51Cr release was determined by plating 50 μl of supernatant on a Lumaplate (Perkin-Elmer) and then measured in an automatic counter (Topcount; Perkin-Elmer). The percentage of specific release was calculated as follows: [(cpm experimental release − cpm spontaneous)/(cpm maximal − cpm spontaneous)]×100.
Release criteria of infusion product
The following release criteria were set for the infusion product:
• Transduction efficiency >10%, as measured by HLA tetramer staining at day 3 posttransduction (i.e., before the start of the lymphodepleting regimen).
• Sterility of a sample withdrawn from the cell bag 48–96 hr before infusion. Sterility was tested using aerobic and anaerobic BactAlert, according to the manufacturer's protocol. In addition, a sample for sterility testing was also taken at the day of infusion, but this was no part of the release criteria.
• Number of transduced cells ≥2.5×108 at the day of infusion, as determined by cell counting using a hemocytometer.
• Cell viability of >70% at the day of infusion, as determined by trypan blue staining using a hemocytometer.
Clinical trial design
In the phase I/IIa study for which the production protocol described here was designed, unresectable stage IIIc and stage IV melanoma patients will be treated with autologous, TCR-modified T-cells. Patients will first undergo an apheresis to harvest autologous lymphocytes. Upon successful transduction, the patient will receive nonmyeloablative lymphodepleting chemotherapy (cyclophosphamide 60 mg/kg/day i.v.×2 days; fludarabine 25 mg/m2/day i.v. ×5 days). After 7 days (day 13 after T-cell activation), the harvested cell product will be infused followed by low-dose IL-2 (2×106 IU/once daily up to 2 weeks). Immunomonitoring will include the tracking of the infused cell product by flow cytometry, and the analysis of cytokine levels in serum (Fig. 1).
FIG. 1.
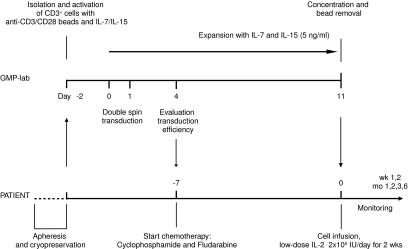
Overview of the GMP T-cell production process and patient conditioning. After thawing of the autologous apheresis product from patients, CD3+ cells are selected and stimulated using anti-CD3/CD28 beads. At days 2 and 3 following activation, cells undergo two cycles of transduction with 1D3HMcys MART-1 TCR pMP71 retroviral supernatant. Subsequently, cells are expanded in the presence of IL-7 and IL-15 (5 ng/ml each) for 11 days. After this expansion phase, cells are concentrated and anti-CD3/CD28 beads are removed. Cells are resuspended in ∼200 ml infusion fluid. Patients will receive a single infusion of autologous, TCR-modified lymphocytes. Before infusion, patients will be treated for 7 days with cyclophosphamide and fludarabine to induce a lymphopenic state. Immediately after cell infusion, patients will be treated with low-dose IL-2 injections subcutaneously for 2 weeks to enhance T-cell survival. GMP, good manufacturing practice.
Results
TCR selection and optimization
For TCR gene therapy of melanoma, we have previously selected a high-affinity, MART-1-specific TCR (1D3), obtained from a MART-1-reactive T-cell population in a patient with metastatic melanoma (Jorritsma et al., 2007). Subsequently, this 1D3 MART-1 TCR was modified in several ways in order to increase expression and induce preferential pairing between the introduced alpha and beta chain (Fig. 2A). First, the constant domains of the TCR alpha and beta chain were substituted with the murine constant domains and an additional cysteine pair was introduced to further facilitate interchain pairing. Second, both chains were codon-optimized and a self-cleaving P2A sequence was used to drive expression of both chains from a single mRNA. These modifications have been shown to result in enhanced expression of introduced TCRs and reduced mixed dimer formation (Cohen et al., 2007; Kuball et al., 2007; Bendle et al., 2010). For transduction of human T-cells, we selected the pMP71 retroviral backbone (Engels et al., 2003) that was demonstrated to be safe in a prior clinical trial (Lunzen et al., 2007). Viral supernatant generated from the resulting vector allowed high-level expression of the 1D3HMcys-specific TCR upon transduction of human peripheral blood T-cells (Fig. 2B).
FIG. 2.

1D3HMcys MART-1 TCR vector for clinical use. (A) 1D3HMcys MART-1 TCR vector design. The constant domains of the 1D3 TCR chains were substituted with their murine counterparts. TCR genes were codon-optimized and an additional cysteine pair was introduced into the constant domains. The TCR was cloned into the pMP71 plasmid containing a P2A motif as a self-cleavable linker. (B) High-level expression of the 1D3HMcys MART-1 TCR. Human peripheral blood lymphocytes were transduced with the 1D3HMcys MART-1 TCR pMP71 retroviral vector. TCR expression was determined using MART-1(26–35 A>L) tetramers and anti-VBeta14 TCR antibody. Numbers in the upper-right corners indicate the percentage of VBeta14 and MART-1 tetramer-positive cells of total lymphocytes. TCR, T-cell receptor.
GMP production of TCR gene-modified T-cells
In most research protocols, spin inoculation on RetroNectin-coated plates is used as a preferred transduction method, leading to high transduction efficiencies (Kotani et al., 1994; Hanenberg et al., 1996). In prior work we demonstrated that retrovirus-coated epoxy beads (Life Technologies) can also be used to achieve high transduction efficiencies, with the added benefit that no centrifugation step is required (Heemskerk et al., 2010). However, with the GMP-grade beads that were commercially available at the time of development of this protocol, high transduction efficiency could no longer be achieved by this method, presumably because of an alteration in their production process (unpublished observations). A procedure was therefore set up in which, following selection and activation with anti-CD3/CD28 beads, T-cells were spin-transduced using retrovirus-coated 24-well plates preloaded with virus. Subsequently, cells were transferred to culture bags and cultured for 11 days in the presence of the cytokines IL-7 and IL-15 (5 ng/ml each). A schematic overview of this GMP production protocol is shown in Fig. 1. This protocol was applied in three validation runs that all met the requirements for release, with a transduction efficiency of more than 10% tetramer+ CD3+ cells (average of 56%) at day 3 posttransduction (see Table 1).
Table 1.
Release Criteria of the Good Manufacturing Practice–Grade Cell Products
| Item | Specification | NKITCR001 | NKITCR002 | NKITCR003 |
|---|---|---|---|---|
| Transduction efficiencya | ≥10% 1D3 TCR+ of CD3+ cells | 20%b | 84% | 65% |
| Microbiological contamination | Negative (48–96 hr before infusion) | Negative | Negative | Negative |
| Transduced cell numberc | 2.5×108–2.5×10101D3 TCR+ CD3+ cells | 3.3×109 | 1.5×109 | 3.4×109 |
| Cell viability | >70% | 97% | 93% | 98% |
TCR, T-cell receptor.
Transduction efficiency measured on day 3 posttransduction.
Transduction only performed once.
The minimum and maximum amount of TCR-transduced cells that will be infused.
At the end of the culture period, a total of 1.5×109–3.4×109 tetramer+ CD3+ cells were obtained, with a viability of more than 93%. In all production runs, no microbiological contamination was detected in the samples taken 48–96 hr before infusion (after the last medium addition), or in the samples taken at the day of infusion. The first validation run, performed with only a single transduction round, resulted in a 20% transduction efficiency of total CD3+ cells at day 3 after transduction, and a 32% and 25% transduction efficiency of CD8+ and CD8− cells at the end of the culture period. We then modified the protocol to perform two subsequent spin transductions, resulting in transduction efficiencies of >50% in both the CD8+ and CD8− cell population (Fig. 3A). Table 2 shows the extended characterization of the TCR-modified cells obtained at the end of the culture period. Cell expansion ranged between 300- and 800-fold, as calculated from the day of transduction. More than 98% of the cells were CD3+ with a 1.7–2.9 ratio of CD4/CD8 cells.
FIG. 3.

TCR expression and phenotype of T-cells in the GMP-grade cell products. (A) TCR expression was determined on the final day of culturing by flow cytometric analysis using anti-CD3, anti-CD8 antibody, and HLA-A*02:01 MART-1(26–35 A>L) tetramers. Cells are gated on CD3+ cells. The number in the upper-right corner indicates the percentage of tetramer+ cells of CD8+ cells. The number in the lower-right corner indicates the percentage of tetramer+ cells of CD8− cells. In the validation run indicated with “*,” only a single cycle of retroviral transduction was performed. (B) Cell products obtained in the three independent production runs were analyzed simultaneously after thawing and overnight recovery. All plots show cells gated on tetramer+ CD8+ lymphocytes. Cells were stained for CD27 and CD28 expression (top row), CD62L and CD45RA expression (middle row), or CD45RA and CD45RO expression (bottom row).
Table 2.
Product Specifications of the Good Manufacturing Practice–Grade Cell Products
| NKITCR001 | NKITCR002 | NKITCR003 | |
|---|---|---|---|
| Fold expansion | 544 | 309 | 800 |
| % CD3+ cells | 99.4 | 99.3 | 98.8 |
| CD4/CD8 ratio | 2.5 | 1.7 | 2.9 |
| Residual beads | 1.1×104 | 1.5×104 | 2.9×104 |
| Fold reduction of supplements based on gentamicin | >212 | >187 | >187 |
| Fold reduction of supplements based on IL-7 | 115 | 181 | 49 |
IL-7, interleukin-7.
To obtain the infusion product, TCR-transduced cells were concentrated, washed, and resuspended in a 0.9% NaCl solution with 2.5% HSA and 200 IU/ml IL-2. In order to determine the amount of residual medium (and medium supplements) left after cell harvesting, we measured residual gentamicin and IL-7 in the final infusion sample as part of our validation process. As shown in Table 2, the amount of IL-7 was reduced 49–115-fold and gentamicin was reduced 187–212-fold in three independent validation runs. Based on the lowest reduction measured (i.e., 49-fold for IL-7), we calculated that the final cell product contains a maximum of 2 ml of culture medium (Table 3). Direct infusion of culture medium such as RPMI, in volumes up to 100 ml, has previously been described and has not resulted in any adverse effects (Ngok et al., 2004). As based on the same calculation, the concentration of gentamycin in the final cell product is reduced to <1.1 mg/liter. This indicates that the amount infused is well below what is normally infused during gentamicin treatment (5 mg/kg). Also, the amounts of medium supplements such as IL-7 and IL-15 are well below the amounts infused in other preclinical trials (Mackall et al., 2011; Waldmann et al., 2011). As such, we consider it safe to use these supplements in the clinical protocol as described here (Table 3).
Table 3.
Residual Compounds Within the Good Manufacturing Practice-Grade Cell Products
| Residual compounds | Starting quantity/concentration | Total amount in final product |
|---|---|---|
| Freezing medium (including DMSO) | 50 ml | 4×10−8 mla |
| RetroNectin | 480 μg | 0.06 μgb |
| 1D3HMcys TCR retroviral supernatant | 130×106 pfu | ≤0.00097 pfuc |
| 50%/50% RPMI/AIM-V medium | 8 liter | ≤2 mld |
| Gentamicin | 50 mg/liter | ≤0.1 mgd |
| Human serum | 5% | ≤0.1 mld |
| Interleukin-7 | 5 ng/ml | ≤10 ngd |
| Interleukin-15 | 5 ng/ml | ≤10 ngd |
DMSO, dimethyl sulfoxide.
Based on 7 wash steps (20-fold reduction per wash [COGEM]) during the whole process (207=1.28×109 reduction).
Based on 3 wash steps. Note: the final amount of RetroNectin will be lower as it will remain attached to the well.
Based on 3 wash steps and 10 days of culture after the second transduction (203×22.4×10=1.3×1011 reduction).
Calculated for a reduction of 49-fold as determined during the validation runs (see Table 2).
The maximum limit for endotoxins in intravenous preparations is 5 IU of endotoxin per kilogram of body mass per hour. With an average patient weight of 70 kg and a final volume of 100 ml in the infusion bag, the limit of endotoxin in the final formulation is <3.5 IU/ml. In all three validation runs, endotoxin levels within the final products were between 0.1 and 1 IU/ml, well within the limits set by the European Pharmacopoeia.
The amount of residual beads found in the final product of the validation runs ranged between 1.1×104 and 2.9×104 (Table 2). In a study in rats, intravenous injection of 8.3×108 beads/kg (corresponding to 5.8×1010 beads in humans) did not result in any adverse effects (White et al., 1995). This amount of beads is 2×106-fold higher than the amount of residual beads in the end product (and in fact 100-fold higher than the amount of beads that is added at the start of the process). Furthermore, clinical studies have been performed with anti-CD3/CD28 bead-stimulated T-cells in which similar or higher numbers of residual beads have been infused without any toxicities observed (Laport, 2003; Thompson et al., 2003; Porter et al., 2006). As such, we consider the use of anti-CD3/CD28 beads in this protocol safe.
Phenotypic properties of TCR-modified T-cells
Having determined that the anti-CD3/CD28 bead- and IL-7/IL-15-based protocol results in the required numbers of gene-modified T-cells, we evaluated the phenotype of the resulting cells. Cells were analyzed for expression of CD27, CD28 CD45RA, CD45RO, and CD62L. CD45RA is expressed on naïve T lymphocytes, replaced by CD45RO upon antigen encounter, but re-expressed in late effector cells (Michie et al., 1992; Hamann et al., 1999). CD62L is a cell adhesion molecule that acts as a homing molecule to enter secondary lymphoid tissues and is lost after T-cell activation, when T-cells acquire effector functions (Sallusto et al., 1999; Garton et al., 2006; Yang et al., 2011). CD27 and CD28 are costimulation markers that are lost during T-cell differentiations with CD28 being lost earlier following antigen exposure than CD27 (Appay et al., 2002; Klebanoff et al., 2006).
In all three final products, almost all tetramer+ CD8+ cells were CD28+, with the majority of these cells (78–92%) also expressing CD27 (Fig. 3B, top row). Furthermore, between 42% and 72% of the cells are double-positive for CD45RA and CD62L (Fig. 3B, second row). The same phenotypic markers were also analyzed on the unmanipulated apheresis material from the same donors (Supplementary Fig.S1; Supplementary Data are available online at www.liebertpub.com/hgtb). This clearly shows that the final cell product is enriched for cells that are both double-positive for CD28 and CD27 and double-positive for CD62L and CD45RA.
Based on these data it can be concluded that this CD3/CD28 bead and IL-7/IL-15 based strategy for the generation of gene-modified cells results in a large fraction of cells that are phenotypically similar to central memory T-cells and memory stem T-cells, an observation that is consistent with the production of TSCM under these conditions in preclinical work by Cieri et al. (2013).
Functional analysis of clinical-scale TCR-transduced cells
We next set out to determine the capacity of the transduced cells to produce the effector cytokines IFNγ, IL-2, and TNFα upon stimulation with MART-1-expressing HLA-A2-positive melanoma cell lines Mel526 and Mel624 (Fig. 4A). Co-incubation with HLA-A2+MART-1+ cell lines resulted in production of all three effector cytokines, especially in the CD8+ cell population. As expected, costaining with the anti-Vb14 antibody (the variable segment used by the 1D3 TCR beta chain) revealed production of cytokines (IL-2 and IFNγ) to be restricted to the transduced cells (data not shown). No cytokine production was observed in the absence of melanoma target cells, or when co-incubation was performed with the HLA-A2− melanoma line Mel938. As additional controls, nontransduced cells from the same donors used for the validation runs showed only background levels of cytokine production upon incubation with melanoma lines, and polyclonal stimulation of the final infusion products led to production of high levels of all three cytokines (Supplementary Fig. S2A and B, respectively).
FIG. 4.

Intracellular cytokine production and cytotoxic activity of TCR-transduced T-cells in the GMP-grade cell products. After thawing and overnight recovery, TCR-transduced cells were incubated with HLA-A2+MART-1+ (mel624 and mel526), HLA-A2−MART-1+ (mel938) melanoma cell lines, or cultured alone (−). Intracellular cytokine production was determined after 5 hr of incubation. (A) For T-cells from each validation run, the percentage of IFNγ-, IL-2-, and TNFα-positive CD8+ cells (top row) and CD4+ cells (bottom row) is shown. (B) For the T-cell products incubated with mel624, Boolean gating is shown for CD8+ cells (left panel) and CD4+ cells (right panel). (C) T-cells were taken into culture 1 day before the assay. HLA-A2+MART-1+ (mel526 and mel624) and HLA-A2−MART-1+ (mel938) melanoma lines were loaded with 51Cr and subsequently added to TCR-transduced cells at the indicated effector to target ratios. Lysis was determined after 5 hr of incubation. The percentage of tetramer+ cells of CD3+ cells measured on the day of the assay was 29%, 57%, and 58% for NKITCR001, NKITCR002, and NKITCR003, respectively.
Prior work has suggested that polyfunctional T-cells (and in particular when including the capacity to produce IL-2) have a less differentiated state and an enhanced functionality (Darrah et al., 2007; Seder et al., 2008). To determine which percentage of cells is triple, double, or single producers for the cytokines analyzed, Boolean gating was applied. Within the CD8+ population, the majority of cells are triple producers (data only shown for Mel624) and only a minority produced only one cytokine. Within the CD4 population, triple producers were likewise observed but at lower frequencies, and accompanied by roughly similar frequencies of double and single producers (Fig. 4B). These results show that the TCR-modified T-cells obtained in this process not only display a phenotype that resembles that of early memory T-cells, but also display a cytokine production profile that is associated with increased functionality.
To also analyze the capability of these TCR-modified T-cells to kill target-expressing tumor cells, cells were incubated with HLA-A201+MART-1+ melanoma cell lines. As shown in Fig. 4C, TCR-transduced cells from all three validation runs were capable of lysing MART-1-expressing melanoma cells, whereas HLA-A2− target cells were not recognized.
Discussion
The GMP production protocol described here results in a drug product consisting of autologous T-cells transduced with a MART-1-specific TCR that is well expressed and that displays phenotypic and functional properties that predict prolonged in vivo engraftment. This protocol has shown to be robust using quality-controlled and GMP-grade reagents with a minimal risk of microbiological contamination through the use of closed culture systems.
Perhaps most importantly, the T-cell culture protocol is based on the use of the IL-7 and IL-15 cytokines that prevent terminal differentiation of T-cells, and that have yielded human T-cells with superior activity in preclinical models (Kaneko et al., 2009; Cieri et al., 2013). In the experiments performed here, CD95 expression was not analyzed on the final infusion products. As such, it is difficult to conclude whether the obtained cells are phenotypically closer to TSCM or TCM cells, but in either case the cell population obtained bears the characteristics of early memory cells.
We do note that the phenotype of the T-cells we obtain is analyzed after a period of in vitro culture that includes T-cell activation. As such, direct comparison with the phenotype of central memory or memory stem T-cells that occur in vivo would not be warranted. Indeed, as shown by Berger and colleagues (2008), T-cells that acquire a similar phenotype in in vitro culture systems can display highly disparate behavior in vivo. Nevertheless, the fact that the majority of the T-cells produced in this GMP protocol are CD27+, CD28+, and CD45RA+/CD62L+, plus the fact that several studies have shown that less differentiated T-cell phenotypes are correlated with a high engraftment and clinical response in ACT (Ochsenbein et al., 2004; Powell et al., 2005; Li et al., 2010), make it reasonable to assume that this GMP protocol can yield T-cell products with a capacity for prolonged engraftment. Immune monitoring of patients in the planned clinical trial should reveal whether the infusion of cell products cultured under the conditions described here does indeed lead to high-level and prolonged engraftment in patients.
In this GMP production setup, in which sterility of the sample taken after the last medium addition is used as a release criterion (with preliminary test results available at the day of infusion), the requirement to freeze the cell product is overcome, thereby preventing loss of cells and decrease in cell quality. Contamination of the cell product at the time of infusion cannot be fully excluded in this procedure, because contamination may either be too low to be detected within 48–96 hr, or the product may potentially become contaminated during the harvesting phase. However, taking into consideration that these involve validated aseptic handling procedures combined with a predominantly closed culture system within a cleanroom environment, we consider this risk acceptable. In all three production runs, microbiological tests were also negative for the sample taken from the infusion product.
In summary, we here describe a GMP production protocol that allows the generation of high numbers of TCR-transduced cells while preventing terminal differentiation of these cells, an approach that may enhance the treatment efficacy of TCR gene therapy.
During the preparation of this article, three patients have been included in the clinical study (MTCR11 NKI-AVL). The T-cell products were produced according to specifications, with a transduction efficiency of 62%, 44%, and 70% of T-cells, a viability of >94.8%, and no microbiological contamination of the infusion samples. A total of 5×109 transduced cells were infused into the first patient. Unfortunately, the patient expired after experiencing an unexpected serious adverse event (SAE) with cerebral hemorrhage, cardiac arrest, sepsis like syndrome, and high levels of circulating cytokines. Analysis of this SAE did not reveal any evidence for TCR-mediated off-target reactivity, as recently seen for two affinity-enhanced MAGE-A3-specific TCRs (Cameron et al., 2013; Morgan et al., 2013). Based on the available data, the clinical protocol was amended (van den Berg et al., in preparation) with respect to patient selection, maximum cell dose, and monitoring of serum cytokine levels as a marker for an overly active antitumor immune response. The second and third patients were infused with 5×107 transduced cells (100-fold lower than patient 1) and did not experience any significant toxicity.
Supplementary Material
Acknowledgments
We would like to thank the NKI FACS facility, radionucleotide facility, S. Michels, and W. van de Kasteele for their technical support. We would like to thank B. Beerman, S. Quaak, D. Meijer, and E. Vermeij (Department of Pharmacy and Pharmacology, Slotervaart Hospital) for assistance and C. Lamers (Erasmus MC, Rotterdam) for generous help and input on GMP matters. This research was supported by ZonMw (Grant 431-00-005) and the Danish Council for Strategic Research (Grant 09-065152).
Author Disclosure Statement
No competing financial interests exist.
References
- Appay V., Dunbar P.R., Callan M., et al. (2002). Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8, 379–385 [DOI] [PubMed] [Google Scholar]
- Bendle G.M., Linnemann C., Hooijkaas A.I., et al. (2010). Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 16, 565–570, 1p following 570 [DOI] [PubMed] [Google Scholar]
- Berger C., Jensen M.C., Lansdorp P.M., et al. (2008). Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 118, 294–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens R.J., Davila M.L., Riviere I., et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron B.J., Gerry A.B., Dukes J., et al. (2013). Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 5, 197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha E., Graham L., Manjili M.H., and Bear H.D. (2009). IL-7+IL-15 are superior to IL-2 for the ex vivo expansion of 4T1 mammary carcinoma-specific T cells with greater efficacy against tumors in vivo. Breast Cancer Res. Treat. 122, 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri N., Camisa B., Cocchiarella F., et al. (2013). IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 121, 573–584 [DOI] [PubMed] [Google Scholar]
- Cohen C.J., Zhao Y., Zheng Z., et al. (2006). Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 66, 8878–8886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen C.J., Li Y.F., El-Gamil M., et al. (2007). Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 67, 3898–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah P.A., Patel D.T., De Luca P.M., et al. (2007). Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat. Med. 13, 843–850 [DOI] [PubMed] [Google Scholar]
- de Witte M.A., Coccoris M., Wolkers M.C., et al. (2006). Targeting self-antigens through allogeneic TCR gene transfer. Blood 108, 870–877 [DOI] [PubMed] [Google Scholar]
- Dietrich P.-Y., Le Gal F.-A., Dutoit V., et al. (2003). Prevalent role of TCR alpha-chain in the selection of the preimmune repertoire specific for a human tumor-associated self-antigen. J. Immunol. 170, 5103–5109 [DOI] [PubMed] [Google Scholar]
- Engels B., Cam H., Schuler T., et al. (2003). Retroviral vectors for high-level transgene expression in T lymphocytes. Hum. Gene Ther. 14, 1155–1168 [DOI] [PubMed] [Google Scholar]
- Garton K.J., Gough P.J., Raines E.W. (2006). Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J. Leukoc. Biol. 79, 1105–1116 [DOI] [PubMed] [Google Scholar]
- Gattinoni L., Lugli E., Ji Y., et al. (2011). A human memory T cell subset with stem cell-like properties. Nat. Med. 17, 1290–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp S.A., Kalos M., Barrett D., et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann D., Roos M.T., Van Lier R.A. (1999). Faces and phases of human CD8 T-cell development. Immunol. Today 20, 177–180 [DOI] [PubMed] [Google Scholar]
- Hanenberg H., Xiao X.L., Dilloo D., et al. (1996). Colocalization of retrovirus and target cells on specific fibronectin fragments increases genetic transduction of mammalian cells. Nat. Med. 2, 876–882 [DOI] [PubMed] [Google Scholar]
- Heemskerk B., Jorritsma A., Gomez-Eerland R., et al. (2010). Microbead-assisted retroviral transduction for clinical application. Hum. Gene Ther. 21, 1335–1342 [DOI] [PubMed] [Google Scholar]
- Hinrichs C.S., Spolski R., Paulos C.M., et al. (2008). IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 111, 5326–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs C.S., Borman Z.A., Cassard L., et al. (2009). Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 106, 17469–17474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M.C., Popplewell L., Cooper L.J., et al. (2010). Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 16, 1245–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorritsma A., Gomez-Eerland R., Dokter M., et al. (2007). Selecting highly affine and well-expressed TCRs for gene therapy of melanoma. Blood 110, 3564–3572 [DOI] [PubMed] [Google Scholar]
- Kaneko S., Mastaglio S., Bondanza A., et al. (2009). IL-7 and IL-15 allow the generation of suicide gene-modified alloreactive self-renewing central memory human T lymphocytes. Blood 113, 1006–1015 [DOI] [PubMed] [Google Scholar]
- Klebanoff C.A., Gattinoni L., Torabi-Parizi P., et al. (2005). Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 102, 9571–9576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff C.A., Gattinoni L., and Restifo N.P. (2006). CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 211, 214–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer J.N., Wilson W.H., Janik J.E., et al. (2010). Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani H., Newton I., Perry B., et al. (1994). Improved methods of retroviral vector transduction and production for gene therapy. Hum. Gene Ther. 5, 19–28 [DOI] [PubMed] [Google Scholar]
- Kuball J., Dossett M.L., Wolfl M., et al. (2007). Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 109, 2331–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers C.H.J., Willemsen R., Van Elzakker P., et al. (2011). Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 117, 72–82 [DOI] [PubMed] [Google Scholar]
- Laport G.G. (2003). Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood 102, 2004–2013 [DOI] [PubMed] [Google Scholar]
- Leisegang M., Engels B., Meyerhuber P., et al. (2008). Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J. Mol. Med. (Berl.) 86, 573–583 [DOI] [PubMed] [Google Scholar]
- Li Y., Liu S., Hernandez J., et al. (2010). MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J. Immunol. 184, 452–465 [DOI] [PubMed] [Google Scholar]
- Louis C.U., Savoldo B., Dotti G., et al. (2011). Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunzen J.V., Glaunsinger T., Stahmer I., et al. (2007). Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol. Ther. 15, 1024–1033 [DOI] [PubMed] [Google Scholar]
- Mackall C.L., Fry T.J., and Gress R.E. (2011). Harnessing the biology of IL-7 for therapeutic application. Nat. Rev. Immunol. 11, 330–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionda F., Fry T.J., Milliron M.J., et al. (2005). Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. J. Clin. Invest. 115, 1177–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie C.A., McLean A., Alcock C., and Beverley P.C. (1992). Lifespan of human lymphocyte subsets defined by CD45 isoforms. Nature 360, 264–265 [DOI] [PubMed] [Google Scholar]
- Morgan R.A., Dudley M.E., Wunderlich J.R., et al. (2006). Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314, 126–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R.A., Chinnasamy N., Abate-Daga D., et al. (2013). Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 36, 133–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris E.C., Tsallios A., Bendle G.M., et al. (2005). A critical role of T cell antigen receptor-transduced MHC class I-restricted helper T cells in tumor protection. Proc. Natl. Acad. Sci. USA 102, 7934–7939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngok F.K., Mitsuyasu R.T., Macpherson J.L., et al. (2004). Clinical gene therapy research utilizing ribozymes: application to the treatment of HIV/AIDS. Methods Mol. Biol. 252, 581–598 [DOI] [PubMed] [Google Scholar]
- Ochsenbein A.F., Riddell S.R., Brown M., et al. (2004). CD27 expression promotes long-term survival of functional effector-memory CD8+ cytotoxic T lymphocytes in HIV-infected patients. J. Exp. Med. 200, 1407–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D.L., Levine B.L., Bunin N., et al. (2006). A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood 107, 1325–1331 [DOI] [PubMed] [Google Scholar]
- Porter D.L., Levine B.L., Kalos M., et al. (2011). Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 365, 725–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell D.J., Dudley M.E., Robbins P.F., and Rosenberg S.A. (2005). Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood 105, 241–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prlic M., Lefrancois L., and Jameson S.C. (2002). Multiple choices: regulation of memory CD8 T cell generation and homeostasis by interleukin (IL)-7 and IL-15. J. Exp. Med. 195, F49–F52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins P.F., Dudley M.E., Wunderlich J., et al. (2004). Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 173, 7125–7130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins P.F., Morgan R.A., Feldman S.A., et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 29, 917–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenko B., Toebes M., Hadrup S.R., et al. (2006). Generation of peptide–MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 1, 1120–1132 [DOI] [PubMed] [Google Scholar]
- Sallusto F., Lenig D., Förster R., et al. (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 [DOI] [PubMed] [Google Scholar]
- Schluns K.S., Kieper W.C., Jameson S.C., and Lefrançois L. (2000). Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat. Immunol. 1, 426–432 [DOI] [PubMed] [Google Scholar]
- Seder R.A., Darrah P.A., and Roederer M. (2008). T-cell quality in memory and protection: implications for vaccine design. Nat. Rev. Immunol. 8, 247–258 [DOI] [PubMed] [Google Scholar]
- Stemberger C., Graef P., Odendahl M., et al. (2014). Lowest numbers of primary CD8+ T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood 124, 628–637 [DOI] [PubMed] [Google Scholar]
- Szymczak A.L., Workman C.J., Wang Y., et al. (2004). Correction of multi-gene deficiency in vivo using a single 'self-cleaving' 2A peptide-based retroviral vector. Nat. Biotechnol. 22, 589–594 [DOI] [PubMed] [Google Scholar]
- Tan J.T. (2002). Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J. Exp. Med. 195, 1523–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.A., Figlin R.A., Sifri-Steele C., et al. (2003). A phase I trial of CD3/CD28-activated T cells (Xcellerated T cells) and interleukin-2 in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 9, 3562–3570 [PubMed] [Google Scholar]
- Toebes M., Coccoris M., Bins A., et al. (2006). Design and use of conditional MHC class I ligands. Nat. Med. 12, 246–251 [DOI] [PubMed] [Google Scholar]
- Topalian S.L., Solomon D., and Rosenberg S.A. (1989). Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J. Immunol. 142, 3714–3725 [PubMed] [Google Scholar]
- Waldmann T.A., Lugli E., Roederer M., et al. (2011). Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood 117, 4787–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.X. (2005). Interleukin-7-dependent expansion and persistence of melanoma-specific T cells in lymphodepleted mice lead to tumor regression and editing. Cancer Res. 65, 10569–10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.D., Glosson J.A., Gordon D.E., and Northup S.J. (1995). Intravenous safety study in rats given paramagnetic, polystyrene beads with covalently bound sheep anti-mouse immunoglobulin G (IgG). Int. J. Toxicol 14, 251–265 [Google Scholar]
- Yang S., Liu F., Wang Q.J., et al. (2011). The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS ONE 6, e22560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Ji Y., Gattinoni L., et al. (2013). Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol. Immunother. 62, 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C., Thompson J.A., Byrd D., et al. (2002). Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. USA 99, 16168–16173 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.