

Abstract
Aim: To evaluate the role of matrix metalloproteinase (MMP)-9 deletion on citrate synthase (CS) activity postmyocardial infarction (MI). Results: We fractionated left ventricle (LV) samples using a differential solubility-based approach. The insoluble protein fraction was analyzed by mass spectrometry, and we identified CS as a potential intracellular substrate of MMP-9 in the MI setting. CS protein levels increased in the insoluble fraction at day 1 post-MI in both genotypes (p<0.05) but not in the noninfarcted remote region. The CS activity decreased in the infarcted tissue of wild-type (WT) mice at day 1 post-MI (p<0.05), but this was not observed in the MMP-9 null mice, suggesting that MMP-9 deletion helps to maintain the mitochondrial activity post-MI. Additionally, inflammatory gene transcription was increased post-MI in the WT mice and attenuated in the MMP-9 null mice. MMP-9 cleaved CS in vitro, generating an ∼20 kDa fragment. Innovation: By applying a sample fractionation and proteomics approach, we were able to identify a novel MMP-9-related altered mitochondrial metabolic activity early post-MI. Conclusion: Our data suggest that MMP-9 deletion improves mitochondrial function post-MI. Antioxid. Redox Signal. 21, 1974–1985.
Introduction
Despite advances in therapeutic approaches for patients with acute myocardial infarction (MI), MI remains the main cause of death in Western countries and is the leading cause of heart failure (43). MI results from the interruption of blood supply to the cardiac tissue; this is commonly due to the occlusion of a coronary artery. An MI leads to decreased oxygen supply and ischemia of the downstream tissue. Myocardial ischemia is characterized by tissue hypoxia that generates acidosis and an altered homeostasis leading to cardiac dysfunction (40). Cardiac tissue has the constant of requiring a high demand for energy. Myocardial energy demand and substrate supply are in continual flux. Thus, ATP-generating pathways must respond to dynamic fluctuations in physiological demands (23). Mitochondria are the energy-generating organelles in cells, and reduced oxygen levels quickly lead to mitochondrial dysfunction. Mitochondrial dysfunction can threaten cellular integrity through the increased generation of reactive oxygen species (ROS), induction of apoptosis, and depletion of high-energy triphosphates due to inhibition of the respiratory chain (6, 25, 46). Metabolic regulation is inextricably linked with cardiac function. The degree of mitochondrial injury defines the cellular fate of the cardiac myocyte (5). Moreover, systolic cardiac function is determined by the cardiomyocyte; therefore, cardiac function depends on mitochondrial function.
Innovation.
By applying a novel differential solubility-based sample fractionation and proteomics approach, we were able to identify genotype-related differences in the mitochondrial metabolic activity early post-myocardial infarction (MI). Post-MI, loss of myocytes translates into loss of mitochondria and low ATP-generating substrate supply. We studied the levels and activity of a step-limiting mitochondrial enzyme post-MI, citrate synthase (CS), which is involved in the cardiac metabolic pathway. CS was identified as a novel in vivo substrate of matrix metalloproteinase (MMP)-9, a protease highly expressed post-MI that modulates myocardial remodeling. In addition to the effects on the early inflammatory response, our study is the first to demonstrate an intracellular role for MMP-9 in the post-MI left ventricle.
Several proteases, including matrix metalloproteinases (MMPs), are activated in different types of cardiomyopathies, such as MI, and can cause extracellular damage. Multiple MMPs have been shown to be altered post-MI in both human and animal studies (59). Of the MMP family, MMP-9 has emerged as a candidate with direct effects on cardiac remodeling post-MI. MMP-9 levels increase early post-MI (31), and MMP-9 deletion reduces cardiac remodeling and stimulates post-MI angiogenesis (32). In a clinical study, Hou et al. assessed the efficacy of MMP-9 and myeloperoxidase (MPO) as biomarkers to predict the presence of coronary artery plaques (22). MMP-9 and MPO levels positively correlate with the Framingham risk score, and the levels of MMP-9 were significantly elevated in patients with nonobstructive coronary artery disease compared with patients with no coronary plaques. This demonstrates that MMP-9 levels are increased during cardiovascular disease, and MMP-9 is a promising therapeutic target. MMP-9 degrades numerous structural extracellular matrix (ECM) proteins, including collagen, fibronectin, and proteoglycans (55, 60). MMPs have been reported to play a role in the cleavage of intracellular matrix proteins (9, 24, 39, 49). For example, MMP-2, -3, -13, and -14 have all been reported to have intracellular substrates (7, 11, 13, 15). Recently, Cauwe et al. have identified 69 MMP-9 intracellular candidate substrates in the cytosol of THP-1 cells (8). However, the intracellular MMP-9 in vivo degradome remains relatively unexplored. Uria and Lopez-Otin have shown that MMP-9 can be activated by MMP-26 (52). Similarly, Zhao et al. showed that active MMP-9 and MMP-26 colocalized intracellularly in ARCaP cells and in human prostate tissue samples (61). These data suggest that active MMP-9 is available intracellularly, but MMP-9 intracellular function has not been studied extensively.
After acute tissue necrosis, the degradation of intracellular proteins may be critical to evade systemic toxicity and immunogenicity. Citrate synthase (CS) is the rate-limiting enzyme of the tricarboxylic acid (TCA) cycle and a commonly used marker of mitochondrial oxidative capacity (38). CS was previously reported to be an in vitro substrate of MMP-9 (8), but whether CS proteolysis also occurs in vivo and in the setting of MI has not been explored. In this study, we compared CS levels and activity in wild-type (WT) and MMP-9 null mice post-MI to assess possible interactions between CS and MMP-9. We hypothesized that MMP-9 cleaves CS intracellularly post-MI as part of the cellular clearing process after extensive necrosis.
Results and Discussion
CS was elevated in the infarcted myocardium of MMP-9 null mice at day 5 post-MI
Mitochondrial CS mediates the first step of the TCA cycle by catalyzing the condensation of acetyl coenzyme A and oxaloacetate to form citrate and coenzyme A. Initially, we compared the infarcted proteome of WT and MMP-9 null mice at day 5 post-MI and of day 0 control animals. Total tissue protein was extracted and fractionated using a differential solubility-based protocol that yields three protein fractions per sample: soluble fraction (step 1), cellular fraction (step 2), and insoluble fraction (step 3). The insoluble proteins (step 3) from the left ventricle infarcted area (LVI) of day 5 post-MI mice (n=4/genotype/time-point) were run in a gel, trypsin digested, analyzed by capillary HPLC-electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS), and compared to the left ventricle (LV) of day 0 control animals. CS was observed only in the day 5 post-MI LVI samples, and spectrum counts showed CS numbers elevated in the MMP-9 null group compared to the WT group (Fig. 1), suggesting that processing of CS by MMP-9 may be a mechanism to control the CS activity and oxidative metabolism post-MI. Interestingly, CS was not observed in control tissue (day 0 control animals). This was likely due to the fact that only the insoluble protein fraction was analyzed. Cardiac myocytes are one of the main cell types present in the healthy heart, and myocytes are rich in mitochondria and CS (21). Protein translocation to a membrane fraction often results from post-translational modifications; this may explain the presence of CS in the insoluble fraction only in the post-MI samples.
FIG. 1.

CS is identified by MS as a possible MMP-9 substrate. The insoluble proteins from day 5 infarcted decellularized LV were analyzed by capillary HPLC-ESI-MS/MS and compared to day 0 controls. CS was present only at day 5 post-MI, and the spectrum analysis showed CS numbers elevated in the MMP-9 null group. (A) CS spectrum. (B) CS sequence coverage. (C) Table showing the observed proteomic differences between the WT and MMP-9 null groups. The thresholds for acceptance of peptide and protein assignments in a scaffold were 90% and 99%, respectively, and minimum of two unique peptides. LV, left ventricle; HPLC-ESI-MS/MS, HPLC-electrospray ionization tandem mass spectrometry; CS, citrate synthase; MI, myocardial infarction; MMP, matrix metalloproteinase; WT, wild type.
CS increased twofold in the LVI at day 1 post-MI
Although the three-step protein extraction protocol allowed us to visualize group differences that otherwise may had be masked by high abundance cellular proteins, a low protein yield was generated in the insoluble fraction. Therefore, we used a two-step protein extraction protocol for immunoblot confirmation of protein differences among the groups. Post-MI, we separated the LV into LVI and noninfarcted remote control (LVC) tissues. Protein samples were fractionated into soluble and insoluble fractions. Tissues were homogenized in phosphate-buffered saline (PBS) solution at a neutral pH with 1×protease inhibitor cocktail (PI) to extract the most water-soluble components, including newly synthesized ECM proteins and degradation products. The insoluble pellet was then treated with a cellular and organelle membrane solubilizing reagent (Sigma Reagent 4). We used immunoblotting to compare the CS protein levels of LVI and LVC to the LV of day 0 control animals. LVI and LVC tissues were harvested and separated under a microscope, after the viable tissue was stained, at days 1, 3, 5, and 7 post-MI. Since the largest statistical differences were observed at day 1 post-MI, we used day 1 samples for our subsequent studies; however the full time-course analysis is presented in the supplemental data (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). There were no differences within the groups based on sex. Male and female results, therefore, were combined. CS protein levels increased approximately twofold in the LVI insoluble fraction 1 day post-MI for both genotypes (p<0.05, Fig. 2). Post-MI, cell death leads to a decrease in mitochondrial function and energy availability. The increase in CS levels in the infarcted region might indicate that CS induction is necessary to respond to new energy demands. In contrast, CS levels were not increased in the soluble and insoluble protein fractions of the remote LVC at day 1 post-MI compared to day 0. To confirm that our results are due to MMP-9 deletion, we also evaluated CS levels after treatment with an MMP-9 inhibitor (MMP-9i). Inhibition of MMP-9 post-MI in WT mice abolished the increase observed in the WT LVI post-MI (p<0.05), providing evidence that MMP-9 is directly or indirectly involved in CS expression post-MI.
FIG. 2.

CS protein levels by immunoblotting. (A) Protein soluble fraction: no differences between genotypes were observed in the soluble CS levels at day 1 post-MI. (B) Protein insoluble fraction: CS levels increased in the infarcted tissue at day 1 post-MI compared to control day 0 values. This increase was abolished with the inhibition of MMP-9. d0, day 0 control; LVI d1, left ventricle infarct at day 1 post-MI; LVC d1, left ventricle remote region at day 1 post-MI; MMP-9i, MMP-9 inhibitor; Null, MMP-9 null; +, positive control human recombinant CS. n=8 per group, mean±SEM; *p<0.05 versus respective day 0; +p<0.05 versus respective LVC day 1; #p<0.05 versus WT LVC day 1, φp<0.05 versus WT LVI day 1. Two loading controls were used, total protein stain and recombinant CS (positive control, 300 ng). The signal intensity of the positive control was used to normalize the data between blots, whereas the protein levels were normalized to the total protein in its respective lane.
In its free state (CSfree), CS exists in open form with its two domains forming a cleft containing the binding site for oxaloacetate (42). When an oxaloacetate molecule binds, the smaller domain rotates, sealing the oxaloacetate binding site (2). This conformational change generates the acetyl-CoA binding site (CSlatent), which results in the acetyl-CoA and oxaloacetate becoming covalently linked and forming citryl-CoA, while still bound to CS (CSactive). Finally, citryl-CoA is hydrolyzed to citrate and CoA (42). Our results indicated that there is differential subcellular localization of CS post-MI compared to day 0 and, probably, different states of CS in the soluble and insoluble protein fractions. We therefore used the CS activity assay to further investigate this observation, as discussed below.
MMP-9 cleaves CS in vitro and in vivo
As can be seen in Figure 3, in vitro exposure of recombinant CS to active MMP-9 resulted in two CS fragments of ∼32 and 20 kDa. The 32 kDa fragment appears to be further cleaved by MMP-9, as evidenced by the much lower protein stain intensity compared to the 20 kDa fragment. A CS fragment with the same apparent molecular weight of 20 kDa has been previously reported by Cauwe et al., implicating MMP-9 in the cleavage of this intracellular protein (8). We performed an immunoblot using the same CS antibody we used for the LV samples. The antibody recognized the recombinant CS full-length protein, but it did not recognize the MMP-9-derived CS fragment. The antibody used was generated against recombinant fragment containing a sequence corresponding to amino acids 44 to 316 of human CS. If MMP-9 cleaves close to the C-terminus region of CS, the antibody will not recognize the C-terminus fragment. This could explain why this MMP-9-derived CS fragment was not observed in the in vivo samples. To investigate CS cleavage by MMP-9 in vivo, we performed dual immunohistochemistry staining in the LV at day 1 post-MI of WT and MMP-9 null animals. The results show that MMP-9 expression was elevated in WT mice in the regions where CS expression was dramatically low, conversely CS expression was observed in the infarcted LV of MMP-9 null mice where MMP-9 is not available. These results suggest that MMP-9 directly affects the levels of CS in the infarcted LV (Fig. 4).
FIG. 3.
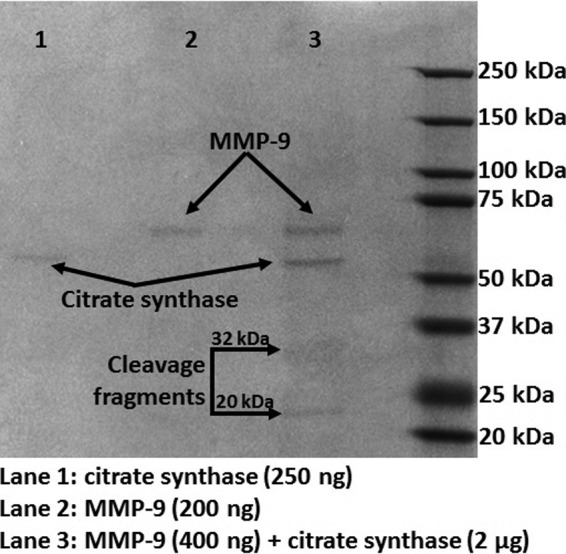
Recombinant CS was cleaved by MMP-9, generating two fragments of ∼32 and 20 kDa. However, the 32 kDa fragment is further cleaved by MMP-9. MMP-9 and CS were added at a ratio of 1:5.
FIG. 4.

Serial sections of the LVI (area within dotted lines) at day 1 post-MI show that CS staining dramatically declines when MMP-9 is present. The left panels show representative pictures of WT animals, with high MMP-9 and low CS expression levels in the LVI. The right panels show serial sections of MMP-9 null mice with CS expression in the LVI area, suggestive of in vivo MMP-9 cleavage of CS. Top panels: Immunostaining for CS (positive staining is dark purple). Bottom panels: Immunostaining for MMP-9 (positive staining is dark brown; arrows show positive staining), 40×magnification.
CS activity was preserved in the MMP-9 null mice
CS activity was assessed in the soluble protein fraction according to the Srere's method at pH 8.0, which is the pH reported for the mitochondrial matrix (33, 48). MI leads to lower pH both extracellularly and intracellularly (29). Rehr et al. measured intracellular myocardial pH by phosphorus-31 nuclear magnetic resonance spectroscopy in ischemia/reperfusion (I/R) and permanent occlusion (PO) MI models (41). Compared to no MI controls, 1 h ischemia and 1 h reperfusion showed a similar pH, indicating that reperfusion brings a quick return to normal intracellular pH values, whereas the PO animals showed reduced pH up to 6 h after occlusion (6.75±0.04 vs. 7.17±0.07 at baseline) (41). The optimal pH for the CS activity has been reported as 8.5; however, this enzyme has a broad pH optimum with decreased activity of only 30% at pH of 6 or 10 (4). The activity assay kit was used at pH 8.0, and the actual CS activities at day 1 post-MI may be different than what was measured in situ. To minimize this limitation, we treated all tissues in the same way to detect differences between the genotype groups. The CS activity was not assessed in the insoluble protein fraction because the presence of the zwitterionic detergent C7BzO interfered with the enzyme activity. Post-MI, the myocardium shows impaired mitochondrial function (44). We observed a pronounced statistically significant decrease in the CS activity 1 day post-MI in the LVI of WT mice, which was expected due to the loss of cardiomyocytes. This decrease in the CS activity was not observed in the MMP-9 null mice (Fig. 5), indicating that the absence of MMP-9 results in preserved CS activity. No significant differences were found in the CS activity in the noninfarcted tissue, paralleling the results obtained for CS protein levels. At day 1 post-MI, MMP-9 levels have been found to increase in LVI tissue but not in the LVC (58). These results are consistent with an MI model in which MMP-9 cleaves active CS contributing to mitochondrial dysfunction at early time points. Here, we show that MMP-9 deletion can improve mitochondrial function by preserving the CS activity.
FIG. 5.

CS activity decreased in the WT infarct region and was maintained in the null mice. (A) Protein soluble fraction LVI: the CS activity decreased at day 1 post-MI in the WT mice, but this decrease was not observed in the MMP-9 null group. (B) Protein soluble fraction LVC: CS activity levels did not significantly changed post-MI in both genotypes. n=8 per group, mean±SEM, *p<0.05 versus respective day 0.
MMP-9 deletion improves mitochondrial function post-MI
The goal of this study was to use proteomics approaches to evaluate the role of MMP-9 deletion on the CS activity and function post-MI. We prepared our samples for MS and immunoblot analysis. Therefore, mitochondria were not isolated, and as such, the samples are not compatible for an oxidation phosphorylation assay. To address the effect of MMP-9 deletion on mitochondria function, we assessed manganese superoxide dismutase (MnSOD), which is an antioxidant enzyme that protects mitochondrial components from superoxide. Increased levels of MnSOD are associated with cardioprotective effects post-I/R (56), and overexpression of MnSOD provides protection to diabetic mitochondria and overall protection to the diabetic heart (47). We quantified protein levels of MnSOD (Fig. 6) in the soluble fraction and found significantly increased values in the MMP-9 null LVI compared to the respective controls (LVC and day 0). This result suggests that MMP-9 deletion confers better mitochondrial function to the myocardium post-MI.
FIG. 6.
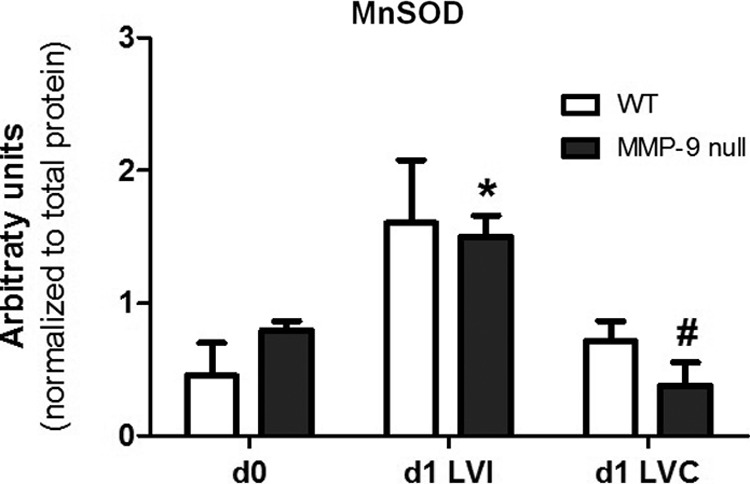
MMP-9 deletion had a protective effect on cardiac mitochondria. Protein levels of the antioxidant protein MnSOD were elevated in the infarcted LV (LVI) of MMP-9 null mice compared to day 0 and remote region (LVC), 1 day post-MI. *p<0.05 versus MMP-9 null d0; #p<0.05 versus MMP-9 null d1 LVC. MnSOD, manganese superoxide dismutase.
MMP-9 deletion does not affect cardiac function at day 1 post-MI
Echocardiographic measurements showed decreased fractional shortening and increased end-diastolic volume (EDV) and end-systolic volume (ESV) at day 1 post-MI (Table 1). Of note, EDV (r2=0.80, p=0.003) and ESV (r2=0.84, p=0.001) values correlated with the CS activity in the MMP-9 null mice, but not in the WT mice (Fig. 7). Loss of the CS activity in WT mice therefore removed the relationship between mitochondrial function and LV geometry. No genotype differences were observed at day 1 post-MI. However, we have previously shown that MMP-9 deletion improves long-term cardiac function (16, 32).
Table 1.
Echocardiographic and Necropsy Measurements
| Day 0 | Day 1 | |||
|---|---|---|---|---|
| WT | MMP-9 null | WT | MMP-9 null | |
| Heart rate (bpm) | 450±13 | 466±13 | 467±13 | 472±11 |
| EDV (μl) | 63.0±3.3 | 56.0±2.7 | 88.0±5.2* | 100.0±6.6* |
| ESV (μl) | 20.0±1.7 | 18.0±1.2 | 78.0±4.9* | 89.0±6.2* |
| Fractional shortening (%) | 37.0±1.3 | 40.0±1.5 | 6.0±0.9* | 6.0±0.8* |
| Infarct area (%) | N/A | N/A | 62±3.0 | 56±3.0 |
| Body weight (g) | 25.1±1.6 | 23.8±1.0 | 23.7±1.8 | 23.8±1.7 |
| LV mass (mg) | 80.3±4.0 | 83.9±3.0 | 82.8±7.0 | 83.9±5.0 |
| Right ventricle mass (mg) | 15.5±1.0 | 16.6±1.0 | 15.9±2.0 | 16.6±1.0 |
| Lung dry weight (mg) | 36.9±1.0 | 41.1±1.0 | 36.0±1.0 | 41.1±2.0 |
| LV remodeling index | 0.78±0.03 | 0.78±0.03 | 1.11±0.09 | 1.19±0.04* |
| LV Hypertrophy index | 4.46±0.11 | 4.82±0.15 | 11.8±1.5* | 9.11±0.95* |
Results are presented as mean±SEM, n=8 per group. *p<0.05 versus respective day 0 control.
N/A, not applicable; WT, wild type; EDD, end-diastolic dimension; ESD, end-systolic dimension; LV remodeling index, EDV/LV mass; LV hypertrophy index, EDD/septum wall thickness in diastole; EDV, end-diastolic volume; ESV, end-systolic volume; LV, left ventricle; MMP, matrix metalloproteinase.
FIG. 7.

EDV (A) and ESV (B) correlated with CS activity in the MMP-9 null group. Data shown corresponds to n=4 per group per time point, days 0 and 1 post-MI. EDV, end-diastolic volume; ESV, end-systolic volume.
MMP-9 deletion increases the levels of plasma proteins associated with improved tissue repair
Quantitative analysis of 58 plasma analytes showed that 13 plasma proteins exhibited significant differences post-MI compared to day 0 (plasma analysis for d0 to d7 time course in supplemental data, Supplementary Fig. S2). For nine of these analytes, differences were only detected in the MMP-9 null group compared to the respective day 0 (all p<0.05): eotaxin, fibrinogen, haptoglobin, monocyte chemotactic protein (MCP)-3, MCP-5, plasminogen activator inhibitor-1 (PAI-1), serum amyloid P component, and tissue inhibitor of metalloproteinase-1 (TIMP-1) increased post-MI and thrombopoietin decreased (Fig. 8). Thrombopoietin was also lower in the MMP-9 null group compared to the WT at day 1 post-MI.
FIG. 8.

The plasma analysis showed differences in nine plasma analyte levels post-MI in the MMP-9 null group. n=8 per group, mean±SEM; *p<0.05 versus respective day 0; #p<0.05 versus day 0 WT; +p<0.05 versus d1 WT.
Increased levels of eotaxin have been associated with improved tissue repair in the MI setting through the CC chemokine-binding receptor 3 (CCR-3) (3). CCR-3 plays a major role in leukocyte traffic and recruitment to sites of tissue damage (53). CCR-3 also mediates the migration of hematopoietic progenitor cells to the ischemic myocardium in response to induced mRNA expression of eotaxin (3). Similarly, MCP-3 has been identified as a homing factor for mesenchymal stem cells (45), which can home to the heart early after MI (34). The increased levels of these ligands in the MMP-9 null mice suggest attenuated LV remodeling at the earlier stages post-MI.
Fibrinogen has been shown to have a chaperone-like activity by specifically binding with partially denatured CS and protecting it from thermal-induced aggregation and inactivation (50). This characteristic of fibrinogen supports the notion that chaperones can redirect the protein aggregation process toward the formation of amorphous deposits, thereby sequestering potentially toxic species from plasma, such as observed after a necrotic event (1). Post-MI, MMP-9 null mice show a robust increase in fibrinogen levels, suggesting a cardioprotective role against systemic inflammation. Haptoglobin and serum amyloid P component have also been reported to have chaperone-like activity (12, 57).
TIMP-1 plasma levels significantly increased at day 1 post-MI and correlated with CS in the MMP-9 null animals (TIMP-1: p<0.01, r2=0.77; Fig. 9). Other groups have reported evidence of an association between TIMP-1 and CS. TIMP-1 mRNA levels increased with exercise in humans and correlated with increased protein levels of CS after 4 weeks of training (20). TIMP-1 is a known MMP-9 inhibitor, which may explain the elevated plasma levels of TIMP-1 in the MMP-9 null group post-MI. The levels of C-reactive protein (CRP) were not different between genotypes and time points; however, CRP levels correlated with the CS activity in the MMP-9 null group (p<0.001, r2=0.87). CRP is a proinflammatory protein that activates the nuclear factor kappa B (NF-κB) pathway (10). Although CRP has never been directly associated with CS activity levels, NF-κB has been reported to activate citrate carrier (CIC) (27). CIC is an integral inner mitochondrial membrane protein that catalyzes the export of citrate from the mitochondrial matrix to the cytosol (28). CIC supplies acetyl-CoA necessary for the production of a number of inflammatory mediators, such as PGE2, NO, and ROS, and is dependent on CS activity (27). To further study how the preserved CS activity observed in the MMP-9 null mice may affect plasma inflammatory markers, we studied the activation of the NF-κB pathway, a key signaling component for early inflammatory activation post-MI.
FIG. 9.
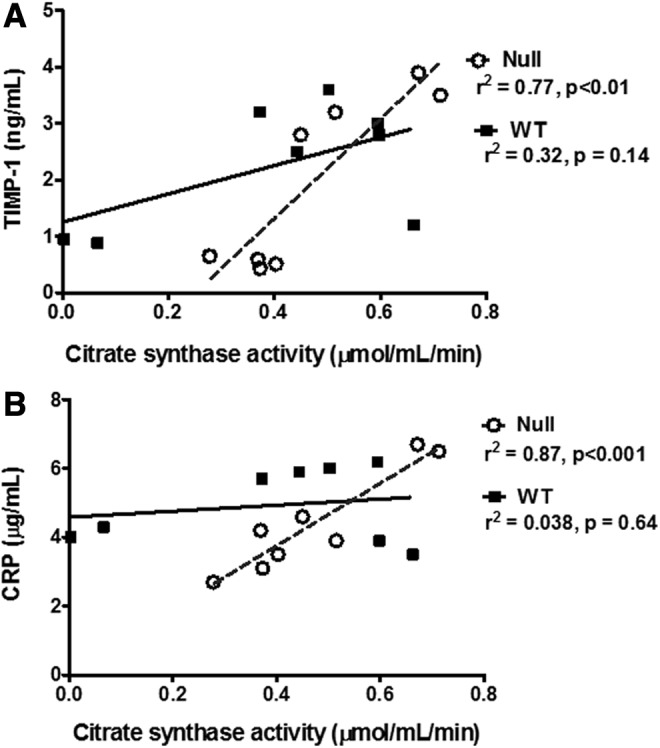
TIMP-1 (A) and CRP (B) correlated with CS activity in the MMP-9 null group. Data shown corresponds to n=4 per group per time point, days 0 and 1 post-MI. CRP, C-reactive protein; TIMP-1, tissue inhibitor of metalloproteinase-1.
NF-κB pathway activation is attenuated with MMP-9 deletion
Post-MI, the infarcted tissue goes through a complex tissue repair process. At day 1 post-MI, the necrotic tissues and cells are removed as part of the inflammatory response. MMP-9 is a key component in this process, and MMP-9 levels increase early post-MI. Accordingly, we assayed the activity of the p50/p65 heterodimer of NF-κB as a measure of the level of involvement of the NF-κB activation pathway in inflammation. At day 1 post-MI, WT mice showed increased activation of the NF-κB pathway in the LVI compared to their respective day 0 (p<0.05, Fig. 10). This increase was not observed in the MMP-9 null mice, suggesting that MMP-9 deletion attenuated the activation of inflammatory gene transcription. Our results are in agreement with previous reports. Other groups have demonstrated that NF-κB activation increases post-MI and peaks at day 3 in a PO model (17, 51). Mice with targeted deletion of the NF-κB subunit p50 showed preserved LV function post-MI and lower tissue levels of MMP-9 (18). The mechanism by which MMP-9 and NF-κB activation interlink has not yet been elucidated. However, our data suggest that preserved mitochondrial function, resulting from MMP-9 deletion, plays an important role in blunting the inflammatory response activation. These results parallel what we observed in plasma, where MMP-9 deletion resulted in increased levels of fibrinogen, which in turn can bind to CS, protecting CS against degradation and therefore preserve the CS activity. Further studies on MMP-9 intracellular substrates are needed to link MMP-9 and regulation of the inflammatory response at the translational level.
FIG. 10.
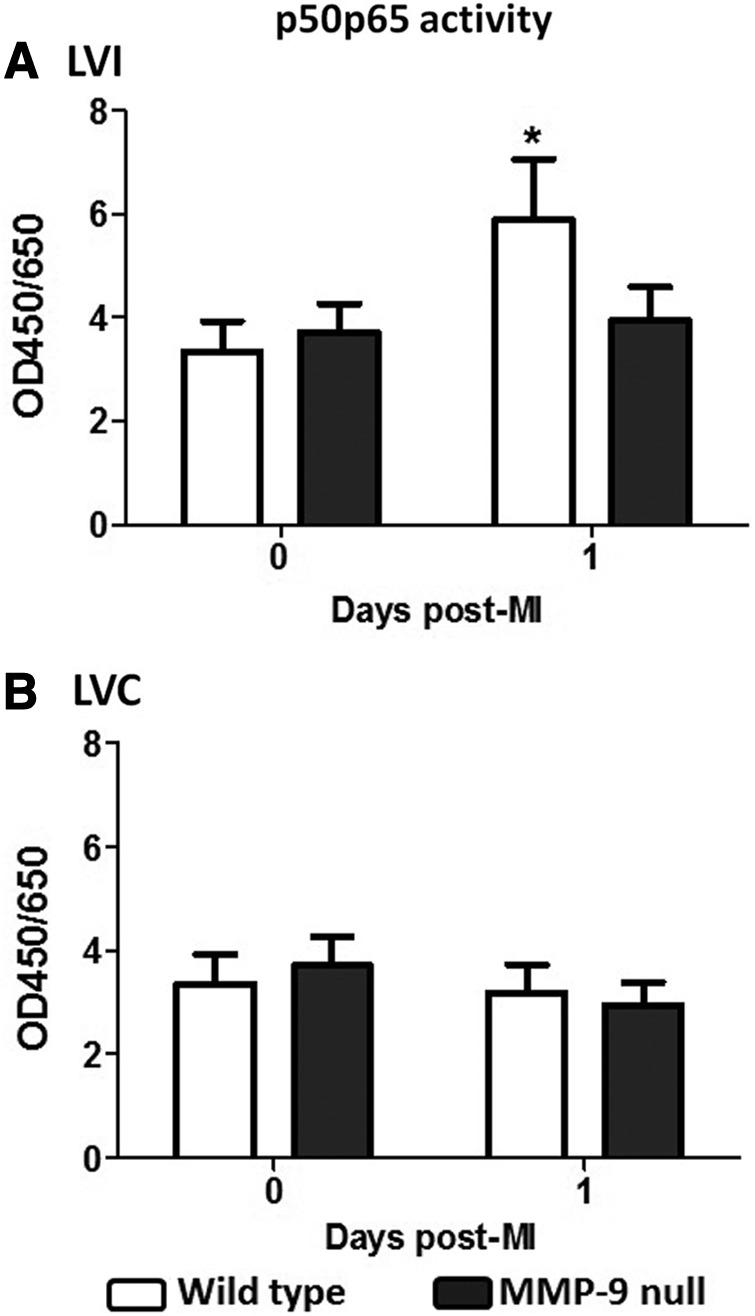
NF-κB pathway activation was attenuated in the MMP-9 null mice in the LVI (A), but not in LVC (B), at day 1 post-MI. n=8 per group, mean±SEM; *p<0.05 versus respective day 0. NF-κB, nuclear factor kappa B.
Conclusion
Our data suggest that MMP-9 deletion improves mitochondrial function post-MI by preventing the decrease in the CS activity levels normally observed in infarcted tissue, which leads to increased energy production and improved mitochondrial function in the infarcted LV. Preserved CS activity resulted in changes in the inflammatory response that translated in increased levels of cardioprotective molecules, such as fibrinogen and eotaxin.
Materials and Methods
Animals and surgery
All animal procedures were conducted according to the “Guide for the Care and Use of Laboratory Animals” (NIH Notice Number: NOT-OD-12-020) and were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio. Male 3- to 7-month-old C57BL6/J WT and MMP-9 null mice were used in this study. Both WT and MMP-9 null colonies were bred in-house as homozygous colonies and were maintained in the same room since birth. The MMP-9 null mice were generated in Zena Werb's laboratory and backcrossed by Lynn Matrisian's laboratory and are on the C57BL/6J background (32, 54). Animals were housed at constant temperature (22°C±2°C) on a 12-h light/dark cycle. They were fed standard laboratory mice chow ad libitum and had free access to tap water. Animals were divided into three main groups: control (no MI, day 0), MI only, and WT MI with MMP-9i. MI was induced by permanent ligation of the left anterior descending coronary artery, as described previously (36). Eight surviving animals were used per genotype (four females and four males) and time point. At 3 h post-MI, infarction was confirmed by echocardiography and osmotic pumps with MMP-9i (0.03 μg/day) were inserted subcutaneously in the WT MMP-9i group. Five time points were analyzed: days 0, 1, 3, 5, and 7 post-MI. MMP-9i was the triple-helical peptide (Gly-Pro-Hyp)4-Gly-mep-Flp-Gly-Pro-Gln-[GlyΨ(PO2H-CH2)Ile]-Tyr-Phe-Gln-Arg-Gly-Val-Arg-Gly-mep-Flp-(Gly-Pro-Hyp)4-Tyr-NH2 with a Ki of 91 nM, where mep=4-methylproline and Flp=4-fluoroproline, synthesized using the methods described previously (30).
Rationale for PO model
In this study, we used an MI model of PO for the following reasons: (i) to dissect the molecular mechanisms of LV remodeling, we induced a robust remodeling response with PO, which has a larger effect size than the I/R model and (ii) ∼20% of MI patients are actually not reperfused, translating to about 250,000 patients each year in the United States that have PO MIs (19). Thus, the PO model is used here as a clinically relevant MI model.
Echocardiography and necropsy evaluations
Echocardiograms were acquired under spontaneous respiration with 1%–2% isoflurane in an oxygen mix. Images were acquired with the use of a Vevo 770™ High-Resolution In Vivo Imaging System (Visual Sonics), at heart rates of >400 bpm. Electrocardiogram and heart rate were monitored throughout the imaging procedure. Echocardiographic measurements were taken from the two-dimensional parasternal long-axis and short-axis (m-mode) recordings of the LV. For each parameter, three images from consecutive cardiac cycles were measured and averaged. The LV remodeling index was calculated as the ratio of EDV to LV mass (26). The hypertrophic index was calculated as the ratio of end-diastolic dimensions to septum wall thickness in diastole.
At necropsy, mice were anesthetized with 1.5%–2.5% isoflurane in an oxygen mix. Heparin (4 U heparin/g body weight) was injected intraperitoneally, and after 5 min, blood was collected from the carotid artery of the mouse. The plasma was isolated by centrifugation and stored with 1×protease inhibitors (Roche Diagnostics) at −80°C. Plasma proteomic profiling of 58 analytes were measured as described previously (35). After blood collection, the coronary vasculature was flushed with the cardioplegic solution (37). The heart was excised, and the LV and right ventricle separated and weighed individually. The LV was sectioned into three transverse sections and stained with 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich) for infarct area determination. The infarct (LVI) and remote (LVC) regions of the apex and base segments were separated under a microscope, individually snap frozen, and stored at −80°C for biochemical analysis. The LV middle section was fixed in 10% zinc formalin, processed in paraffin, and sectioned at 5 μm for immunohistochemistry studies.
Texas three-step protein fractionation
LV samples are initially incubated in a neutral low salt, nondenaturing buffer (0.5 M NaCl, 10 mM Tris base, pH 7.5, and 1×protease inhibitor cocktail [PI]) for the extraction of soluble proteins, including newly synthesized ECM proteins and degradation products (step 1). The remaining tissue is then decellularized by the treatment with sodium dodecyl sulfate (SDS) (1% SDS in PBS and 1×PI, step 2). Finally, the insoluble protein fraction that remains is exposed to acid extraction (4 M GnHCl, 50 mM sodium acetate, pH 5.8, and 1×PI), deglycosylation (150 mM NaCl, 50 mM sodium acetate [pH 6.8], 1×PI, and 0.05 U of each of the following deglycosylation enzymes: chondroitinase ABC from Proteus vulgaris, endo-β-galactosidase from Bacteroides fragilis, and heparinase II from Flavobacterium heparinum), and solubilization with dimethyl sulfoxide (step 3).
Mass spectrometry
Insoluble proteins (step 3; 12.75 μg) were separated in a short 4%–12% Bis-Tris gel. The top 1 cm of the gel, which contained the proteins, was excised and the proteins digested in situ with trypsin. The digests were analyzed by capillary HPLC-ESI-MS/MS on a Thermo Fisher LTQ Orbitrap Velos mass spectrometer, using a top-6 protocol as previously described (14). Mascot (version 2.3.02; Matrix Science) was used to search the MS results against a combination of the mouse subset of the NCBInr database (Mus. [145,083 sequences]) and a database of common contaminants (179 sequences). Methionine oxidation was considered a variable modification; trypsin was specified as the proteolytic enzyme, with one missed cleavage allowed. The Mascot data files were combined in a scaffold (Proteome Software; version 3) for a subset search of the CID spectra using X! Tandem, cross-correlation of the X! Tandem, Mascot results, determination of protein, and peptide identity probabilities. The thresholds for the acceptance of peptide and protein assignments in a scaffold were 90% and 99%, respectively, and minimum of two unique peptides. The data are reported as normalized spectrum counts (protein's relative abundance across biosamples).
Protein extraction for immunoblotting
The LV was homogenized in 1×PBS with 1×protease inhibitors (16 μl for every mg of wet LV) using the Power Gen 1000 Homogenizer (Fisher Scientific). The homogenate was centrifuged at 4700 rpm. The supernatant (the soluble protein fraction) was extracted and placed in a fresh tube to be stored at −80°C. The remaining pellet (insoluble protein fraction) was homogenized in Protein Extraction Reagent 4 (Sigma-Aldrich) with 1×protease inhibitor, using the same volume as before. The insoluble protein fraction was then transferred to a clean tube, and both fractions were stored at −80°C.
Immunoblotting
Total protein (10 μg) from each fraction (soluble and insoluble) were run on 26-well, 4%–12% criterion Bis–Tris gel (Bio-Rad), and transferred onto nitrocellulose membranes (Bio-Rad). Human recombinant CS protein (300 ng) was added as a loading control to normalize the densitometry values between blots. The membranes were stained with MemCode™ Reversible Protein Stain Kit (Thermo Scientific) to verify protein concentration and loading accuracy. Membranes were blocked with 5% nonfat milk (Bio-Rad) and incubated overnight at 4°C with a polyclonal anti-citrate synthetase antibody (1:1000 dilution, ab96600; Abcam). An anti-rabbit secondary antibody (Vector Laboratories) was used, and signal detection was accomplished by SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) (35). Protein levels were quantified by densitometry using the IQ-TL image analysis software (GE Healthcare, Waukesha, WI). Densitometric units were normalized to the densitometry of the total protein stain for the entire lane. The groups were subanalyzed to determine if there were any differences based on sex, genotype, and time point.
CS cleavage assay
Human recombinant CS (2 μg) was incubated with MMP-9 in a 1:5 enzyme/substrate ratio for 18 h at 37°C, in the presence of 1×zymogram buffer. The reaction was stopped with 2.5 mM EDTA, and the cleavage products were run in a 4%–12% Bis-Tris gel for visualization. The gel was stained with InstantBlue (Expedeon), and an image was acquired with ImageQuant 4000 (GE Healthcare).
Immunohistochemistry
Dual staining for CS and MMP-9 was performed in serial sections of zinc–formalin-fixed LV. Tissue sections were deparaffinized, dehydrated, and heat-incubated with the antigen retrieval solution (Dako) for ∼2 min. Tissue was blocked with horse serum (Vector Laboratories) and incubated overnight at 4°C with primary antibody diluted 1:100 in blocking serum (CS, ab96600; MMP-9, ab38898; Abcam). After 30 min incubation with an anti-rabbit secondary antibody (Vector Laboratories), positive signal was visualized using HistoMark BLACK Peroxidase substrate (KPL) for CS and NovaRED substrate (Vector Laboratories) for MMP-9. Representative images were acquired at 40×magnification using an Olympus BX43 microscope.
CS activity assay
We measured CS activity levels with a CS assay kit (Sigma-Aldrich) according to the manufacturer's instructions (n=4/group).
NF-κB pathway
The NF-κB pathway activation was assessed with the colorimetric NF-κB p50/p65 Transcription Factor Assay (#70–510; Millipore) according to the manufacturer's instructions.
Statistical analysis
Data are reported as mean±SEM. The Kruskal–Wallis nonparametric test was used to assess the differences among the groups, and the Dunn's multiple comparison post-test was used when differences were observed. A p<0.05 was considered significant. Linear regressions were performed of the plasma analytes, echocardiographic data, and CS activity.
Supplementary Material
Abbreviations Used
- CCR-3
CC chemokine-binding receptor 3
- CIC
citrate carrier
- CRP
C-reactive protein
- CS
citrate synthase
- ECM
extracellular matrix
- EDV
end-diastolic volume
- ESV
end-systolic volume
- HPLC-ESI-MS/MS
HPLC-electrospray ionization tandem mass spectrometry
- I/R
ischemia/reperfusion
- LV
left ventricle
- LVC
left ventricle remote region (control, noninfarcted tissue)
- LVI
left ventricle infarcted area
- MCP
monocyte chemotactic protein
- MI
myocardial infarction
- MMPs
matrix metalloproteinases
- MMP-9i
MMP-9 inhibitor
- MnSOD
manganese superoxide dismutase
- MPO
myeloperoxidase
- MS
mass spectrometry
- NF-κB
nuclear factor kappa B
- NO
nitric oxide
- PAI-1
plasminogen activator inhibitor-1
- PBS
phosphate-buffered saline
- PGE2
prostaglandin E2
- PI
protease inhibitor cocktail
- PO
permanent occlusion
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
- TCA
tricarboxylic acid
- TIMP-1
tissue inhibitor of metalloproteinase-1
- WT
wild type
Acknowledgments
We acknowledge the support from AHA for POST14350034 and NIH/NHLBI T32 HL074464 to K.Y.D.-P., from NIH-NCCAM-AT006704 to G.V.H., from NIH/NHLBI HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center and R01 HL075360, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505 to M.L.L. Mass spectrometry analyses were conducted in the UTHSCSA Institutional Mass Spectrometry Laboratory. The expert technical assistance of Kevin W. Hakala and the support from the University of Texas Health Science Center at San Antonio are gratefully acknowledged for these analyses.
Author Disclosure Statement
No competing financial interests exist
References
- 1.Barral JM, Broadley SA, Schaffar G, and Hartl FU. Roles of molecular chaperones in protein misfolding diseases. Semin Cell Dev Biol 15: 17–29, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Bayer E, Bauer B, and Eggerer H. Evidence from inhibitor studies for conformational changes of citrate synthase. Eur J Biochem 120: 155–160, 1981 [DOI] [PubMed] [Google Scholar]
- 3.Bonaros N, Sondermeijer H, Schuster M, Rauf R, Wang SF, Seki T, Skerrett D, Itescu S, and Kocher AA. CCR3- and CXCR4-mediated interactions regulate migration of CD34+ human bone marrow progenitors to ischemic myocardium and subsequent tissue repair. J Thorac Cardiovasc Surg 136: 1044–1053, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Bond DR, Mester T, Nesbo CL, Izquierdo-Lopez AV, Collart FL, and Lovley DR. Characterization of citrate synthase from Geobacter sulfurreducens and evidence for a family of citrate synthases similar to those of eukaryotes throughout the Geobacteraceae. Appl Environ Microbiol 71: 3858–3865, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borutaite V. Mitochondria as decision-makers in cell death. Environ Mol Mutagen 51: 406–416, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Bugger H, Chemnitius JM, and Doenst T. Differential changes in respiratory capacity and ischemia tolerance of isolated mitochondria from atrophied and hypertrophied hearts. Metabolism 55: 1097–1106, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Butler GS, Dean RA, Tam EM, and Overall CM. Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol Cell Biol 28: 4896–4914, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cauwe B, Martens E, Proost P, and Opdenakker G. Multidimensional degradomics identifies systemic autoantigens and intracellular matrix proteins as novel gelatinase B/MMP-9 substrates. Integr Biol (Camb) 1: 404–426, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Cauwe B, Martens E, Van den Steen PE, Proost P, Van Aelst I, Blockmans D, and Opdenakker G. Adenylyl cyclase-associated protein-1/CAP1 as a biological target substrate of gelatinase B/MMP-9. Exp Cell Res 314: 2739–2749, 2008 [DOI] [PubMed] [Google Scholar]
- 10.Chang JW, Kim CS, Kim SB, Park SK, Park JS, and Lee SK. C-reactive protein induces NF-kappaB activation through intracellular calcium and ROS in human mesangial cells. Nephron Exp Nephrol 101: e165–e172, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Choi DH, Kim EM, Son HJ, Joh TH, Kim YS, Kim D, Flint Beal M, and Hwang O. A novel intracellular role of matrix metalloproteinase-3 during apoptosis of dopaminergic cells. J Neurochem 106: 405–415, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Coker AR, Purvis A, Baker D, Pepys MB, and Wood SP. Molecular chaperone properties of serum amyloid P component. FEBS Lett 473: 199–202, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Cuadrado E, Rosell A, Borrell-Pages M, Garcia-Bonilla L, Hernandez-Guillamon M, Ortega-Aznar A, and Montaner J. Matrix metalloproteinase-13 is activated and is found in the nucleus of neural cells after cerebral ischemia. J Cereb Blood Flow Metab 29: 398–410, 2009 [DOI] [PubMed] [Google Scholar]
- 14.de Castro Bras LE, Deleon KY, Ma Y, Dai Q, Hakala K, Weintraub ST, and Lindsey ML. Plasma fractionation enriches post-myocardial infarction samples prior to proteomics analysis. Int J Proteomics 2012: 397103, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dean RA, Butler GS, Hamma-Kourbali Y, Delbe J, Brigstock DR, Courty J, and Overall CM. Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/Connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol Cell Biol 27: 8454–8465, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, and Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 106: 55–62, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frantz S, Fraccarollo D, Wagner H, Behr TM, Jung P, Angermann CE, Ertl G, and Bauersachs J. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res 57: 749–756, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, and Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. FASEB J 20: 1918–1920, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Gharacholou SM, Alexander KP, Chen AY, Wang TY, Melloni C, Gibler WB, Pollack CV, Jr., Ohman EM, Peterson ED, and Roe MT. Implications and reasons for the lack of use of reperfusion therapy in patients with ST-segment elevation myocardial infarction: findings from the CRUSADE initiative. Am Heart J 159: 757–763, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Hoier B, Nordsborg N, Andersen S, Jensen L, Nybo L, Bangsbo J, and Hellsten Y. Pro- and anti-angiogenic factors in human skeletal muscle in response to acute exercise and training. J Physiol 590(Pt 3): 595–606, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holmuhamedov EL, Oberlin A, Short K, Terzic A, and Jahangir A. Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PLoS One 7: e44667, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou ZH, Lu B, Gao Y, Cao HL, Yu FF, Jing N, Chen X, Cong XF, Roy SK, and Budoff MJ. Matrix metalloproteinase-9 (MMP-9) and myeloperoxidase (MPO) levels in patients with nonobstructive coronary artery disease detected by coronary computed tomographic angiography. Acad Radiol 20: 25–31, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Huss JM. and Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest 115: 547–555, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang IK, Park SM, Kim SY, and Lee ST. A proteomic approach to identify substrates of matrix metalloproteinase-14 in human plasma. Biochim Biophys Acta 1702: 79–87, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Iijima T, Mishima T, Akagawa K, and Iwao Y. Mitochondrial hyperpolarization after transient oxygen-glucose deprivation and subsequent apoptosis in cultured rat hippocampal neurons. Brain Res 993: 140–145, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Illman SA, Keski-Oja J, Parks WC, and Lohi J. The mouse matrix metalloproteinase, epilysin (MMP-28), is alternatively spliced and processed by a furin-like proprotein convertase. Biochem J 375(Pt 1): 191–197, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, and Iacobazzi V. The mitochondrial citrate carrier: a new player in inflammation. Biochem J 438: 433–436, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Kaplan RS, Mayor JA, and Wood DO. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J Biol Chem 268: 13682–13690, 1993 [PubMed] [Google Scholar]
- 29.Kemi OJ, Arbo I, Hoydal MA, Loennechen JP, Wisloff U, Smith GL, and Ellingsen O. Reduced pH and contractility in failing rat cardiomyocytes. Acta Physiol 188: 185–193, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Lauer-Fields JL, Whitehead JK, Li S, Hammer RP, Brew K, and Fields GB. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J Biol Chem 283: 20087–20095, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael L, and Entman M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 103: 2181–2187, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM, Jr., Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, and Spinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol 290: H232–H239, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, and Tsien RY. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci U S A 95: 6803–6808, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma J, Ge J, Zhang S, Sun A, Shen J, Chen L, Wang K, and Zou Y. Time course of myocardial stromal cell-derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats with experimental myocardial infarction. Basic Res Cardiol 100: 217–223, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Ma Y, Chiao YA, Zhang J, Manicone AM, Jin YF, and Lindsey ML. Matrix metalloproteinase-28 deletion amplifies inflammatory and extracellular matrix responses to cardiac aging. Microsc Microanal 18: 81–90, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, and Lindsey ML. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting m2 macrophage activation. Circ Res 112: 675–688, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michael LH, Ballantyne CM, Zachariah JP, Gould KE, Pocius JS, Taffet GE, Hartley CJ, Pham TT, Daniel SL, Funk E, and Entman ML. Myocardial infarction and remodeling in mice: effect of reperfusion. Am J Physiol 277: H660–H668, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Moreira JB, Bechara LR, Bozi LH, Jannig PR, Monteiro AW, Dourado PM, Wisloff U, and Brum PC. High- versus moderate-intensity aerobic exercise training effects on skeletal muscle of infarcted rats. J Appl Physiol 114: 1029–1041, 2013 [DOI] [PubMed] [Google Scholar]
- 39.Park SM, Hwang IK, Kim SY, Lee SJ, Park KS, and Lee ST. Characterization of plasma gelsolin as a substrate for matrix metalloproteinases. Proteomics 6: 1192–1199, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Penna C, Perrelli MG, and Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxid Redox Signal 18: 556–599, 2013 [DOI] [PubMed] [Google Scholar]
- 41.Rehr RB, Clarke G, Tatum J, Hirsch J, Fatouros P, Lower R, and Wetstein L. Intracellular myocardial pH measured in vivo with sustained and reperfused coronary occlusion. Surgery 102: 178–185, 1987 [PubMed] [Google Scholar]
- 42.Remington S, Wiegand G, and Huber R. Crystallographic refinement and atomic models of two different forms of citrate synthase at 2.7 and 1.7 A resolution. J Mol Biol 158: 111–152, 1982 [DOI] [PubMed] [Google Scholar]
- 43.Roe MT, Messenger JC, Weintraub WS, Cannon CP, Fonarow GC, Dai D, Chen AY, Klein LW, Masoudi FA, McKay C, Hewitt K, Brindis RG, Peterson ED, and Rumsfeld JS. Treatments, trends, and outcomes of acute myocardial infarction and percutaneous coronary intervention. J Am Coll Cardiol 56: 254–263, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Sanbe A, Tanonaka K, Hanaoka Y, Katoh T, and Takeo S. Regional energy metabolism of failing hearts following myocardial infarction. J Mol Cell Cardiol 25: 995–1013, 1993 [DOI] [PubMed] [Google Scholar]
- 45.Schenk S, Mal N, Finan A, Zhang M, Kiedrowski M, Popovic Z, McCarthy PM, and Penn MS. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem Cells 25: 245–251, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Schild L, Reinheckel T, Wiswedel I, and Augustin W. Short-term impairment of energy production in isolated rat liver mitochondria by hypoxia/reoxygenation: involvement of oxidative protein modification. Biochem J 328(Pt 1): 205–210, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen X, Zheng S, Metreveli NS, and Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 55: 798–805, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Srere PA. Controls of citrate synthase activity. Life Sci 15: 1695–1710, 1974 [DOI] [PubMed] [Google Scholar]
- 49.Sung MM, Schulz CG, Wang W, Sawicki G, Bautista-Lopez NL, and Schulz R. Matrix metalloproteinase-2 degrades the cytoskeletal protein alpha-actinin in peroxynitrite mediated myocardial injury. J Mol Cell Cardiol 43: 429–436, 2007 [DOI] [PubMed] [Google Scholar]
- 50.Tang H, Fu Y, Cui Y, He Y, Zeng X, Ploplis VA, Castellino FJ, and Luo Y. Fibrinogen has chaperone-like activity. Biochem Biophys Res Commun 378: 662–667, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tillmanns J, Carlsen H, Blomhoff R, Valen G, Calvillo L, Ertl G, Bauersachs J, and Frantz S. Caught in the act: in vivo molecular imaging of the transcription factor NF-kappaB after myocardial infarction. Biochem Biophys Res Commun 342: 773–774, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Uria JA. and Lopez-Otin C. Matrilysin-2, a new matrix metalloproteinase expressed in human tumors and showing the minimal domain organization required for secretion, latency, and activity. Cancer Res 60: 4745–4751, 2000 [PubMed] [Google Scholar]
- 53.Vandervelde S, van Luyn MJ, Tio RA, and Harmsen MC. Signaling factors in stem cell-mediated repair of infarcted myocardium. J Mol Cell Cardiol 39: 363–376, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, and Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93: 411–422, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson EM. and Spinale FG. Myocardial remodelling and matrix metalloproteinases in heart failure: turmoil within the interstitium. Ann Med 33: 623–634, 2001 [DOI] [PubMed] [Google Scholar]
- 56.Yamashita N, Hoshida S, Otsu K, Asahi M, Kuzuya T, and Hori M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J Exp Med 189: 1699–1706, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yerbury JJ, Kumita JR, Meehan S, Dobson CM, and Wilson MR. alpha2-Macroglobulin and haptoglobin suppress amyloid formation by interacting with prefibrillar protein species. J Biol Chem 284: 4246–4254, 2009 [DOI] [PubMed] [Google Scholar]
- 58.Zamilpa R, Kanakia R, Cigarroa IV J, Dai Q, Escobar GP, Martinez H, Jimenez F, Ahuja SS, and Lindsey ML. CC chemokine receptor 5 deletion impairs macrophage activation and induces adverse remodeling following myocardial infarction. Am J Physiol Heart Circul Physiol 300: H1418–H1426, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zamilpa R. and Lindsey ML. Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. J Mol Cell Cardiol 48: 558–563, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zamilpa R, Lopez EF, Chiao YA, Dai Q, Escobar GP, Hakala K, Weintraub ST, and Lindsey ML. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics 10: 2214–2223, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao YG, Xiao AZ, Newcomer RG, Park HI, Kang T, Chung LW, Swanson MG, Zhau HE, Kurhanewicz J, and Sang QX. Activation of pro-gelatinase B by endometase/matrilysin-2 promotes invasion of human prostate cancer cells. J Biol Chem 278: 15056–15064, 2003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.