

Abstract
Background
Analysis of large datasets produced by mass spectrometry-based proteomics relies on database search algorithms to sequence peptides and identify proteins. Several such scoring methods are available, each based on different statistical foundations and thereby not producing identical results. Here, the aim is to compare peptide and protein identifications using multiple search engines and examine the additional proteins gained by increasing the number of technical replicate analyses.
Methods
A HeLa whole cell lysate was analyzed on an Orbitrap mass spectrometer for 10 technical replicates. The data were combined and searched using Mascot, SEQUEST, and Andromeda. Comparisons were made of peptide and protein identifications among the search engines. In addition, searches using each engine were performed with incrementing number of technical replicates.
Results
The number and identity of peptides and proteins differed across search engines. For all three search engines, the differences in proteins identifications were greater than the differences in peptide identifications indicating that the major source of the disparity may be at the protein inference grouping level. The data also revealed that analysis of 2 technical replicates can increase protein identifications by up to 10-15%, while a third replicate results in an additional 4-5%.
Conclusions
The data emphasize two practical methods of increasing the robustness of mass spectrometry data analysis. The data show that 1) using multiple search engines can expand the number of identified proteins (union) and validate protein identifications (intersection), and 2) analysis of 2 or 3 technical replicates can substantially expand protein identifications. Moreover, information can be extracted from a dataset by performing database searching with different engines and performing technical repeats, which requires no additional sample preparation and effectively utilizes research time and effort.
Keywords: Mass spectrometry, proteomics, search engine
Introduction
High throughput proteomics methods can generate large volumes of data that precludes manual validation. One of the most commonly used mass spectrometry-based techniques, termed shotgun proteomics, involves the digestion of sample proteins typically by trypsin or another specific protease and subsequent peptide sequencing using tandem mass spectrometry aiming to catalogue all proteins in a particular sample. The mass spectrometer provides the mass-to-charge ratios of the precursor peptides (the MS1 measurement), as well as mass-to-charge ratios of fragments produced from these peptides (the MS2 measurement) allowing for the subsequent determination of the protein(s) from which these peptides originated. Search engines attempt to match peptides from in silico digested proteins to those measured by the mass spectrometer. With the availability of various search engines powered by different algorithms, each producing unique sets of protein identifications, data analysis can be a daunting task.
Database-searching algorithms assign mass spectra to peptide sequences in protein databases and provide scores for each assignment. A number of software applications (e.g., Mascot [1], SEQUEST [2], and MaxQuant/Andromeda [3]) are available for identifying peptides from mass spectra. These applications rely on algorithm-dependent measures to determine the quality of peptide and protein identifications. Peptide identification is largely statistically-based and as such, an inherent risk exists of obtaining false positives. Currently, no independent measure is universally available, yet several applications can reliably access similarities and differences among a variety of search engines [4-7].
The threshold of protein detection is commonly determined by a false positive rate (FDR) [4]. The FDR is generally calculated by searching a decoy database with the same protein entries as the search database, but consisting of reversed or scrambled sequences and dividing the false positives by the total proteins identified. The FDR is typically fixed at 1% to 5% at the protein level, meaning that 10 to 50 proteins are false positives per 1000 proteins that may have been identified. It follows that if a peptide or protein is identified by a series of search engines (each with a 1% protein FDR), fewer false positives will be observed in the region of intersection. Inversely, taking the union of these search engines will increase the protein FDR over 1%. Recently, linear discriminatory analysis methods are gaining popularity as a result of the robust, multi-dimensional analysis of peptide and protein identification [5]. The goal of these methods, however, remains to identify a comprehensive set of proteins while minimizing false positives.
In this study, peptide and protein identifications were compared using multiple search engines - Mascot, SEQUEST, and MaxQuant – to investigate the overlap among the identifications as determined by each algorithm. In addition, the percentage of additional proteins gained by increasing the number of technical replicate analyses are examined. For these investigations, a whole cell lysate of HeLa cells, which has been analyzed via a 2 hr liquid chromatography gradient on an Orbitrap-based mass spectrometer [6] for 10 technical replicates is used. These results may not be generalizable to all samples, but the aim is to present a framework, which other researchers can use and expand for their particular sample sets or applications.
Materials and Methods
Materials
Dulbecco's modified Eagle's-F12 medium (DMEM/F12; 11330) was purchased from Gibco (Carlsbad, CA). Fetal bovine serum (FBS; F0392) was purchased from Sigma (St. Louis, MO). CellStripper (25-056-CL) was purchased from Mediatech (Manassas, VA). Sequencing-grade modified trypsin (V5111) was obtained from Promega (Madison, WI). Other reagents and solvents were from Sigma-Aldrich and Burdick & Jackson, respectively.
Cell growth and harvesting of HeLa cells
In brief, HeLa cells were propagated in Dulbecco's modified Eagle's-F12 medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Upon achieving >90% confluency, the growth media was aspirated from the 15 cm dish and the cells were washed 3 times with ice-cold phosphate-buffered saline (PBS). The cells were dislodged with non-enzymatic CellStripper, harvested by scraping following the addition of 10 mL PBS and pelleted by centrifugation at 3,000 × g for 5 min at 4°C, after which the supernatant was removed.
Cell lysis and protein extraction
One milliliter of TBSp (50 mM Tris, 150 mM NaCl, pH 7.4 supplemented with 1X Roche Complete protease inhibitors), 1% Triton X-100, and 0.5% SDS was added to each cell pellet. Cells were homogenized with 12 passages through a 27 gauge (1.25 inches long) needle and incubated on ice with gentle agitation for 1 hour. The homogenate was sedimented by ultracentrifugation at 100,000 × g for 60 minutes at 4°C. Protein concentration of the supernatant was determined using the bicinchoninic acid (BCA) assay (23225, ThermoFisher Scientific). Protein concentration was adjusted to 2 mg/mL using TBSp. Protein was reduced in 20 mM TCEP and alkylated with 1% acrylamide for 30 minutes at room temperature.
Acetone precipitation
Ice-cold 100% acetone (four sample volumes; 500 μL total) was added to 125 μL of sample (∼250 μg of protein), vortexed briefly, and incubated at -20°C for 3 hours. The samples were then centrifuged at 20,000×g at 4°C for 30 minutes. The supernatants were carefully aspirated and the pellets allowed to air dry at room temperature.
Tryptic digestion
For tryptic digestion, the sample was resuspended in 50 mM ammonium bicarbonate, pH 8.1. The sample was incubated with 2.5 μg of trypsin for 16 hrs. Following the incubation, the reaction was acidified with formic acid to a final concentration of 0.1% and evaporated via vacuum centrifugation. To remove interfering substances prior to mass spectrometry analysis, peptides were isolated with C18 spin columns following manufacturer's instructions. Samples were again vacuum centrifuged to dryness and stored at -80°C until analysis. Prior to mass spectrometric analysis, the peptides were resuspended in sample loading buffer (5% formic acid, 5% acetonitrile, 90% water).
Mass spectrometry
The peptides were subjected to fractionation using reversed-phase high performance liquid chromatography (HPLC; Thermo Scientific, Waltham, MA) and the gradient-eluted peptides were analyzed by a hyphenated Orbitrap hybrid mass spectrometer (Thermo Scientific, Waltham, MA). The liquid chromatography columns (15 cm × 100 μm ID) were packed in-house. Samples were analyzed with a 90 minute linear gradient (5-35% acetonitrile with 0.2% formic acid) and data were acquired in a data dependent manner, in which MS/MS fragmentation was performed using the 10 most intense peaks of every full MS scan.
Database searching
RAW files were downloaded from the Proteome Commons tranche using: asb+K4xwG/XVIc4hR8kuZ46pdR3LClQa/RgOI+4/FJ9TsXHge/m97AHzBhh1c1Vbn9kDsNg+/gmowO p0AF2EHc3jUMkAAAAAAABu4Q = = [11]. Data were searched against the UniProt human database (downloaded May 1, 2012) using Mascot, SEQUEST, and MaxQuant. For Mascot (v2.4) and SEQUEST searches, data were processed through ProteomeDiscoverer (v 1.3; ThermoFisher Scientific). For MaxQuant (v. 1.2.2.5), the Andromeda search engine was used. Search parameters are listed in Table 1. Two miscleavages were allowed per peptide and mass tolerances of ± 10 ppm for precursor and of ± 0.8 Da for fragment ions were used. Amino acid modifications: fixed: propionamide (Cys); variable: deamidation (Asn/Gln), oxidation (Met), and Acetylation (N-term). The false discovery rate (FDR) of 1% at the protein level was determined by searching the same dataset against the target database and a decoy database; the latter featured the reversed amino acid sequences of all the entries in the database above [7, 8].
Table 1. Parameters for Mascot, SEQUEST, and MaxQuant Andromeda.
| Parameter | Setting |
|---|---|
| Database | Human SwisProt (Downloaded May 1, 2012) |
| Missed cleavages | 2 |
| Enzyme specificity | Typsin |
| Precursor mass tolerance | 20 ppm |
| Fragment mass tolerance | 0.8 Da |
| Dynamic modifications | Deamidated (NQ), Oxidation (M), Acetylation (N-term) |
| Static modifications | Propionamide (C) |
| Protein False Discovery Rate | 1% |
Venn diagrams
The VENNY on-line Venn diagram plotter was used to obtain lists of proteins exclusive to or in common among the sample types investigated [9].
Results
Peptide overlap among the search engines was greater than that of proteins
The data from a total of 10 technical replicates of HeLa whole cell lysates were searched with three different engines: Mascot, SEQUEST, and MaxQuant. Each of the three search engines identified several thousand peptides corresponding to several hundred proteins in the queried database (Table 2). A total of 2152, 2283, and 2019 proteins were identified for Mascot, SEQUEST and MaxQuant searches, respectively. These proteins were determined from 13235, 14543, and 14892 peptides for Mascot, SEQUEST and MaxQuant, respectively. The peptide corresponded to 100749, 116262, and 121653 peptide-spectra matches (PSM) for Mascot, SEQUEST and MaxQuant, respectively.
Table 2. Protein Summary.
| No. of proteins | No. of unique peptides | No. of peptide-spectra matches | |
|---|---|---|---|
| Mascot | 2152 | 13235 | 100749 |
| SEQUEST | 2283 | 14543 | 116262 |
| MaxQuant | 2019 | 14892 | 121653 |
No., number.
At the peptide level, 69% overlapped in all 3 search engines, while an additional 12% overlapped in 2 of the 3 search engines, leaving 19% of the peptides identified in only one search engine (Figure 1A). According to these data, MaxQuant differed the most among the three search engines, as approximately 11% of the total peptides identified were exclusive to MaxQuant. At the protein level, less overlap was apparent among the search engines (Figure 1B). Several hundred protein groups were identified by each search engine. Of these proteins, 47% overlapped in all 3 search engines, while an additional 28% overlapped in 2 of 3 search engines, and 25% of the peptides were identified in only one search engine. The proportions of redundancies of protein identifications differed somewhat from those of peptide identifications (Figure 1C). Proportionately fewer proteins (47%) than peptides (69%) were identified by all 3 search engines, and accordingly more proteins (25%) than peptides (19%) were identified by only a single search engine. It follows that binary comparisons may indicate more clearly the degree by which the peptides and protein identifications of the three search engines differ.
Figure 1. Search engine comparison.
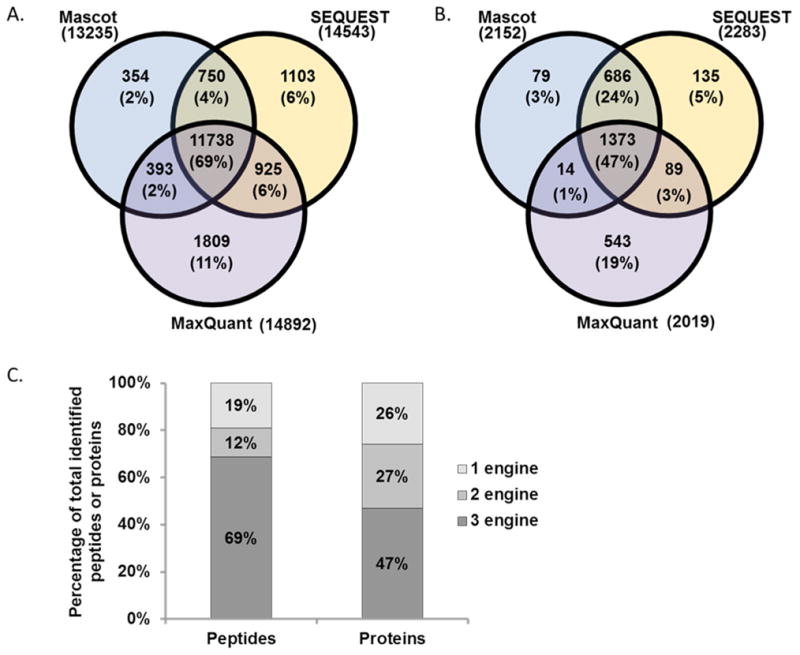
Venn diagrams comparing A) peptide identifications and B) protein identifications. C) Bar graph illustrating the redundancy of peptides and proteins identified by one, two, and three search engines
Binary comparison of search engines revealed differences in peptide and protein identifications between search engines
Binary comparisons were performed to compare the similarities between pairs of search engine results. At the peptide level, comparing the Mascot versus SEQUEST search engines, revealed an overlap of 82% (Figure 2A), Mascot versus MaxQuant, gave an overlap of 76% (Figure 2B), while SEQUEST and MaxQuant showed an overlap of 76% (Figure 2C). Overall, all binary comparisons showed overlap of greater than 75% between the two search engines compared.
Figure 2. Binary comparisons of peptides and proteins identified by Mascot, SEQUEST, and MaxQuant.
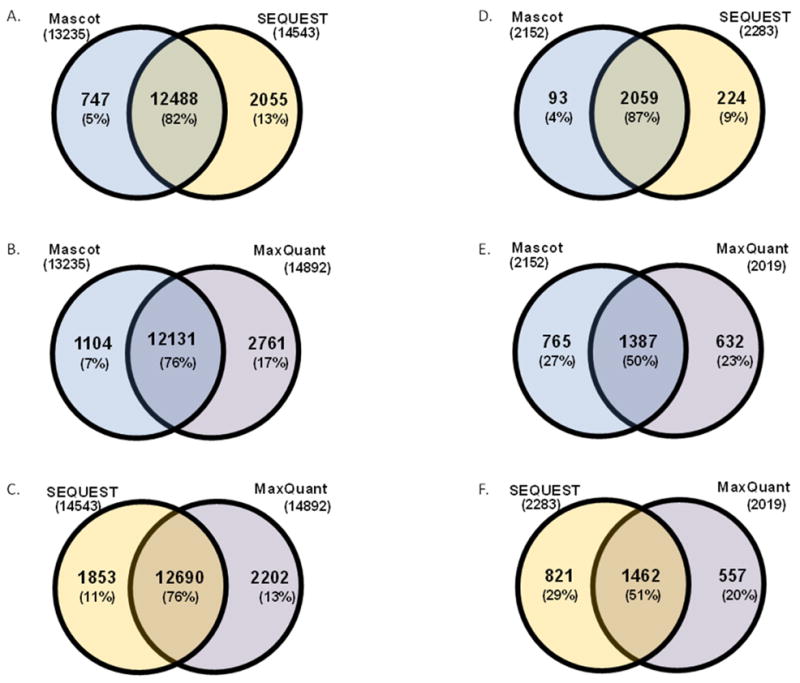
Peptide overlap for A) Mascot versus SEQUEST, B) Mascot versus MaxQuant, and C) SEQUEST versus MaxQuant. Protein overlaps for D) Mascot versus SEQUEST, E) Mascot versus MaxQuant, and F) SEQUEST versus MaxQuant.
Similarly, the search engines were compared at the level of protein identifications. Comparing the Mascot and SEQUEST search engines, revealed an overlap of 87% (Figure 2D), Mascot versus MaxQuant, gave an overlap of 50% (Figure 2E), while SEQUEST and MaxQuant showed an overlap of 51% (Figure 2F). Unlike the peptide comparison in which the range of overlap was between 76% and 82%, the overlap between proteins was relatively wider at 50% to 87%. Mascot and SEQUEST showed the highest overlap of peptides; similarly, these search engines had the greatest overlap in regard to protein identifications.
A greater overlap between Mascot and MaxQuant may have been expected, particularly as Andromeda – the search engine behind MaxQuant – has shown strong correlation with Mascot [3], however, this occurrence may have been the result of protein grouping to reduce redundancy, as performed post-search by each application as the Mascot data were processed via ProteomeDiscoverer. If peptides are shared among proteins, protein grouping methods basically reduce the number of identified proteins by assigning each peptide to the protein with the highest total probability. Algorithms investigating this “protein inference” problem have been intensively studied [10, 11]. Moreover, although the identical database was used for all three search engines, the protein grouping for both Mascot and SEQUEST was performed via ProteomeDiscoverer, while Andromeda had its own unique method of protein grouping. Without the availability of a MaxQuant/Andromeda node in ProteomeDiscoverer, a proper comparison of the protein overlaps between Mascot or SEQUEST with MaxQuant is not currently possible.
Technical replicate analyses had a greater impact on the number of unique peptides than on the number proteins identified, although the trend tapers off after 3 replicates
A total of 10 replicates of HeLa whole cell lysates were analyzed on an Orbitrap mass spectrometer, after which database searches were performed, adding one replicate to each subsequent search (i.e., the first search consisted of only a single run, the second search was of two replicates, the third three, and so forth). The sequence of replicate addition mirrors that of mass spectrometric acquisition (i.e., the “2 replicates” consisted of the first and second mass spectrometry analyses, while the “3 replicates” consisted on the first, second, and third mass spectrometry analyses, and so forth). The ten searches were performed using the three search engines. For each of the 10 searches, the files were searched together so that a list of protein groups with a 1% FDR was obtained (i.e., files were not simply searched individually at a 1% FDR threshold and the results combined), as doing so will inflate the FDR. The number of additional peptides and proteins identified with each additional replicate added to the search were then determined.
At both the peptide (Figure 3A) and protein (Figure 3B) level, results show an expected overall decrease in the number of additional proteins identified with the subsequent addition of another data file. After the third replicate, the gains in the numbers of both peptides and proteins began to stabilize. When comparing among search engines, this trend was consistent for both peptides and proteins. Analysis of the peptide results demonstrated that the addition of a single replicate increased the number of peptides identified by 25-30%, which resulted in an 11-15% increase in protein identification when using either search engine. In general, relatively fewer additional proteins were identified with subsequent replicates. Three replicates resulted in less of an increase in peptides (∼10%) and proteins (∼5%), while the gains were minimal for 4 or more replicates (approaching the FDR of 1%).
Figure 3. Additional peptides and proteins acquired with technical replicates.

A total of 10 replicate analyses were performed. A) Additional peptides identified with subsequent technical replicates. B) Additional proteins identified with subsequent technical replicates.
Discussion
The data show shown that using multiple search engines can 1) expand the number of identified proteins and 2) provide evidence supporting the validity of such identifications from single search engines which can effectively limit targets for downstream analysis. The data also show that although disparities exist among search engines, the majority of these differences can be attributed to the protein grouping level. The increase in additional peptide and protein identifications upon searching with more technical replicates was also investigated. From the analysis, it was determined that substantial increases in additional peptide and protein identifications were evident when searches consist of 2 or more replicates compared to just a single run. However, the increases in the identification of previously-unidentified peptides and proteins tapered off with 4 or more technical replicates. In essence, more robust data sets can be obtained if the increase of proteins identified in additional replicates justifies the increase in instrument time needed to collect these replicate data.
Analyzing technical and biological replicates leads to a greater number of protein identifications, albeit at the expense of reagents, sample processing, and increased mass spectrometer usage. From the data, the analysis of two replicates resulted in approximately 25-30% increase in peptides for each search engine. At the protein level, these increases resulted in 11-15% more protein identifications. However, these additional proteins were obtained with the doubling of instrument acquisition time. Adding a third technical replicate, triples the time of acquisition, but adds only a fraction of additional peptides and proteins. With the third technical replicate, approximately 10% additional peptides are identified, which corresponds to a concurrent protein identification increase of 5%. With the quadrupling of acquisition time for 4 replicates (and subsequent replicates), the gain of additional peptides and proteins is much less. Approximately 5% additional peptides are identified when searching four or more replicates regardless of the search engine used, which correspond to fewer than 5% additional proteins. These data indicate that the analysis of at least 2, but no more than 3, replicates should be performed to maximize proteome coverage, while efficiently using mass spectrometer acquisition time.
For all search engines investigated, the search space is limited by parameters, such as mass tolerance of the peptide and fragments, enzyme specificity, number of missed cleavages, and amino acid modifications. Consistent parameters were maintained for all search engines investigated. However, the generalizability of the results described herein to other datasets may be limited by various factors specific to the dataset under investigation. Therefore, many factors that are not taken into account here may influence the results of consensus database searching and subsequent amalgamation of search results from different datasets. Such factors include, but are not limited to, sample and data quality, sample complexity, the type of instrument used for the analysis, the number of replicates and/or fractions combined for the analysis, the enzyme(s) used for digestion, number of missed cleavages, choice of fixed and variable modifications, and the mass tolerances of peptide and fragment ions.
Differences in peptide and protein identifications may be expected when using a particular search engine. Even with matching search parameters, algorithms for peak picking, peptide sequencing, and assignments of peptides to proteins differ among the various search engines. Moreover, typically, less than 30% of the spectra collected are successfully mapped to a peptide [11]. As such, the lack of identification of the complete set of spectra contributes to the disparity among search engines insofar as some spectra may be assigned to a peptide by a particular search engine, while these spectra may not pass the thresholds of other search engines. The different search engines consider unique spectrum models based on the selected database and each have unique methods to score against MS/MS spectra. SEQUEST ranks its results according to a cross-correlation of the measured and theoretical spectra obtained from an in silico digest [1]. Mascot calculates the likelihood of matching peaks in common with a model and the original spectrum using statistical and geometric-based methods for scoring and ranking peptides [2]. Andromeda, similar to Mascot, uses a probability based approach, which is built on the binominal distribution probability to rank the peptides [3]. Following peptide identification, the database searching application assigns peptides to proteins, typically parsimoniously in the form of protein groups to reduce redundancies and prevent inflated lists of proteins. Many such algorithms are application specific, such as those implemented in ProteomeDiscoverer interface or ProteinPilot [12] and require proprietary input. As described above, some of the disparity in the protein identifications between Mascot or SEQUEST and MaxQuant, may be due partially to this post-identification protein grouping, which is the present case is unavoidable due to the current lack of a MaxQuant node in ProteomeDiscoverer.
Presently, applications are available that attempt to integrate results of the various search engines. Packages such as Scaffold [13], ProteoIQ (https://www.bioinquire.com/index.php), FDRAnalysis [14], ROVER [15], and MSblender [16] are freely or commercially available to compare and combine search engine results. Scaffold uses an alternative to the FDR as a metric to combine search engine results, as it uses PeptideProphet algorithm [17] to determine the probability of a peptide being correct. Similarly, ProteoIQ incorporates the false discovery rate and protein probability approaches in efforts to maximize the number of proteins identified and minimize the number of false positives [17-19]. Not all these applications at the moment are compatible with all search engine outputs, but typically the more commons search engines, such as Mascot and SEQUEST, are supported by many of these applications.
Although to the author's knowledge, this is the first directly comparison of replicate analyses of HeLa whole cell lysates among the Mascot, SEQUEST, and MaxQuant search engine results, many prior studies have investigated the consensus of search engines in regard to protein and peptide identifications. Previously, several studies have investigated the benefits and caveats associated with searching the same data with multiple search engines. It has been shown previously that the union of search engine results provided higher sensitivity, but that the intersection produced better specificity [20]. In fact, one study showed that the correct combination of search algorithms (Mascot, OMSSA, and X!Tandem), not the number of search engines used, maximized the accuracy of peptide identification while minimizing false positives [21]. Approximately 35% more peptide identifications have been obtained when combining results from Mascot, OMSSA, and X!Tandem using the combined FDR score [22]. In addition, improved sensitivity of protein identifications was obtained by combining the results of several search engines -Mascot, OMSSA, and X!Tandem – in one study [14]. In an earlier study, combining protein identifications from SEQUEST, X!Tandem, and Mascot also improved sensitivity of protein identifications [23]. Using different datasets, an analogous comparison of SEQUEST, Mascot, and X!Tandem showed that the intersection performed better in accuracy, but sensitivity was greater when the union of the search engines was considered [20]. Similar results were obtained by integrating peptide identifications obtained from four search engines in two separate studies, one using SEQUEST, X!Tandem, MyriMatch, and InsPecT [16], and the second using Mascot, OMSSA, SEQUEST, and X!Tandem [24]. In a comprehensive investigation, results from over 50 combinations of seven different search engines – including Mascot and SEQUEST, but not MaxQuant – were compared and the combination of certain search methods improved accuracy of protein identifications [25]. A previous study which investigated five different search engines –including Mascot and SEQUEST, but again not MaxQuant- revealed that at the individual search engine level, SEQUEST performed well in terms of sensitivity, but specificity was greater in Mascot [26]. This result agrees with the findings herein as more peptides and proteins were identified in SEQUEST compared to Mascot. However, in a 1.5 hr data-dependent mass spectrometry analyses of whole cell lysates, under-sampling is inherent in this study [27], and as such sensitivity and specificity are difficult to gauge without comprehensive knowledge of the proteome present in the sample. It follows that analysis of more dilute samples merit further investigation. These data reveal the advantage of using multiple search engines to obtain both comprehensive and consensus peptide and protein identifications.
In summary, the data demonstrate two practical methods of increasing the robustness of mass spectrometry data analysis. The data show that 1) using multiple search engines can expand the number of identified proteins (the union of the data) and validate protein identifications (the intersection of the data), and 2) analysis of 2 or 3 technical replicates can substantially expand protein identifications. Basically, more data was extracted from a collected data set via in silico methods without the additional, and potentially costly, sample preparation or mass spectrometry time. Using the proteins at the intersection of the search engine results will narrow targets for downstream analysis and follow-up experiments.
However, using the union of search engine results allows for the casting of a wider net, producing a more comprehensive dataset at the expense of a higher number of false positives. The data show that among search engines, protein grouping may be the source of greater disparity in the results among search engines than peptide sequence assignment. From the analysis, it was determined that increases in additional peptide and protein identifications compared to just a single run, stabilize after 3 replicates, after which mass spectrometry analysis time may be better spent performing biological replicates or analyzing additional samples. Performing technical and biological replicates can lead to a larger number of protein identifications, however at the expense of reagents, sample processing, and increased mass spectrometer usage. The effects of biological replicates with a similar comparison as performed herein merit further investigation. In conclusion, the extraction of information from prepared samples can be maximized with repeated analysis of mass spectrometry data by performing technical repeats and database searching with different search engines, requiring no additional sample preparation and effectively utilizing research time and effort.
Acknowledgments
I would also like to thank members of the Steen Lab at Children's Hospital Boston, in particular John FK Sauld and Ali Ghoulidi, as well as the Center for Pancreatic Disease at Brigham and Women's Hospital for their technical assistance and critical reading of the manuscript.
Sources of funding: Funds were provided by the following NIH grant: 1 F32 DK085835-01A1 (JP).
Footnotes
Conflicts of interests: The author declares no competing interests.
Author contributions: JP conceived of the study and wrote the original manuscript.
References
- 1.Eng J, Mccormack AL, Yates JR., Iii An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 2.Perkins DN, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Cox J, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10(4):1794–805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 4.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du X, et al. Linear discriminant analysis-based estimation of the false discovery rate for phosphopeptide identifications. J Proteome Res. 2008;7(6):2195–203. doi: 10.1021/pr070510t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Q, et al. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40(4):430–43. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 7.Elias JE, et al. Intensity-based protein identification by machine learning from a library of tandem mass spectra. Nat Biotechnol. 2004;22(2):214–9. doi: 10.1038/nbt930. [DOI] [PubMed] [Google Scholar]
- 8.Moore RE, Young MK, Lee TD. Method for screening peptide fragment ion mass spectra prior to database searching. J Am Soc Mass Spectrom. 2000;11(5):422–6. doi: 10.1016/S1044-0305(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 9.Oliveros J. VENNY: An interactive tool for comparing lists with Venn diagrams. 2007 Available from: http://bioinfogp.cnb.csic.es/tools/venny/index.html.
- 10.Claassen M, et al. Generic comparison of protein inference engines. Mol Cell Proteomics. 2012;11(4):O110 007088. doi: 10.1074/mcp.O110.007088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics. 2005;4(10):1419–40. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Shilov IV, et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6(9):1638–55. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Searle BC. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics. 2010;10(6):1265–9. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]
- 14.Wedge DC, et al. FDRAnalysis: a tool for the integrated analysis of tandem mass spectrometry identification results from multiple search engines. J Proteome Res. 2011;10(4):2088–94. doi: 10.1021/pr101157s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colaert N, et al. Rover: a tool to visualize and validate quantitative proteomics data from different sources. Proteomics. 2010;10(6):1226–9. doi: 10.1002/pmic.200900379. [DOI] [PubMed] [Google Scholar]
- 16.Kwon T, et al. MSblender: A probabilistic approach for integrating peptide identifications from multiple database search engines. J Proteome Res. 2011;10(7):2949–58. doi: 10.1021/pr2002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller A, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 18.Nesvizhskii AI, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 19.Weatherly DB, et al. A Heuristic method for assigning a false-discovery rate for protein identifications from Mascot database search results. Mol Cell Proteomics. 2005;4(6):762–72. doi: 10.1074/mcp.M400215-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Sultana T, Jordan R, Lyons-Weiler J. Optimization of the Use of Consensus Methods for the Detection and Putative Identification of Peptides via Mass Spectrometry Using Protein Standard Mixtures. J Proteomics Bioinform. 2009;2(6):262–273. doi: 10.4172/jpb.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dagda RK, Sultana T, Lyons-Weiler J. Evaluation of the Consensus of Four Peptide Identification Algorithms for Tandem Mass Spectrometry Based Proteomics. J Proteomics Bioinform. 2010;3:39–47. doi: 10.4172/jpb.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones AR, et al. Improving sensitivity in proteome studies by analysis of false discovery rates for multiple search engines. Proteomics. 2009;9(5):1220–9. doi: 10.1002/pmic.200800473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Searle BC, Turner M, Nesvizhskii AI. Improving sensitivity by probabilistically combining results from multiple MS/MS search methodologies. J Proteome Res. 2008;7(1):245–53. doi: 10.1021/pr070540w. [DOI] [PubMed] [Google Scholar]
- 24.Balgley BM, et al. Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol Cell Proteomics. 2007;6(9):1599–608. doi: 10.1074/mcp.M600469-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Alves G, et al. Enhancing peptide identification confidence by combining search methods. J Proteome Res. 2008;7(8):3102–13. doi: 10.1021/pr700798h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapp EA, et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5(13):3475–90. doi: 10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, et al. Comparison of extensive protein fractionation and repetitive LC-MS/MS analyses on depth of analysis for complex proteomes. J Proteome Res. 2010;9(2):1032–40. doi: 10.1021/pr900927y. [DOI] [PMC free article] [PubMed] [Google Scholar]