

Abstract

Protectin D1 (PD1 (3)), a C22-dihydroxylated polyunsaturated fatty acid biosynthesized from all-Z-docosahexaenoic acid, belongs to the new family of endogenous mediators referred to as specialized pro-resolving lipid mediators. PD1 (3) is a natural product that displays potent anti-inflammatory properties together with pro-resolving actions including inhibition of polymorphonuclear leukocyte (PMN) infiltration and promotion of macrophage phagocytosis and efferocytosis. Given its potent endogenous actions, this compound has entered several clinical development programs. Little has been reported on the metabolism of PD1 (3). The synthesis and biological evaluations of the ω-22 monohydroxylated metabolite of PD1 (3), named herein 22-OH-PD1 (6), are presented. LC-MS/MS data of the free acid 6, obtained from hydrolysis of the synthetic methyl ester 7, matched data for the endogenously produced 22-OH-PD1 (6). Compound 6 exhibited potent pro-resolving actions by inhibiting PMN chemotaxis in vivo and in vitro comparable to its precursor PD1 (3) and decreased pro-inflammatory mediator levels in inflammatory exudates. The results reported herein provide new knowledge of the metabolism of the protectin class of specialized pro-resolving mediators.
Several naturally occurring chemical mediators have been identified that have the capacity to initiate, modulate, and reduce acute inflammation as well as stimulate resolution.1,2 Recent efforts have established that the return to homeostasis, i.e., catabasis,3 is mediated by active biosynthesis and termination programs directed by natural products named specialized pro-resolving mediators (SPMs).3 The SPMs are derived from the dietary ω-3 polyunsaturated fatty acids (PUFAs) eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) during the resolution phase of acute inflammation.4,5 Examples of SPMs are the resolvins, protectins, and maresins, which resolve inflammation, protect organs, and stimulate tissue regeneration.5 Resolvin E1 (1) is biosynthesized from EPA,6 while resolvin D1 (2),7,8 protectin D1 (PD1, 3),7−10 also known as neuroprotectin D1 (NPD1) when produced in neural systems,9 and maresin 1 (4)11 are all enzymatic products of DHA. PD1n-3 DPA (5), derived from ω-3 DPA, is a recent addition to the SPM family of compounds.12,13 The molecular understanding of the resolution of many inflammatory diseases has been elucidated with these endogenously formed compounds as pharmacological tools.14

The lipid mediator PD1 (3) has been the subject of many pharmacological studies for the development of potential new anti-inflammatory drugs and has entered clinical trials.5,14 In 2003 initial investigations on the metabolism of PD1 (3) were reported.8 These experiments yielded picogram amounts of a 10,17,22-trihydroxy C22 polyunsaturated fatty acid, which was structurally assigned based on biosynthetic considerations and data from LC-MS/MS experiments. Biological evaluation and the full stereochemical assignment of this 22-hydroxylated metabolite of PD1 (3) were not communicated due to the minute amounts obtained.8 Due to the attention SPMs have attracted from biologists and pharmacologists, it is also of interest to investigate if metabolites of SPMs exhibit anti-inflammatory or pro-resolving actions. Herein a stereoselective total synthesis of 22-OH-PD1 (6) is reported. This allowed, based on data from matching experiments, the exact structure elucidation and biological evaluation of this metabolite of PD1 (3).
Results and Discussion
The total organic synthesis of 22-OH-PD1 (6) commenced with an Evans–Nagao acetate aldol reaction15 between the chiral auxiliary 8 and aldehyde 9 as previously reported (Scheme 1).16 The alcohol 10 was formed in a 15.3:1 diastereomeric ratio when 8 was treated with (i-Pr)2NEt and TiCl4 in CH2Cl2 at −78 °C followed by the addition of freshly prepared aldehyde 9. Protection of the hydroxy group in 10 was performed using standard conditions (TBSOTf, 2,6-lutidine), affording compound 11 in 84% yield over the two steps. Reductive removal of the auxiliary in 11 was achieved with DIBAL-H, yielding aldehyde 12, which was used directly in the next step (Scheme 1).
Scheme 1. Synthesis of Stereochemically Pure Ester 15.

Next, pent-4-ynoic acid (13) was converted into the Wittig salt 14 in five steps and 42% overall yield as previously reported.16 Then aldehyde 12 was added to the ylide of 14; the latter formed after reaction with NaHMDS in THF and HMPA at −78 °C. Stereochemically pure ester 15 was obtained in 47% yield from 12 after purification by column chromatography (Scheme 1).
The alkyne 18 was prepared using a stereoselective boron-mediated allylation reaction17 between the chiral boron-auxiliary 16 and 3-(trimethylsilyl)propiolaldehyde (17) using conditions reported by Pietruszka and Schöne.18 This afforded the alkyne 18 in 72% yield. Removal of the TMS group with MeOH/K2CO3 gave the terminal alkyne 19, that was reacted with the vinylic-bromide ester 15 in a Sonogashira reaction (Pd(PPh3)4, CuI, Et2NH). This yielded ester 20, containing all stereogenic centers and carbon atoms present in 22-OH-PD1 (6), in 65% yield over two steps. Removal of the TBS group went smoothly with excess tetra-n-butylammonium fluoride in THF to produce the triol-ester 21 in 86% yield. The Boland protocol19 was employed in the reduction of the internal conjugated triple bond in 21, providing the methyl ester 7 in 40% yield after chromatography. In addition, <1% of remaining 21 and approximately 2–3% of the over-reduced material of 7 were obtained. All attempts to improve the yield of 7 while retaining a high stereoselectivity were unsuccessful. Moreover, other methods explored did not give a stereoselective and concurrent high-yielding reduction of alkyne 21.20,21 The spectroscopic data (UV, IR, NMR, and MS) for the methyl ester 7 of 22-OH-PD1 (6) were in accord with the structure (Supporting Information).
Scheme 2. Synthesis of Methyl Ester 7.

Matching experiments using synthetic and biologically produced products were then conducted. First, the ester 7 was hydrolyzed to the acid 6 under basic aqueous conditions.16 In order to determine whether synthetic material matched biologically formed 6, authentic 6 was obtained from human neutrophils.8 Figure 1A shows the multiple reaction monitoring (MRM) chromatograms of 22-OH-PD1 (6) from human neutrophils, which gave a sharp peak at tR = 10.4 min. Figure 1B depicts the MRM chromatogram of synthetic 22-OH-PD1 (6), also with retention time tR = 10.4 min. Figure 1C illustrates the co-injection of synthetic and endogenous material added at essentially equal amounts. Overall, Figure 1 demonstrates that synthetic 6 coelutes with the material produced by human neutrophils. Prominent ions of diagnostic value were assigned at m/z 375 = [M – H]−, m/z 357 = [M – H – H2O]−, m/z 339 = [M – H – 2H2O]−, m/z 331 = [M – H – CO2]−, m/z 314 = [M – H – 2H2O – CO2]−, m/z 313 = [M – H – H2O – CO2]−, and m/z 295 = [M – H – CO2 – 2H2O]− (see the Supporting Information). The UV and MS spectra for both natural and synthetic 6 were essentially identical and in accord with the literature.8
Figure 1.

Human neutrophil 22-OH-PD1 (6) matches synthetic material. Representative MRM chromatograms. n = 3. Selected ion chromatogram (m/z 375–153) depicting (A) 22-OH-PD1 (6) obtained from human peripheral blood neutrophils (5 × 107/mL) incubated with 0.1 mg of opsonized zymosan and PD1 (3) (10 ng/mL, 37 °C, 30 min, DPBS+/+, pH = 7.45). n = 3 human neutrophil preparations. (B) Synthetic material (inset: characteristic UV-absorption spectrum in MeOH, ±1 nm). (C) Co-injection of human neutrophil 22-OH-PD1 (6) with synthetic 6.
During the acute phase of inflammation, circulating polymorphonuclear leukocytes (PMN) extravasate into the inflamed tissue. Inhibition of PMN infiltration and a decrease of pro-inflammatory mediators are key processes in the resolution of inflammation and are defining actions of SPMs.4 Therefore, the anti-inflammatory actions of 22-OH-PD1 (6) were investigated using an in vivo zymosan-induced peritonitis mouse model. Compound 6 was administrated 10 min prior to zymosan challenge, and peritoneal inflammatory exudates were collected after 4 h. At 4 h 22-OH-PD1 (6) decreased PMN infiltration by ∼43%, to a similar extent as PD1 (3) (∼54%) itself (Figure 2). Synthetic material of PD1 (3), matched with biologically produced 3, was employed.16
Figure 2.
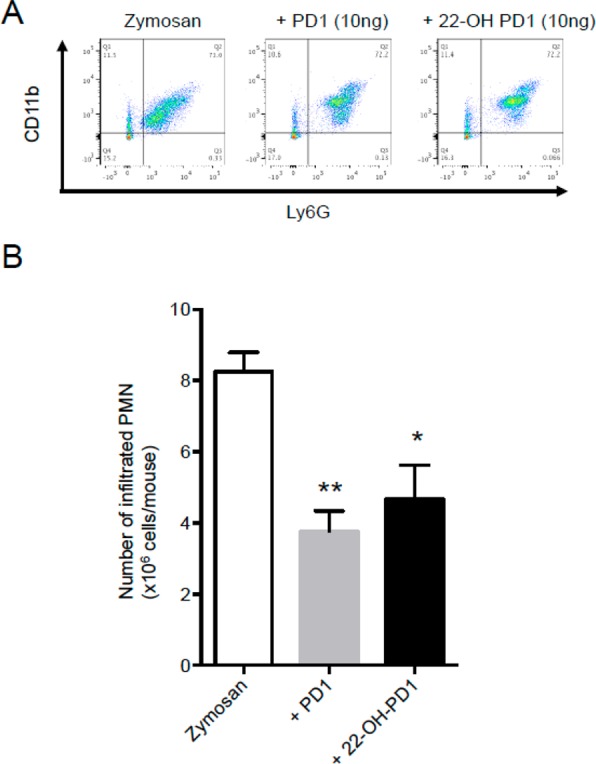
PD1 (3) and 22-OH-PD1 (6) limit leukocyte recruitment in vivo. Mice were administered vehicle (saline containing 0.1% EtOH), PD1 (3) (10 ng/mouse), or 22-OH-PD1 (6) (10 ng/mouse) i.p. 10 min prior to zymosan (1 mg/mouse, i.p.). At 4 h peritoneal lavages were collected and cells enumerated using light microscopy and flow cytometry. (A) Representative flow cytometry dot plots. (B) Total leukocyte counts in murine peritoneal lavages. Results for A are representative of n = 4 mice per treatment. Results for B are mean ± SEM; n = 4 mice per treatment. *p < 0.05, **p < 0.01 versus zymosan-treated mice.
Because pro-inflammatory eicosanoid levels, including LTB4 and PGF2α, increase during acute inflammation,2 we next assessed the regulation of local pro-inflammatory eicosanoids by 22-OH-PD1 (6) using LC-MS/MS-based lipid mediator metabololipidomics. Mediators were identified in accordance with published criteria22 that include matching retention times (Figure 3A) and MS-MS fragmentation patterns (Figure 3B). Quantification of exudate lipid mediators by MRM demonstrated a significant reduction in exudate eicosanoid levels including PGD2 (∼27% decrease), PGF2α (∼43% decrease), and TXB2 (∼48% decrease; Figure 3C), following 22-OH-PD1 (6) administration, actions that are also shared with PD1 (3). Together these results demonstrate that 22-OH-PD1 (6) displays potent anti-inflammatory actions in vivo.
Figure 3.

PD1 (3) and 22-OH-PD1 (6) reduce local pro-inflammatory eicosanoid levels in acute inflammation. Lipid mediators in peritoneal exudates collected 4 h after zymosan administration (1 mg/mouse, i.p.) were assessed using LC/MS-MS metabololipidomics following solid-phase extraction. (A) Representative MRM chromatograms of selected ion pairs for arachidonic acid-derived eicosanoids, a = 6-trans-LTB4 and b = 6-trans-12-epi-LTB4. (B) Representative MS-MS spectra with diagnostic ions employed for the identification of LTB4 and PGE2. M–H, molecular ion. m/z, mass-to-charge ratio, Da, dalton. (C) Quantification of exudate lipid mediators following PD1 (3) and 22-OH-PD1 (6) administration (10 ng i.p., 10 min prior to zymosan challenge). Results are mean ± SEM; n = 4 mice per group. *p < 0.05, **p < 0.01 versus zymosan-treated mice.
SPMs regulate cell adhesion and transmigration to the site of inflammation.23 As 22-OH-PD1 (6) displayed potent actions on neutrophils in vivo, we next tested whether these findings could be translated to humans by investigating the actions of synthetic 22-OH-PD1 (6) on human PMN chemotaxis and adhesion (Figure 4). With primary human PMN, 22-OH-PD1 (6) was found to inhibit PMN chemotaxis to LTB4 (∼19%) and IL-8 (∼23%); see Figure 4A and B, respectively. Compound 6 also inhibited PMN adhesion to fibronectin under flow (Figure 4C). These findings demonstrate that 22-OH-PD1 (6) displays potent biological functions in vivo with mice and in vitro with human cells that are comparable to its precursor PD1 (3).
Figure 4.

PD1 (3) and 22-OH-PD1 (6) inhibit human PMN chemotaxis and adhesion. Purified human PMN were pretreated with 22-OH-PD1 (6), PD1 (3), or vehicle control (15 min, 37 °C). PMN were then cultured on a ChemoTx chamber (90 min, 37 °C in 5% CO2), and PMN chemotaxis toward (A) LTB4 (10 nM) or (B) IL-8 (100 ng/mL) was measured. Cell chemotaxis was quantified, and samples were normalized to vehicle control. Results show four independent experiments, presented as the mean ± SEM. (C) PMN were pretreated with 22-OH-PD1 (6), PD1 (3), or vehicle control (10 min, RT) and stimulated with 10 nM LTB4 (10 min, RT). PMN were then passed through a flow chamber at 0.5 mL/min for 5 min. Six random fields of view were recorded, and the number of adhered PMN was quantified. Representative images of adhered PMN (left panel) and quantification (right panel) are shown. Results were obtained from three independent experiments, shown as the mean ± SEM. All results were compared to vehicle control using a paired t test; statistical significance defined as *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
In conclusion, the complete structure of 22-OH-PD1 (6) has been reported. A stereoselective synthesis of the methyl ester of 6 has been achieved in nine steps and 6% overall yield. Furthermore, 22-OH-PD1 (6) was shown to maintain bioactive properties resembling those of PD1 (3), as it decreases leukocyte recruitment in vivo, dampens production of classical pro-inflammatory eicosanoids, and inhibits human PMN chemotaxis and adhesion. The ω-oxidation of PD1 (3) to 22-OH-PD1 (6) is most likely, as reported for some other SPMs,24,25 mediated by CYP1 monooxygenases. In many cases ω-oxidation reactions reduce the bioactivity of many local autacoids, i.e., LTB4. Of interest, the further metabolism of PD1 (3) gives a product, namely, 22-OH-PD1 (6), that retains the potent actions of PD1 (3). The results presented for 22-OH-PD1 (6) will be useful for future developments toward new pro-resolving and anti-inflammatory drug candidates.
Experimental Section
General Experimental Procedures
Optical rotations were measured using a 1 mL cell with a 1.0 dm path length on a PerkinElmer 341 polarimeter. The UV/vis spectra from 190 to 900 nm were recorded using a Biochrom Libra S32PC spectrometer using quartz cuvettes. The CD spectrum from 200 to 360 nm was recorded using a DSM 1000 CD instrument. IR spectra (4000–600 cm–1) were obtained on a PerkinElmer Spectrum BX series FT-IR spectrophotometer. NMR spectra were recorded on a Bruker AVII400 spectrometer at 400 MHz for 1H NMR and at 101 MHz for 13C NMR. Spectra are referenced relative to the central residual protium solvent resonance in 1H NMR (CDCl3 δ = 7.27 and MeOH-d4 δ = 3.31) and the central carbon solvent resonance in 13C NMR (CDCl3 δ = 77.00 ppm and MeOH-d4 = δ 49.00). Mass spectra were recorded at 70 eV on a Waters Prospec Q spectrometer using EI, ES, or CI as the method of ionization. High-resolution mass spectra were recorded on a Waters Prospec Q spectrometer using EI or ES as the method of ionization. Thin-layer chromatography was performed on silica gel 60 F254 aluminum-backed plates fabricated by Merck. Flash column chromatography was performed on silica gel 60 (40–63 μm) produced by Merck. HPLC analyses for chemical purities were performed on an Agilent Technologies 1200 Series instrument with diode array detector set at 254 nm and equipped with a C18 stationary phase (Eclipse XDB-C18 5 μm 4.6 × 150 mm), applying the conditions stated. Unless stated otherwise, all commercially available reagents and solvents were used in the form they were supplied without any further purification. The stated yields are based on isolated material. Diastereomeric ratios reported in this paper have not been validated by calibration; see Wernerova and Hudlicky for discussions and guidelines.26 Liquid chromatography (LC)-grade solvents were purchased from Fisher Scientific. The Eclipse Plus C18 column (100 × 4.6 mm × 1.8 μm) was obtained from Agilent, and C18 SPE columns were from Waters. The synthetic standards for LC-tandem mass spectrometry (MS-MS) quantitation and deuterated (d) internal standards (d4-PGE2 and d4-LTB4) were purchased from Cayman Chemicals. Zymosan A was purchased from Sigma. Anti-mouse CD11b and anti-mouse Ly6G were purchased from BioLegend.
(S,Z)-8-(Trimethylsilyl)oct-3-en-7-yne-1,6-diol (18)
Allylboronate 16 was prepared as described by Pietruszka et al.18 Compound 16 (275 mg, 0.50 mmol, 1.0 equiv) was dissolved in CH2Cl2 (0.25 mL) and stirred at 0 °C for 30 min before TMS-propargylaldehyde (17) (0.1 mL, 0.60 mmol, 1.2 equiv) was added. The solution was stirred at 4 °C for 24 h and another 4 days at rt before being concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes/EtOAc, 1:1) to afford the title compound 18 as a colorless oil. Yield: 76 mg (72%); [α]D20 −13 (c 0.2, MeOH); 1H NMR (400 MHz, CDCl3) δ 5.69–5.54 (m, 2H), 4.40 (t, J = 5.9 Hz, 1H), 3.64 (ddd, J = 6.5, 5.5, 2.1 Hz, 2H), 3.06 (bs, 2H), 2.48 (dd, J = 7.0, 5.9 Hz, 2H), 2.37–2.27 (m, 2H), 0.15 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 129.9, 127.2, 106.5, 89.3, 61.9, 61.7, 35.5, 30.7, 0.0 (3C); HRESTOFMS m/z 235.1133 [M + Na]+ (calcd for C11H20O2SiNa, 235.1130); TLC (hexanes/EtOAc, 1:1, CAM stain) Rf = 0.27.
(S,Z)-Oct-3-en-7-yne-1,6-diol (19)
The TMS-alkyne (18) (130 mg, 0.61 mmol, 1.0 equiv) was dissolved in dry MeOH (20 mL), and K2CO3 (80 mg) was added. After 3 h, phosphate buffer (20 mL, pH = 7.2) was added, and the aqueous phase was extracted with EtOAc (4 × 30 mL). The combined organic layers were dried (Na2SO4), before being concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes/EtOAc, 4:6) to afford the title compound 19 as a yellow oil. Yield: 81 mg (94%); [α]D20 −9 (c 0.2, MeOH); 1H NMR (400 MHz, CDCl3) δ 5.72–5.58 (m, 2H), 4.46–4.41 (m, 1H), 3.67 (t, J = 5.9 Hz, 2H), 3.46 (bs, 1H), 2.60 (bs, 1H), 2.55–2.49 (m, 2H), 2.46 (d, J = 2.2 Hz, 1H), 2.38–2.31 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 130.3, 126.9, 84.6, 73.0, 61.7, 61.4, 35.4, 30.7; HRESTOFMS m/z 163.0739 [M + Na]+ (calcd for C8H12O2Na, 163.0734); TLC (hexanes/EtOAc, 4:6, CAM stain) Rf = 0.17.
Methyl (4Z,7Z,10R,11E,13E,17S,19Z)-10-((tert-Butyldimethylsilyl)oxy)-17,22-dihydroxydocosa-4,7,11,13,19-pentaen-15-ynoate (20)
To a solution of vinyl bromide 15 (253 mg, 0.57 mmol, 1.0 equiv) in Et2NH (1.2 mL) and benzene (0.4 mL) was added Pd(PPh3)4 (20 mg, 0.02 mmol, 3 mol %), and the reaction was stirred for 45 min in the dark. CuI (5 mg, 0.03 mmol, 5 mol %) dissolved in a minimum amount of Et2NH was added, followed by dropwise addition of alkyne 19 (80 mg, 0.57 mmol, 1.0 equiv) in Et2NH (1.0 mL). After 20 h of stirring at ambient temperature, the reaction was quenched by saturated NH4Cl solution (15 mL). Et2O (15 mL) was added, and the phases were separated. The aqueous phase was extracted with Et2O (2 × 15 mL), and the combined organic layers were dried (Na2SO4), before being concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes/EtOAc, 95:5) to afford the title compound 20 as a yellow oil. Yield: 199 mg (69%); [α]D20 −9 (c 0.1, MeOH); 1H NMR (400 MHz, CDCl3) δ 6.56 (dd, J = 15.6, 10.9 Hz, 1H), 6.19 (dd, J = 15.3, 11.0 Hz, 1H), 5.78 (dd, J = 15.1, 5.9 Hz, 1H), 5.73–5.62 (m, 2H), 5.58 (d, J = 15.6 Hz, 1H), 5.46–5.32 (m, 4H), 4.63–4.55 (m, 1H), 4.20 (q, J = 6.2 Hz, 1H), 3.72–3.65 (m, 5H), 2.83–2.73 (m, 2H), 2.59–2.50 (m, 2H), 2.42–2.34 (m, 6H), 2.28 (q, J = 6.9, 6.1 Hz, 2H), 0.89 (s, 9H), 0.03 (d, J = 9.1 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 173.7, 141.9, 139.8, 130.1, 129.9, 129.4, 128.5, 128.0, 127.2, 125.7, 110.1, 92.0, 84.3, 72.7, 62.3, 62.0, 51.7, 36.4, 35.7, 34.2, 30.8, 26.0 (3C), 25.9, 23.0, 18.4, −4.4, −4.6; HRESTOFMS m/z 525.3012 [M + Na]+ (calcd for C29H46O5SiNa, 525.3008); TLC (hexanes/EtOAc, 1:1, CAM stain) Rf = 0.21.
Methyl (4Z,7Z,10R,11E,13E,17S,19Z)-10,17,22-Trihydroxydocosa-4,7,11,13,19-pentaen-15-ynoate (21)
TBAF (253 mg, 0.97 mmol, 2.5 equiv, 1.0 M in THF) was added to a solution of TBS-protected alcohol 20 (195 mg, 0.39 mmol, 1.0 equiv) in THF (5.5 mL) at 0 °C. The reaction was stirred for 20 h before it was quenched with phosphate buffer (pH = 7.2, 2.5 mL). Brine (20 mL) and EtOAc (20 mL) were added, and the phases were separated. The H2O phase was extracted with EtOAc (2 × 20 mL), and the combined organic layer was dried (Na2SO4) before being concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes/EtOAc, 4:6) to afford the title compound 21 as a pale yellow oil. Yield: 129 mg (86%); [α]D20 25 (c 0.1, MeOH); 1H NMR (400 MHz, CDCl3) δ 6.56 (dd, J = 15.5, 10.9 Hz, 1H), 5.83 (dd, J = 15.2, 6.0 Hz, 1H), 5.74–5.58 (m, 3H), 5.58–5.50 (m, 1H), 5.44–5.32 (m, 3H), 4.62–4.54 (m, 1H), 4.24 (q, J = 5.7 Hz, 1H), 3.74–3.64 (m, 5H), 2.90–2.72 (m, 3H), 2.61–2.48 (m, 2H), 2.42–2.30 (m, 8H), 2.08 (bs, 1H), 1.97 (bs, 1H); 13C NMR (101 MHz, CDCl3) δ 173.8, 141.5, 138.3, 131.6, 130.1, 129.4, 129.1, 128.2, 127.2, 124.8, 110.9, 92.4, 84.2, 71.6, 62.3, 61.9, 51.8, 35.6, 35.4, 34.1, 30.8, 25.9, 23.0; HRESTOFMS m/z 411.2147 [M + Na]+ (calcd for C23H32O5Na, 411.2156); TLC (hexanes/EtOAc, 4:6, CAM stain) Rf = 0.12.
Methyl (4Z,7Z,10R,11E,13E,15Z,17S,19Z)-10,17,22-Trihydroxydocosa-4,7,11,13,15,19-hexaenoate or 22-OH-PD1 (7)
Alkyne 21 (25 mg, 0.064 mmol, 1.0 equiv) was dissolved in H2O and MeOH (1:1, 7.6 mL), to which a freshly prepared batch of Zn(Cu/Ag) (Boland reagent)19 (∼2.3 g) was added in one portion. The flask was evacuated and filled with argon (3×), and the reaction mixture was stirred for 5 h. The suspension was vacuum filtrated through a pad of Celite, and the filter cake was washed with additional fresh MeOH. The filtrate was concentrated in vacuo, to which brine and EtOAc were added. The phases were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product was purified by column chromatography on silica (heptane/EtOAc/MeOH, 28:70:2) to afford the title compound 7 as a colorless oil. Yield: 10 mg (40%); [α]D20 −31 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 262 (4.53), 271 (4.60), 282 (4.54) nm; CD (0.1 μg/mL, MeOH), λmax (Δε) 343 (12.05), 272 (15.09), and 220 (6.20) nm; IR (neat) νmax 3356, 3014, 2924, 1734, 1663, 1436, 1045, 997 cm–1; 1H NMR (400 MHz MeOH-d4) δ 5.30–5.18 (m, 1H), 5.06–4.89 (m, 2H), 4.79 (t, J = 11.1 Hz, 1H), 4.46 (dd, J = 14.2, 6.6 Hz, 1H), 4.25–4.04 (m, 7H), 3.35–3.23 (m, 1H), 2.85 (q, J = 6.5 Hz, 1H), 2.36 (s, 3H), 2.26 (t, J = 6.8 Hz, 2H), 1.59–1.44 (m, 2H), 1.15–0.90 (m, 10H); 13C NMR (101 MHz, MeOH-d4) δ 175.3, 138.0, 135.0, 134.8, 131.4, 130.9, 130.6, 130.3, 129.1, 129.0, 128.9, 128.1, 126.5, 73.0, 68.5, 62.6, 52.1, 36.6, 36.4, 34.8, 32.0, 26.7, 23.8; HRESTOFMS m/z 413.2505 [M + Na]+ (calcd for C23H34O5Na+, 413.2298); TLC (heptane/EtOAc/MeOH, 28:70:2, CAM stain) Rf = 0.30. The chemical purity (>96%) was determined by HPLC analysis (Eclipse XDB-C18, MeOH/H2O, 65:35, 1.0 mL/min): tR(minor) = 7.52 and 8.02 min, and tR(major) = 9.83 min.
Sterile Peritonitis
Male FVB mice (6 to 8 weeks old) purchased from Charles River Laboratories were fed ad libitum laboratory rodent diet 20-5058 (Lab Diet, Purina Mills). Animal experimental procedures were approved by the Standing Committee on Animals of Harvard Medical School (protocol no. 02570) and complied with institutional and U.S. National Institutes of Health (NIH) guidelines. Zymosan (1 mg/mL; Sigma-Aldrich) was injected intraperitoneally (i.p.) 10 min after i.p. administration of PD1 (3) or 22-OH-PD1 (6) (10 ng) or vehicle (0.1% EtOH in 100 mL of saline). At 4 h peritoneal lavages were collected in PBS without calcium and magnesium, cells were enumerated as described by Dalli et al.27 using light microscopy and flow cytometry, and local eicosanoid levels were assessed by targeted lipid mediator metabololipidomics as described below.
Targeted Lipid Mediator Metabololipidomics
All samples for LC/MS-MS-based metabololipidomics were extracted with solid-phase extraction columns as previously reported.28,29 Prior to sample extraction, lavages were placed in two volumes of ice-cold MeOH containing d4-PGE2 and d4-LTB4, representing each region in the chromatographic analysis (500 pg each) to facilitate quantification. Extracted samples were analyzed by an LC/MS-MS system, Qtrap 6500 (AB Sciex) equipped with a Shimadzu SIL-20AC autoinjector and LC-20AD binary pump (Shimadzu Corp.). An Agilent Eclipse Plus C18 column (100 × 4.6 mm × 1.8 μm) was used with a gradient of MeOH/H2O/acetic acid of 55:45:0.01 (vol:vol:vol) that was ramped to 85:15:0.01 (vol:vol:vol) over 10 min and then to 98:2:0.01 (vol:vol:vol) for the next 8 min. This was subsequently maintained at 98:2:0.01 (vol:vol:vol) for 2 min. The flow rate was maintained at 0.4 mL/min. To monitor and quantify the levels of PGD2, PGE2, PGF2α, TXB2, and LTB4, a multiple reaction monitoring method was developed with signature ion fragments (m/z) for each molecule. Identification was conducted using published criteria where a minimum of six diagnostic ions were employed.28 Calibration curves were determined using a synthetic lipid mediator mixture. A linear calibration curve for each compound was obtained with r2 values ranging from 0.98 to 0.99. The detection limit was ∼0.1 pg in the matrix. Quantification was carried out as reported by Dalli and Serhan.28
PMN Isolation and Chemotaxis
Peripheral blood human PMN were purified as previously described.12 In brief, using density-based Ficoll-Histopaque 1077-1 (Sigma), PMN were isolated from human whole blood. Red blood cells were lysed by hypotonic lysis. Purified PMN were resuspended in DPBS containing 0.1% bovine serum albumin and incubated with either vehicle (<0.03% EtOH by volume), PD1 (3), or 22-OH-PD1 (6) (15 min, 37 °C). For the chemotaxis assays, 1 × 105 PMN were plated on the upper chamber of a ChemoTx System plate (3 μm pore size filter, Neuro Probe), and chemotaxis toward LTB4 (10 nM) or IL-8 (100 ng/mL, R&D Systems) was measured after a 90 min incubation period (37 °C in 5% CO2). The number of migrated PMN was measured using fluorescence-based PrestoBlue quantification (Life Technologies) and read on a SpectraMax M3 plate reader (Molecular Devices Inc.).
PMN Flow Chamber
Purified human PMN were pretreated for 10 min with 22-OH-PD1 (6), PD1 (3), or vehicle control; cells were then stimulated with 10 nM LTB4. PMN (1 × 106 cells/mL) were perfused over fibronectin-coated (R&D Systems) 1 μ-Slide VI0.4 ibiTreat microscopy chambers (Ibidi) at 0.5 mL/min flow rate. Six fields of view were recorded per slide for 10 s. Adherent PMN were quantified using Image ProPlus 7 (MediaCybernetics).12
Acknowledgments
We are grateful to Dr. A. Bjørkøy, Department of Physics, Norwegian University of Science and Technology, for recording the CD spectrum of compound 7. The Norwegian Research Council (KOSK II) and the School of Pharmacy, University of Oslo, are gratefully acknowledged for Ph.D. scholarships to M.A. and J.E.T., respectively. T.V.H. is grateful for a Leiv Eriksson travel grant from The Norwegian Research Council. S.R., R.C., and J.D. are supported by the National Institutes of Health GM Grant PO1GM095467 (C.N.S.).
Supporting Information Available
Experimental procedures and characterization data of the methyl ester 7 and synthetic intermediates 10–21, 1H and 13C NMR spectra, HRMS and UV/vis spectra, HPLC analyses of synthetic compounds as well as LC/MS-MS data, and GC/MS chromatograms of endogenous 22-OH-PD1 (6). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ J. E. Tungen and M. Aursnes contributed equally.
The authors declare the following competing financial interest(s): C.N.S. has filed patents on PD1 (3), 22-OH-PD1 (6), and related compounds. C.N.S. and J.D. have filed patents on PD1n-3 DPA (5) and related compounds. C.N.S.’s interests are reviewed and are managed by BWH and Partners HealthCare in accordance with their conflict of interest policies.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tabas I.; Glass C. K. Science 2013, 339, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P. A. In Fundamentals of Inflammation; Serhan C. N.; Ward P. A.; Gilroy D. W., Eds.; Cambridge University Press: Cambridge, 2010; pp 1–16. [Google Scholar]
- Serhan C. N.; Savill J. Nat. J. Immunol. 2005, 6, 1191–1197. [DOI] [PubMed] [Google Scholar]
- Serhan C. N. Annu. Rev. Immunol. 2007, 25, 101–137. [DOI] [PubMed] [Google Scholar]
- Serhan C. N.; Petasis N. A. Chem. Rev. 2011, 111, 5922–5943and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N.; Clish C. B.; Brannon J.; Colgan S. P.; Chiang N.; Gronert K. J. Exp. Med. 2000, 192, 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N.; Hong S.; Gronert K.; Colgan S. P.; Devchand P. R.; Mirick G.; Moussignac R. L. J. Exp. Med. 2002, 196, 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.; Gronert K.; Devchand P. R.; Moussignac R.-L.; Serhan C. N. J. Biol. Chem. 2003, 278, 14677–14687. [DOI] [PubMed] [Google Scholar]
- Mukherjee P. K.; Marcheselli V. L.; Serhan C. N.; Bazan N. G. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 8491–8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel A.; Pin-Lan L.; Wang W.; Tang W. X.; Fredman G.; Hong S.; Gotlinger K. H.; Serhan C. N. J. Biol. Chem. 2005, 280, 43079–43086. [DOI] [PubMed] [Google Scholar]
- Serhan C. N.; Yang R.; Martinod K.; Kasuga K.; Pillai P. S.; Porter T. F.; Oh S. F.; Spite M. J. Exp. Med. 2009, 206, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J.; Colas R. A.; Serhan C. N. Sci. Rep. 2013, 3, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aursnes M.; Tungen J. T.; Vik A.; Collas R.; Cheng C.-Y.; Dalli J.; Serhan C. N.; Hansen T. V. J. Nat. Prod. 2014, 77, 910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N.; Chiang N. Curr. Opin. Pharmacol. 2013, 13, 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tello-Aburto R.; Ochoa-Teran A.; Olivo H. F. Tetrahedron Lett. 2006, 47, 5915–5917. [Google Scholar]
- Aursnes M.; Tungen J. T.; Vik A.; Dalli J.; Hansen T. V. Org. Biomol. Chem. 2014, 12, 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemler S. R.; Roush W. R. In Modern Carbonyl Chemistry; Otera J., Ed.; Wiley-VCH: Weinheim, Germany, 2000; pp 403–490. [Google Scholar]
- Pietruszka J.; Schöne N. Synthesis 2006, 24–30. [Google Scholar]
- Boland W.; Schroer N.; Sieler C.; Feigel M. Helv. Chim. Acta 1987, 70, 1025–1040. [Google Scholar]
- Nicolaou K. C.; Ladduwahetty T.; Taffer I. M.; Zipkin R. E. Synthesis 1986, 344–347. [Google Scholar]
- Oger C.; Balas L.; Durand T.; Galano J. M. Chem. Rev. 2013, 113, 1313–1350. [DOI] [PubMed] [Google Scholar]
- Colas R. A.; Shinohara M.; Dalli J.; Chiang N.; Serhan C. N. Am. J. Physiol. Cell. Physiol. 2014, 307, C39–C54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheselli V. L.; Hong S.; Lukiw W. J.; Tian X. H.; Gronert K.; Musto A.; Hardy M.; Gimenez J. M.; Chiang N.; Serhan C. N.; Bazan N. G. J. Biol. Chem. 2003, 278, 43807–43817. [DOI] [PubMed] [Google Scholar]
- Sumimoto H.; Minakami S. J. Biol. Chem. 1990, 265, 4348–4353. [PubMed] [Google Scholar]
- Divanovic S.; Dalli J.; Jorge-Nebert L. F.; Flick L. M.; Gálvez-Peralta M.; Boespflug N. D.; Stankiewicz T. E.; Fitzgerald J. M.; Somarathna M.; Karp C. L.; Serhan C. N.; Nebert D. W. J. Immunol. 2013, 191, 3347–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernerova M.; Hudlicky T. Synlett 2010, 18, 2701–2707. [Google Scholar]
- Dalli J.; Winkler W.; Colas R. A.; Arnardottir H.; Cheng C. Y.; Chiang N.; Petasis N. A.; Serhan C. N. Chem. Biol. 2013, 20, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J.; Serhan C. N. Blood 2012, 120, e60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y.; Chiang N.; Oh S. F.; Serhan C. N.. Current Protocols in Immunology; John Wiley & Sons, 2011, Chapter 14: Unit 14.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.