

SUMMARY
The antiepileptic drugs (AEDs) introduced during the past two decades have provided several benefits: they offered new treatment options for symptomatic treatment of seizures, improved ease of use and tolerability, and lowered risk for hypersensitivity reactions and detrimental drug– drug interactions. These drugs, however, neither attenuated the problem of drug-refractory epilepsy nor proved capable of preventing or curing the disease. Therefore, new preclinical screening strategies are needed to identify AEDs that target these unmet medical needs. New therapies may derive from novel targets identified on the basis of existing hypotheses for drug-refractory epilepsy and the biology of epileptogenesis; from research on genetics, transcriptomics, and epigenetics; and from mechanisms relevant for other therapy areas. Novel targets should be explored using new preclinical screening strategies, and new technologies should be used to develop medium- to high-throughput screening models. In vivo testing of novel drugs should be performed in models mimicking relevant aspects of drug refractory epilepsy and/or epileptogenesis. To minimize the high attrition rate associated with drug development, which arises mainly from a failure to demonstrate sufficient clinical efficacy of new treatments, it is important to define integrated strategies for preclinical screening and experimental trial design. An important tool will be the discovery and implementation of relevant biomarkers that will facilitate a continuum of proof-of-concept approaches during early clinical testing to rapidly confirm or reject preclinical findings, and thereby lower the risk of the overall development effort. In this review, we overview some of the issues related to these topics and provide examples of new approaches that we hope will be more successful than those used in the past.
Keywords: Drug development, Antiseizure drug, Epileptogenesis, Disease modification, Comorbidities, Biomarkers
Preclinical screening for new antiepileptic drugs (AEDs) was first introduced by Merritt and Putnam in 1937, when they tested a number of compounds from Parke Davis against maximal electroshock (MES) seizures in cats, leading to the discovery of phenytoin. The subcutaneous (s.c.) pentylenetetrazol (PTZ) test was later used in mice and revealed the anticonvulsant properties of trimethadione (Everett & Richards, 1944). Phenytoin was found to be inactive in this model, proving that these two seizure tests identify AEDs with different clinical profiles. For this reason, these two models have for several decades constituted the two key preclinical tests for random screening aimed at detection of new AEDs. Together with structural variation of known AEDs and rational drug discovery, preclinical screening tests have led to the identification of a significant number of new drugs, in particular of third-generation AEDs introduced during the last two decades. This has expanded the armamentarium of AEDs, providing more treatment options, improved ease of use and tolerability, and lowered risk of hypersensitivity reactions and detrimental drug–drug interactions. However, none of these AEDs have been able to significantly reduce the prevalence of drug-refractory epilepsy or to provide a preventive and/or curative pharmacologic treatment for the disease (Löscher & Schmidt, 2011). This highlights the need for future preclinical screening strategies to focus on identifying AEDs that target key unmet medical needs by new treatment paradigms that prevent or reverse drug-refractory epilepsy, epileptogenesis, and/or comorbidity.
New treatment paradigms may derive from novel targets identified by creative scientists taking inspiration from existing hypotheses for drug-refractory epilepsy and the biology of epileptogenesis, or from research on genetics, transcriptomics, and epigenetics, or from mechanisms relevant for other therapy areas. These targets should be explored using new preclinical screening strategies (Galanopoulou et al., 2012). To find better drugs, it will probably be necessary to expand the traditional “antiseizure” approach based on the screening models employed in the past because, most likely, the continued use of existing screening models will not favor the development of drugs (or, more generally, therapeutic approaches) more effective than those already available. Instead, in vivo testing should incorporate relevant models that mimick different aspects of drug refractory epilepsy and epileptogenesis. Moreover, it is essential to optimize the experimental design and outcome measures to permit valid conclusions with reference to the multiple forms of the human epilepsies. To minimize the high attrition rate associated with the drug development pipeline resulting from the failure to demonstrate sufficient clinical efficacy, it is important to define new integrated strategies for preclinical screening and experimental trial design. These should focus on identifying drug candidates for viable patient populations and end points permitting conduct of pivotal trials targeting indications in drug refractory epilepsy, disease modification, and epileptogenesis. An important tool for successful execution of integrated preclinical screening and experimental trial design strategies will be the availability of relevant biomarkers that will enable the preclinical research community to assess to what extent a drug candidate engages with its target and modulates associated biological processes, disease activity, and regulatory end points. This will permit a continuum of proof-of-concept approaches during early clinical testing to more rapidly confirm or reject preclinical findings, important to de-risk the development efforts. In this review, based on the discussion generated by our presentations at the XI Workshop on the Neurobiology of Epilepsy (WONOEP XI) organized by the Neurobiology Commission of the International League Against Epilepsy (ILAE) in Grottaferrata, Italy (August 2011), we will discuss some of the issues related to these topics and provide examples of new approaches that will hopefully accelerate the search for improved therapies of epilepsy.
Finding New Antiseizure Drugs
So far, AEDs have been identified either by serendipity, screening in simple acute-seizure models, evaluating structural modifications to existing AEDs, or “rational” drug design approaches based on the concept of increasing inhibition or reducing excitation. Because serendipity is not an approach, and given the limited success of the “rational” yet simplified approach based on decreasing an abstract ratio of excitation to inhibition in a synaptic network, current screening models testing the ability to prevent a single seizure in a nonepileptic brain remain the only practical option, unless a more useful rational approach can be developed. As stated above, however, there is growing pessimism that the acute, induced seizure models employed thus far (i.e., MES and PTZ) will lead to identification of drugs more effective than those currently in use. A growing literature exists that suggests that the epileptic brain has substantially altered structure and function and this could lead to novel targets for drug development, but also suggests that preclinical tests should use epileptic tissue. Therefore, new concepts and models of drug-resistant epilepsy should be employed (Margineaunu & Klitgaard, 2009; Löscher, 2011). In this respect, both animal (e.g., the phenytoin-resistant or the lamotrigine-resistant kindled rats; post–status epilepticus models with spontaneous recurrent seizures) and human models are available (hippocampal slices prepared from surgical resection in drug-resistant patients) (Klitgaard et al., 2008; Löscher, 2011).
An alternative, new approach may be based on systems biology (Loeb, 2011). Although deductive approaches to drug target identification use our current state of knowledge, based mostly on animal models, systems biology takes advantage of newer high-throughput technologies to profile large numbers and types of molecules simultaneously. This approach can be used to link human brain anatomy, histology, and electrophysiology from patients who have undergone epilepsy surgery with functional genomics, proteomics, and metabolomics as a means to hone in on new biomarkers and therapeutic targets. This requires developing computational tools for storing, integrating, and “mining” diverse types of data. Proposed experimental paradigms include identification of pathways important for seizures or of pathways unique to regions of human brain with frequent interictal spiking. Because this approach does not profile the acute effects of seizures, but the chronic differences in regions of the brain prone to seizures, it may produce biomarkers (and targets) that differentiate these critical regions in the chronic state, and which might even reflect events that have occurred during epileptogenesis. By identifying common pathways shared by many patients with epilepsy, this approach has led to the identification of biomarkers of epileptic activity that implicate specific cellular populations (Rakhade et al., 2005). As expected, some of the most important of these relate to pathways involved in synaptic plasticity, inflammation, and metabolism.
The next steps will be to translate biomarkers into therapeutic targets through a combination of both human tissue and animal model validation studies followed by the identification and testing of compounds that specifically disrupt these pathways in animal models, as a prelude to clinical testing. This will also permit preparation of ligands for positron emission tomography (PET) that can determine target engagement at therapeutic doses in preclinical models. By conduct of similar PET studies in humans, this will enable optimal translation of dose selection for clinical trials—an issue that has been haunting several previous development programs with antiseizure drugs.
Finding New Antiepileptogenic Drugs
Prevention of epileptogenesis remains a significant unmet medical need in the field of epilepsy. The currently available AEDs have been developed primarily in models of seizures (not of epileptogenesis) and, as one might predict, they do not have antiepileptogenic effects. Available animal models of epileptogenesis include kindling models, genetic models, status epilepticus models, insult specific models (posttraumatic brain injury, poststroke, and so on). However, it is cumbersome to screen for drugs that prevent epilepsy in these models, because spontaneous recurrent seizures begin gradually and the interval between seizures varies widely, requiring long-term, intensive seizure monitoring to determine whether a drug alters epileptogenesis. A rigorous yet rapid screen would enable us to test many potentially antiepileptogenic compounds.
One promising option could be a multistep approach, in which putative drugs are screened in predictive in vitro models before being tested in expensive and time-consuming in vivo models. Indeed, an in vitro model of epileptogenesis has been developed, where brain slices are placed on glass coverslips in culture (organotypic slice culture). The model maintains much of the normal circuitry of the brain slice, and allows one to record electrical activity (including epileptiform discharges and seizures) and administer drugs efficiently to multiple tissue samples simultaneously. Early reports regarding epileptogenesis in this preparation (McBain et al., 1989; Bausch & McNamara, 2000) have led to the development of a promising new model of posttraumatic epilepsy. These organotypic brain-slice cultures undergo a rapid, predictable process of epileptogenesis, and respond to anticonvulsant drugs just as human patients do, including suppression of seizures (but not interictal spiking) and gradual development of drug resistance (Dyhrfjeld-Johnsen et al., 2010; Berdichevsky et al., 2012). Computer algorithms have been developed for continuously recording and quantifying electrographic spikes and seizures (White et al., 2006). These algorithms have been validated in two in vivo models of epileptogenesis as well as in the in vitro model (Williams et al., 2009; Kadam et al., 2010; White et al., 2010).
Screening of the NINDS compound library for antiepileptogenic activity is currently ongoing, based on a multistage protocol (Berdichevsky et al., 2011). In the first stage, compounds are screened using the parallel cultured brain slice assays described. Compounds that prevent, reduce, or reverse epileptogenesis, or are neuroprotective, are evaluated in a second stage comprising blinded replication with larger numbers of brain slices, additional concentrations of the compound, and more precise measures of neuroprotection. Compounds that demonstrate sufficient promise in the second stage are then subjected to a third, more rigorous albeit much slower assay, such as the in vivo kainate model of experimental epileptogenesis. All compounds are subjected to at least two stages in which electrographic seizure activity is assayed quantitatively using continuously recorded and analyzed electroencephalography (EEG) data. This three-stage protocol is demonstrating considerable utility as a means to screen compound libraries for antiepileptogenic activity. Costs for the first 100 compounds screened were nearly $1,000 per compound, but this could be reduced to approximately $100 per compound with appropriate scaling.
Numerous in vivo studies have now been undertaken with the goal of testing hypotheses about therapeutic strategies for blocking the development of epilepsy after brain injury (for review see Löscher & Brandt, 2010; Pitkänen, 2010). Most work is now focusing on postinsult interventions (vs. interventions before the insult), because this is the clinically relevant approach. The major focus in these studies is reduction of spontaneous recurrent seizures (as opposed to increasing seizure threshold). Because of the need for continuous video-EEG monitoring of spontaneous seizures, in vivo antiepileptogenesis (or disease modification) studies are resource-intensive and fraught with potential problems.
Continuous versus discontinuous recording protocols with either wired or wireless systems have been used for testing antiepileptogenic or disease-modifying treatment. The progressive development of epilepsy has also been analyzed in two animal models of acquired epilepsy (Williams et al., 2009; Kadam et al., 2010), and after different hypothetical treatments to suppress seizures and/or the development of epilepsy (Dudek et al., 2010; Pouliot et al., 2011). The data from the different approaches and animal models provide insights concerning advantages and disadvantages of different protocols and recording configurations. First, nearly continuous recordings strongly suggest that the development of chronic epilepsy, as defined by the measurement of the frequency and severity of spontaneous chronic seizures, is a sigmoid function of time, and not a step function; therefore, the measurement of the “latent” period between the epileptogenic insult and the first spontaneous seizure as the basis for assessing changes in epileptogenesis seems highly problematic on theoretical grounds (Fig. 1). From a practical perspective, accurate determination of the latent period requires continuous video-EEG recordings with virtually no breaks in the recordings; the details of such measurements have generally not been provided in published studies. Because the earliest epileptic seizures are almost always nonconvulsive (clinically inapparent), both video and EEG recordings must be done continuously to accurately measure the latent period. These data argue that other measurements of epileptogenesis might be more accurate and informative. Although intermittent recordings (e.g., 2 weeks of recording with 6 weeks between recording epochs) may appear useful in terms of optimizing equipment usage and reducing labor-intensive analyses, the trade-off in terms of accuracy and statistical power is a concern (Dudek et al., 2010; Pouliot et al., 2011).
Figure 1.
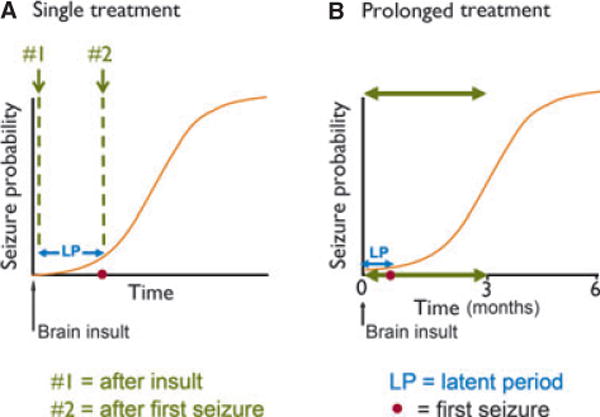
Diagram of the time course of acquired epileptogenesis and hypothetical times to administer a disease-modification therapy. Sigmoid curves illustrate the time course of epileptogenesis after a brain insult (vertical arrow). The occurrence of the first clinical seizure (closed circle) marks the duration of the latent period (LP). Possible therapeutic interventions could hypothetically involve a single drug treatment or may require a prolonged treatment schedule. (A) Single administration of a disease-modification therapy. One obvious possibility is therapy administration after the insult, but before the first spontaneous seizure (see no. 1). However, a more realistic treatment protocol might be administration of the therapy after the first spontaneous seizure (see no. 2). A disease-modification therapy, however, may require prolonged administration (e.g., 3 months), well after the onset of spontaneous recurrent seizures (B). Modified from Williams et al. (2009) and Dudek and Staley (2011).
Long-term, continuous monitoring from the onset of brain injury has theoretical and practical advantages over other approaches, such as measuring the latent period or assessing seizure frequency weeks or months after the brain injury. However, neither approach is easy or inexpensive. Protocols and analytical tools are under development to balance the accuracy of measurement and resource utilization in preclinical antiepileptogenesis studies.
Another critical consideration in a clinical study is the timing of the treatment protocol for the prevention or modification of acquired epileptogenesis, and these issues are obviously just as important in the design of preclinical studies. It is generally agreed that preclinical experiments involving administration of a hypothetical therapy before the brain insult are unrealistic and could alter the severity of the insult. Administration of a therapy during the brain insult would obviously be a realistic approach for some insult types (e.g., complex febrile seizures, brain infections, status epilepticus), but, again, would include insult-modification along with disease-modification. The issue under consideration here are treatments that would alter the process of epileptogenesis when administered after the brain insult, recognizing that the duration of many epileptogenic insults is not yet clear. In status epilepticus models in the adult rodent, for example, there is often a very gradual transition from status epilepticus to intermittent seizures to interictal spiking over the first 24–48 h (White et al., 2010), although 3 h after onset of status is generally considered to be “after the brain insult.” Is it practical to administer a therapy immediately after the brain injury, or should treatment be given only after the occurrence of the first spontaneous recurrent seizure (Fig. 1A)? The occurrence of the first clinical seizure marks the duration of the latent period, and most clinical epileptologists and basic scientists would argue that epileptogenesis has already occurred by the time the first seizure has been detected. The likelihood that a patient will develop epilepsy after a particular brain insult and the degree that the hypothetical therapy has potential to induce adverse effects are among the important factors that would dictate when a therapy might be realistically administered to a brain-injured patient. For example, it might well be necessary to administer the therapy after the first unprovoked or spontaneous seizure if (1) the probability of developing epilepsy after a brain insult was low, (2) the hypothetical therapy carried a distinct risk for adverse effects, and (3) the therapy had suboptimal efficacy. This dilemma highlights the importance of identifying biomarkers for acquired epileptogenesis, since they could determine the nature and time course of therapy. Finally, it is possible that some therapies might need to be administered for prolonged periods (Fig. 1B); and furthermore, these therapies could have both a disease-modifying effect and may also symptomatically suppress seizures, as proposed for levetiracetam (Löscher & Brandt, 2010). These considerations determine directly how realistic preclinical studies will need to be performed.
Therefore, the discovery of disease-modifying and antiepileptogenic therapies remains a challenging issue for both epilepsy research community and pharmaceutical industry. Rat models in which spontaneous recurrent seizures develop after brain insults such as status epilepticus (e.g., the kainate or pilocarpine models) and genetically defined rat models of spontaneous recurrent seizures may offer an opportunity for compounds testing and preclinical demonstration of their potential for disease modification. One promising example is represented by the success of early preclinical ethosuximide in rat strains with spontaneous spike-wave seizures (Blumenfeld et al., 2008). Unfortunately, developing similar reliable models in mice has proven difficult, mainly as a result of genetic interstrain and intrastrain differences in sensitivity to chemical or electrical induction of epilepsy (Schauwecker, 2002; Müller et al., 2009; Schauwecker, 2011). Although such differences may help to further explain genetic factors that contribute to the pharmacology of seizure disorders, there is a considerable risk that using certain strains of mice for preclinical testing may lead to false-negative data. However, the utility of transgenic mice in epilepsy research is undisputable. Mouse models may also offer other advantages such as smaller amounts of compounds needed for testing and potentially shorter treatment duration due to faster development of epileptogenesis. These factors will eventually lead to a significant reduction of study costs and higher throughput.
Recently, a model has been developed of self-sustained status epilepticus (SSSE) induced by electrical stimulation via bipolar electrode unilaterally implanted into the amygdala of adult C57 black mice (Niespodziany et al., 2010). A few days after SSSE mice start to develop spontaneous recurrent seizures, as documented by 8-week–long continuous video-EEG monitoring. Of interest, as observed in rats, most mice display clustering of spontaneous recurrent seizures with seizure-free periods between clusters. Histopathologic assessment reveals neuronal loss that is confined to the hilus of the dentate gyrus (DG). This is a novel model that has great potential because it provides a means to address a longstanding question: the relevance of endfolium sclerosis to limbic epileptogenesis. Field potential recordings were performed in hippocampal slices taken from ipsilateral brain hemispheres 12–14 weeks after SSSE induction and were compared to sham-implanted mice. Field excitatory postsynaptic potentials (fEPSPs) were evoked via electrical stimulation in CA3, CA1, and DG by electrical stimulation of fimbria, Schaffer collaterals, and perforant path, respectively. The fEPSPs evoked in the CA3 and CA1 areas showed a significantly higher number of population spikes in SSSE mice than in sham mice, whereas no change was observed in the DG. Combining in vivo video-EEG monitoring with in vitro electrophysiologic and histopathologic end points in this SSSE mouse model offers a unique approach for the discovery and preclinical testing of future disease-modifying treatments.
Preclinical Study Optimization
Optimization of preclinical study protocols is essential to improve success in the translation to the clinics. Many important parameters should be carefully controlled, including the choice of the species, strains, age, and gender of animals; the methodology for in vivo seizure initiation, termination, recording, and typing; the determination of pharmacokinetics and pharmacodynamics; and the sample size. Equally important are the parameters used to reach valid conclusions: the primary end point should be the therapeutic gain (i.e., efficacy, not potency). The therapeutic window should also be determined, as should the therapeutic index, based on tolerability and safety measures. This will enable one to determine if a drug candidate may hold a potential therapeutic benefit.
It is not the aim of this review to discuss in detail each of these points; however, we will provide some examples of how modeling specific syndromes, as well as appreciation of age and gender, can be very important in projecting results in translational terms. One example is infantile spasms. A model of cryptogenic infantile spasms has been developed and validated, consisting of prenatal priming with betamethasone and postnatal trigger of developmentally specific spasms with N-methyl-D-aspartic acid (NMDA) (Velíšek et al., 2007; Chachua et al., 2011). The spasms in this model respond to adrenocorticotropic hormone (ACTH); therefore the model offers an opportunity to investigate the mechanisms of ACTH effects against the spasms and may be used to identify new therapeutic approaches, specific for this disease. Another example is catamenial epilepsy. Epileptic female rats have been examined and interictal spikes monitored in awake animals in their home cages every day (Scharfman et al., 2009; D’Amour et al., 2010). Every 4 days (i.e., the duration of the estrous cycle in the female rat) animals exhibit a dramatic rise in interictal spikes during all behavioral states, including exploration, quiet immobility, and sleep. These data support a neurobiologic mechanism for catamenial epilepsy in women, which may be underestimated because women with epilepsy may have robust cyclic EEG activity even when convulsive seizures are not reported.
Clinical Trial Optimization
Other major problems with AED development are implicit in clinical trial design and conduction that may lead to “false negatives,” that is, useful discoveries in the preclinical phase that are lost because not properly tested in clinical trials. Most of randomized controlled trials for AEDs are carried out on patients with “complex partial seizures with or without secondarily generalized seizures.” This description undoubtedly includes a wide variety of ictal events, representing diverse epileptogenic and ictogenic mechanisms. Extraordinary efficacy of a potential antiseizure compound against one subset of these seizure types would go unnoticed if it were not effective against any of the others, in which case a potentially important antiseizure medication would never reach the market. Therefore, research is needed to more carefully characterize fundamental neuronal mechanisms underlying different types of ictogenesis that might influence their response to specific antiseizure compounds.
Another problem is that most trials of new antiseizure drugs are conducted in treatment resistant or medically refractory patients with a baseline seizure frequency of at least one per week. Many of these patients were evaluated for epilepsy surgery and rejected, often due to multifocality. Although there are both practical and ethical reasons for defining trial populations in this fashion, the population may have biologic characteristics that render it poorly representative of the general epilepsy population. For example, animal studies suggest that treatment resistance may be in part genetically determined (Cramer et al., 1998). Moreover, clinical experience and epidemiologic data suggest that although some individuals are predisposed to treatment resistance from the time of onset of their epilepsy (Kwan & Brodie, 2000), at the very least seizure frequency and severity often worsen over time, implying a progressive component of the illness. These observations raise several important issues related to clinical trial design. First, it is possible that the individuals commonly studied have background characteristics that render them treatment resistant that would not be relevant to others in the target population. Second, it is possible these individuals have developed secondary mechanisms of ictogenesis due to disease progression (e.g., gliosis, depletion of specific classes of interneurons or principle cells, or use dependent facilitation of pathways and connections mediating pathological activity) that distinguish them from the general epilepsy population. Finally, it is possible that clinical trials are preferentially enrolling individuals in whom treatment may be futile due to some combination of the features enumerated above as well as due to the underlying etiology and pathophysiology of their disease. The net result of these factors is that current standard clinical trial designs may perversely generate false-negative results regarding the efficacy of antiepileptic treatments in the broader epilepsy patient population. Because existing treatments, even if efficacious, carry a significant adverse effect burden reducing their effectiveness, development of new antiseizure agents must target the entire spectrum of patients with epilepsy, not just those who are most refractory. Although pharmacogenomics may provide valuable insights in how subsets of epilepsies can be more effectively treated, it remains essential to overcome the practical and ethical issues and develop new trial designs aimed at recruiting patients earlier in the course of their disease, perhaps with lower seizure frequencies in order to test new agents in a more representative clinical population. For example, a time to nth seizure design might be acceptable to individuals with milder epilepsy due to the ability to rapidly escape from ineffective experimental treatment.
Even more difficult are the clinical trials of antiepileptogenic treatments, which must overcome major conceptual, practical, and financial challenges to successfully answer the question of whether a specific treatment is antiepileptogenic. Conceptual challenges include identifying the optimal clinical population for study, resolving the confound between antiseizure and antiepileptogenic properties of the study treatment, differentiating between delay and prevention of the emergence of epilepsy, and defining an adequate duration of follow-up after which success can be declared given the highly variable latent interval between initial insult and appearance of epilepsy in clinical practice. Practical challenges include enrolling an adequate number of subjects to provide the required power, determining the length of treatment prior to commencement of follow-up, defining the optimal time for initiation of antiepileptogenic treatment, minimizing loss to follow-up, and separating the appearance of genuine epilepsy from the occurrence of isolated symptomatic or provoked seizures during the follow-up interval. Financial challenges include the inevitably high cost of conducting large-scale long-duration clinical trials, the issue of reconciling the duration of antiepileptogenesis studies with the limitations on patent life that affect return on investment for industrial sponsors on the one hand, and the current constraints that limit government-financed clinical trials on the other. This highlights the importance of defining new integrated strategies for the discovery and development of antiepileptogenic treatments in order to counteract the risk of a high attrition rate. Important for optimal translation of preclinical findings will be the availability of relevant biomarkers that should also permit determination of target engagement of new potential antiepileptogenic treatments at therapeutic doses in preclinical models, improving translation to human studies and enabling conduct of clinical trials with optimal doses. In addition, availability of biomarkers that permit measurement of the consequence of target modulation on associated biologic processes, disease activity, and regulatory end points will enable proof-of-concept studies already during early clinical testing, to rapidly confirm or reject preclinical findings. This holds the potential to markedly de-risk the development efforts by terminating them before initiation of financially demanding clinical trials.
Patients who have sustained head injury offer the best-characterized population in which to study the process of epileptogenesis, in part because the timing of the inciting event can be precisely established in most cases, the incidence of posttraumatic epilepsy as a function of severity of injury has been well characterized, and dropout rates and loss to follow-up can be reliably estimated based on previous studies. Power calculations indicate that approximately 750 patients would have to be randomized to treatment or placebo to have an 80% chance of detecting a 50% reduction in the appearance of epilepsy, defined as two spontaneous seizures without clearly identifiable provocative cause during a 3-year follow-up period. The estimated cost of a clinical trial of this scale and duration ranges from $25,000,000 to $40,000,000.
This analysis strongly supports the need for rigorous target validation and preclinical studies as well as new integrated discovery and development strategies in order to sufficiently de-risk the investment in multimillion dollar clinical development programs. In particular, information on the timing and duration of treatment, as well as on the extent of target engagement, will be critical for successful translation of preclinical findings and optimization of clinical trial design. Again, this highlights that identification of relevant biomarkers and PET ligands that determine target engagement will favor productive outcome of future development of antiepileptogenic treatments.
Usefulness of Biomarkers
Therefore, clinical trials could be greatly facilitated if there were biomarkers or surrogate end points that could reliably predict the efficacy of a potential antiepilepsy therapy without the need to wait for another seizure to occur (antiseizure effects in diagnosed patients) or of spontaneous seizures to occur (antiepileptogenic effect). A biomarker can be defined as an objectively measured characteristic of a normal or pathologic biologic process, or a biologic response to a therapeutic intervention (Biomarkers Definitions Working Group, 2001, Engel, 2011). A surrogate end point is a biomarker that can substitute for a clinical end point (Biomarkers Definitions Working Group, 2001) and can therefore provide an indirect measure of disease presence or progression (Engel, 2011). Noninvasive biomarkers that could predict the risk of future ictal events would facilitate development of antiseizure and antiepileptogenic compounds as well as clinical trials, reducing their costs. Possible biomarkers of epileptogenesis include hippocampal changes in MRI, interictal (or preictal) spikes, pathologic high-frequency oscillations, changes in excitability in response to transcranial magnetic stimulation (TMS), PET imaging with alpha-methyl-tryptophan (AMT), and molecular markers of gene expression pathways.
Conclusions
The preclinical development of new antiepilepsy therapies will require intense efforts in many directions. An important feature that should distinguish the future from the past is the use of integrated discovery and development strategies. These will be mandatory to ensure optimal translation of preclinical findings and to de-risk costly development efforts. Future preclinical screening strategies can be strengthened by adopting a rational drug discovery approach that focuses on novel targets and includes relevant animal models of drug refractory epilepsy and epileptogenesis. Optimized experimental design of the screening models will be important to permit valid conclusions. In vivo pharmacodynamic testing should be associated with proper pharmacokinetic analysis. Most importantly, there is a need to identify and validate biomarkers and/or surrogate end points, especially for antiepileptogenesis, and to develop screening models relevant to epilepsies affecting the young and the old population. Pharmacogenomics may provide valuable insights in how subsets of epilepsies can be more effectively treated. There is an urgent need to identify viable patient populations and end points for pivotal trials that target drug refractory epilepsies, disease modification, and epileptogenesis. Targeting the intermediate pathology in epileptic brain, cell death for example, may represent an option to explore.
Several points remain open to debate. For instance: Is “rational” drug development working? Is the concept of inhibition/excitation imbalance useful to drug discovery? Are there targetable final common pathways in epileptogenesis (e.g., “master regulators”)? What is the optimal time window for intervention, and will cocktails of drugs (rather than monotherapy) be useful for antiepileptogenesis?
In conclusion, to target unmet medical needs for treatments that prevent or reverse drug-refractory epilepsy and/or epileptogenesis, new integrated preclinical screening and experimental trial design strategies with relevant biomarkers are needed to rapidly confirm or reject preclinical findings within an experimental trial design. This will require significant experimental effort and discussion among preclinical and clinical scientists, with the promise of the ultimate reward—better treatments for people with epilepsy.
Footnotes
Disclosures
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- Bausch SB, McNamara JO. Synaptic connections from multiple subfields contribute to granule cell hyperexcitability in hippocampal slice cultures. J Neurophysiol. 2000;84:2918–2932. doi: 10.1152/jn.2000.84.6.2918. [DOI] [PubMed] [Google Scholar]
- Berdichevsky Y, Saponjian Y, Mail M, Staley KJ. Organotypic culture model of post-traumatic epileptogenesis used as a medium-throughput screen of antiepileptic drugs. American Epilepsy Society Annual Meeting. 2011 3.026. [Google Scholar]
- Berdichevsky Y, Dzhala V, Mail M, Staley KJ. Interictal spikes, seizures and ictal cell death are not necessary for post-traumatic epileptogenesis in vitro. Neurobiol Dis. 2012;45:774–785. doi: 10.1016/j.nbd.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, Klein JP, Schridde U, Vestal M, Rice T, Khera DS, Bashyal C, Giblin K, Paul-Laughinghouse C, Wang F, Phadke A, Mission J, Agarwal RK, Englot DJ, Motelow J, Nersesyan H, Waxman SG, Levin AR. Early treatment suppresses the development of spike-wave epilepsy in a rat model. Epilepsia. 2008;49:400–409. doi: 10.1111/j.1528-1167.2007.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chachua T, Yum M-S, Velíšková J, Velíšek L. Validation of the rat model of cryptogenic infantile spasms. Epilepsia. 2011;52:1666–1677. doi: 10.1111/j.1528-1167.2011.03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer S, Ebert U, Löscher W. Characterization of phenytoin-resistant kindled rats, a new model of drug-resistant partial epilepsy: comparison of inbred strains. Epilepsia. 1998;39:1046–1053. doi: 10.1111/j.1528-1157.1998.tb01289.x. [DOI] [PubMed] [Google Scholar]
- D’Amour J, Friedman D, Poveda A, Radman T, LaFrancois J, Schevon C, Emerson R, Schroeder C, MacLusky N, Scharfman H. EEG correlates of catamenial epilepsy in intact female rats. American Epilepsy Society Annual Meeting. 2010 3.050. [Google Scholar]
- Dudek FE, Staley KJ. The time course of acquired epilepsy: implications for therapeutic intervention to suppress epileptogenesis. Neurosci Lett. 2011;497:240–246. doi: 10.1016/j.neulet.2011.03.071. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Pouliot WA, Rossi CA, Staley KJ. The effect of the cannabinoid-receptor antagonist, SR141716, on the early stage of kainate-induced epileptogenesis in the adult rat. Epilepsia. 2010;51(Suppl. 3):126–130. doi: 10.1111/j.1528-1167.2010.02626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Berdichevsky Y, Swiercz W, Sabolek H, Staley KJ. Interictal spikes precede ictal discharges in an organotypic hippocampal slice culture model of epileptogenesis. J Clin Neurophysiol. 2010;6:418–424. doi: 10.1097/WNP.0b013e3181fe0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J., Jr Biomarkers in epilepsy: foreword. Biomark Med. 2011;5:529–530. doi: 10.2217/bmm.11.63. [DOI] [PubMed] [Google Scholar]
- Everett GM, Richards RK. Comparative anticonvulsive action of 3,5,5-trimethyloxazolidine-2,4-dione (Tridione), Dilantin and phenobarbital. J Pharmacol Exp Ther. 1944;81:402–407. [Google Scholar]
- Galanopoulou AS, Buckmaster PS, Staley KJ, Moshé SL, Perucca E, Engel J, Jr, Löscher W, Noebels JL, Pitkänen A, Stables J, White SH, O’Brien TJ, Simonato M. Identification of new treatments for epilepsy: issues in preclinical methodology. Epilepsia. 2012;53:571–582. doi: 10.1111/j.1528-1167.2011.03391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam SD, White AM, Staley KJ, Dudek FE. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J Neurosci. 2010;30:404–415. doi: 10.1523/JNEUROSCI.4093-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klitgaard H, Matagne A, Schachter SC, White HS. Animal and translational models of the epilepsies. In: McArthur RA, Borsini F, editors. Animal and translational models for CNS drug discovery (Volume 2, Neurological disorders) Elsevier Academic Press; San Diego, CA: 2008. pp. 311–335. [Google Scholar]
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- Loeb JA. Identifying targets for preventing epilepsy using systems biology. Neurosci Lett. 2011;497:205–212. doi: 10.1016/j.neulet.2011.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20:359–368. doi: 10.1016/j.seizure.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Löscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev. 2010;62:668–700. doi: 10.1124/pr.110.003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52:657–678. doi: 10.1111/j.1528-1167.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- Margineaunu DG, Klitgaard H. Mechanisms of drug resistance in epilepsy: relevance for antiepileptic drug discovery. Expert Opin Drug Discov. 2009;4:23–32. doi: 10.1517/17460440802611729. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Boden P, Hill RG. Rat hippocampal slices ‘in vitro’ display spontaneous epileptiform activity following long-term organotypic culture. J Neurosci Methods. 1989;27:35–49. doi: 10.1016/0165-0270(89)90051-4. [DOI] [PubMed] [Google Scholar]
- Müller CJ, Gröticke I, Hoffmann K, Schughart K, Löscher W. Differences in sensitivity to the convulsant pilocarpine in substrains and sublines of C57BL/6 mice. Genes Brain Behav. 2009;8:481–492. doi: 10.1111/j.1601-183X.2009.00490.x. [DOI] [PubMed] [Google Scholar]
- Niespodziany I, Leclere N, Neuveux M, Kaminski R. In vitro field potential recording reveals enhanced neuronal excitability after the development of spontaneous seizures in the mouse model of self-sustained status epilepticus. Society for Neuroscience Annual Meeting. 2010 55.6.Q7. [Google Scholar]
- Pitkänen A. Therapeutic approaches to epileptogenesis – hope on the horizon. Epilepsia. 2010;51(Suppl. 3):2–17. doi: 10.1111/j.1528-1167.2010.02602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouliot WA, Ricks K, Roach B, Nelson C, Dudek FE. An ultra low-dose repeated kainate injection model of repetitive seizures and subsequent epileptogenesis. American Epilepsy Society Annual Meeting. 2011 3.065. [Google Scholar]
- Rakhade SN, Yao B, Ahmed S, Asano E, Beaumont TL, Shah AK, Draghici S, Krauss R, Chugani HT, Sood S, Loeb JA. A common pattern of persistent gene activation in human neocortical epileptic foci. Ann Neurol. 2005;58:736–747. doi: 10.1002/ana.20633. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Malthankar-Phatak GH, Friedman D, Pearce P, McCloskey DP, Harden CL, Maclusky NJ. A rat model of epilepsy in women: a tool to study physiological interactions between endocrine systems and seizures. Endocrinology. 2009;150:4437–4442. doi: 10.1210/en.2009-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE. Complications associated with genetic background effects in models of experimental epilepsy. Prog Brain Res. 2002;135:139–148. doi: 10.1016/s0079-6123(02)35014-3. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. The relevance of individual genetic background and its role in animal models of epilepsy. Epilepsy Res. 2011;97:1–11. doi: 10.1016/j.eplepsyres.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšek L, Jehle K, Asche S, Velíšková J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann Neurol. 2007;61:109–119. doi: 10.1002/ana.21082. [DOI] [PubMed] [Google Scholar]
- White AM, Williams PA, Ferraro DJ, Clark S, Kadam SD, Dudek FE, Staley KJ. Efficient unsupervised algorithms for the detection of seizures in continuous EEG recordings from rats after brain injury. J Neurosci Methods. 2006;152:55–266. doi: 10.1016/j.jneumeth.2005.09.014. [DOI] [PubMed] [Google Scholar]
- White A, Williams PA, Hellier JL, Clark S, Dudek FE, Staley KJ. EEG spike activity precedes epilepsy after kainate-induced status epilepticus. Epilepsia. 2010;51:371–383. doi: 10.1111/j.1528-1167.2009.02339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, Dudek FE. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]