

Abstract
Autophagy as an immune mechanism controls inflammation and acts as a cell-autonomous defense against intracellular microbes including Mycobacterium tuberculosis. An equally significant role of autophagy is its anti-inflammatory and tissue-sparing function. This combination of antimicrobial and anti-inflammatory actions prevents active disease in animal models. In human populations, genetic links between autophagy, inflammatory bowel disease, and susceptibility to tuberculosis provide further support to these combined roles of autophagy. The autophagic control of M. tuberculosis and prevention of progressive disease provide novel insights into physiological and immune control of tuberculosis. It also offers host-based therapeutic opportunities because autophagy can be pharmacologically modulated.
Autophagy enables cells to capture pathogens (e.g., Mycobacterium tuberculosis) and kill them in lysosomes. It can also suppress excessive inflammatory responses that contribute to disease progression.
Autophagy in a broader sense is a collection of homeostatic processes (macroautophagy, chaperone mediate autophagy, microautophagy) in the eukaryotic cell that deliver cytoplasmic portions or specific cytosolic targets to lysosomes for degradation or removal (Mizushima et al. 2011). Macroautophagy, commonly referred to as autophagy (and in this text), is a pathway defined in genetic terms as dependent on autophagy-related (Atg) genes (Mizushima et al. 2011) and in morphological terms as the appearance in the cytoplasm of double-membrane organelles termed autophagosomes that capture cytosolic cargo and fuse with lysosomes (Deter and De Duve 1967; Deter et al. 1967). Autophagy affects human health and diseases including aging, neurodegeneration, cancer, and metabolic disorders (Mizushima et al. 2008). The recognition that autophagy plays an antimicrobial role against pathogens (Deretic 2005) when they invade the mammalian cell interior came from two nearly simultaneous reports in 2004 (Gutierrez et al. 2004; Nakagawa et al. 2004). One of these studies reported on the autophagic elimination of virulent Mycobacterium tuberculosis and the vaccine strain Mycobacterium bovis BCG (Bacillus Calmette–Guérin) in infected murine and human macrophages (Gutierrez et al. 2004). These and additional studies have established that autophagy is a bona fide immunological process (Deretic 2005; Levine et al. 2011). Today we recognize that autophagy is widely integrated with immunity starting from cell-autonomous defense against invading bacteria and viruses to regulation of innate and adaptive immunity in general (Deretic et al. 2013). The initial observations uncovering the role of autophagy against M. tuberculosis (Gutierrez et al. 2004; Singh et al. 2006) have been followed by a gradual increase in studies supporting and extending these early findings (Alonso et al. 2007; Harris et al. 2007; Xu et al. 2007; Biswas et al. 2008; Jagannath et al. 2009; Yuk et al. 2009; Ghadimi et al. 2010; Kumar et al. 2010; Ponpuak et al. 2010; Shin et al. 2010b; Singh et al. 2010; Fabri et al. 2011a,b; Campbell and Spector 2012; Juarez et al. 2012; Petruccioli et al. 2012; Watson et al. 2012; Zullo and Lee 2012; Anandaiah et al. 2013; Klug-Micu et al. 2013; Manzanillo et al. 2013). In this article we briefly cover autophagy as a pathway and its broad roles in immunity and summarize what has been learned about autophagy in tuberculosis thus far.
AUTOPHAGY PATHWAY
Autophagy is a cytoplasmic quality and quantity control pathway ubiquitous in eukaryotes. The core autophagy pathway is depicted in its minimalistic rendition in Figure 1. The membranes forming autophagic organelles come primarily from the endoplasmic reticulum, but additional compartments provide sources of membrane or lipids for the growing autophagosomal isolation membrane (phagophore) (Fig. 1). Autophagosome formation is dependent on a suite of Atg factors, which are consecutively numbered: for example, Atg1–Atg35 in yeast, with alternative names in mammals such as the Atg1 paralogs in mammals ULK1 and 2 (ULK1 being more studied) and the Atg6 ortholog Beclin 1. Autophagy is often equated with the emergence or modifications of endomembranes decorated with “LC3” (LC3B), which is one of six mammalian Atg8s (LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABRAPL2) (Mizushima et al. 2011), with a key event being the carboxy-terminal lipidation of LC3 to generate LC3-II (LC3-phosphatidylethanolamine). Autophagosomes capture cytosolic or cytoplasmic cargo destined for elimination, most commonly through lysosomal degradation (Deter and De Duve 1967; Deter et al. 1967; Itakura et al. 2012), although other modes of cargo disposal have been observed (Jiang et al. 2013). Autophagy can be modulated by metabolic, physiological, immunological (cytokines and innate immunity signaling), and pharmacological (e.g., rapamycin) agonists and antagonists. There are two forms of autophagy—nonselective (bulk or generalized) autophagic degradation of the cytoplasm, usually as a response to starvation, and selective autophagy, whereby specific targets in the cytosol are recognized by autophagic receptors and captured by autophagosomes (Birgisdottir et al. 2013).
Figure 1.

Autophagy pathway. Shown are a simplified macroautophagy pathway, protein factors, and membrane sources for the formation of autophagosomes in mammalian cells. (Top) Different sources of membranes contributing to the formation of autophagosomes. ER, endoplasmic reticulum; PM, plasma membrane. Within the cartoon: Crescent, autophagic phagophore or isolation membrane; double-membrane, closed autophagosome; red circle, lysosome; hatched circle delimited by a single membrane, autolysosome; green dots, LC3B (one of six mammalian Atg8s listed on the right side; typically used as a marker for autophagosomes). Other key factors are shown using their mammalian nomenclature.
INDUCTION OF BULK AUTOPHAGY BY NUTRITIONAL SIGNALS
The classical signal for generalized autophagy is starvation, which can be caused by low levels of amino acids, low energy, or absence of growth factors. Starvation can induce autophagy to kill virulent M. tuberculosis in macrophages (Fig. 2A) (Gutierrez et al. 2004). During starvation, portions of the cytosol are corralled into autophagosomes to generate amino acids (Lum et al. 2005) and energy sources, for example, via lipolysis (Settembre et al. 2013). These upstream metabolic events leading to autophagy activation are controlled via mTOR (mammalian target of rapamycin) and AMPK (AMP-activated Ser/Thr protein kinase). The mTOR Ser/Thr protein kinase negatively controls the most upstream of the Atg factors, ULK1, by phosphorylating it at inactivating sites (e.g., Ser 757), whereas AMPK positively regulates ULK1 by phosphorylating ULK1 at activating sites (e.g., Ser 317 and Ser 777) (Egan et al. 2011; Kim et al. 2011). ULK1 phosphorylates a number of key substrates, most pertinently Beclin 1 (mammalian Atg6) at Ser 15 (Russell et al. 2013). Beclin 1 works with other subunits of class III phosphatidylinositol 3 kinase hVPS34 to generate phosphatidylinositol 3-phosphate (PI3P) on donor (e.g., ER [Axe et al. 2008]) membranes. PI3P marks the spot for initiation of autophagic phagophore formation. A phagophore expands into a full autophagosome. This is concomitant with or preceding the acquisition of SNARE Syntaxin 17 (Itakura et al. 2012; Hamasaki et al. 2013), which promotes fusion with lysosomes whereby a terminal degradative organelle, autolysosome, is formed where the captured cargo is degraded.
Figure 2.

Autophagy protects against M. tuberculosis infection and pathogenesis. (A) Autophagy induction by starvation or rapamycin kills virulent M. tuberculosis in macrophages. (B) Increased gross lung pathology in mice defective for autophagy in the myeloid lineage (Cre+) relative to autophagy-competent (Cre−) mice infected with M. tuberculosis H37Rv. (C) Increased lung tissue necrosis and bacillary load in the lungs of mice defective for autophagy in the myeloid lineage (Cre+) relative to their autophagy-competent (Cre−) littermates. (Bottom panels) Acid-fast stain. Arrows, M. tuberculosis H37Rv bacilli. (A, Reprinted, with permission, from Gutierrez et al. 2004; B,C, reprinted, with permission, from Castillo et al. 2012.)
SELECTIVE AUTOPHAGY
Selective autophagy is driven by the recognition of cargos for selective autophagy via autophagic receptors (Fig. 3). The spectrum of selective autophagy targets includes defunct organelles such as depolarized mitochondria, protein aggregates, and intracellular microbes in contact with the cytosol. A subset of autophagic receptors involved in antimicrobial defense is termed SLRs, for Sequestosome 1/p62-like receptors (Fig. 3). The cargo, such as a cytosolic bacterium or a depolarized mitochondrion (bacteria and mitochondria share recognition machinery [Deretic 2010]), is often tagged by ubiquitin (Bjorkoy et al. 2005; Kirkin et al. 2009), galectins (Thurston et al. 2012; Li et al. 2013), and possibly other modifications (Deretic et al. 2013). Ubiquitin is placed on or around bacteria via specific E3 ligases such as LRSAM1, which possesses a leucine-rich repeat characteristic of pattern recognition receptors and a RING domain conferring its E3 ubiquitin ligase activity (Huett et al. 2012), or another E3 ligase, Parkin (Manzanillo et al. 2013). Parkin is well known for ubiquitination of depolarized or damaged mitochondria en route for autophagic elimination via mitophagy (Youle and Narendra 2011). Ubiquitin and other earmarks are recognized by autophagic receptors that bind the tags at one end and interact at the other end with mammalian Atg8s via an LC3-interacting region (LIR) (Birgisdottir et al. 2013). Whether and how cargo capture triggers signaling and execution of membrane growth during selective autophagy are not well understood, but many of the factors participating in generalized/nonselective autophagy are engaged.
Figure 3.

Sequestosome 1/p62–like receptors (SLRs), a new class of pattern-recognition receptors involved in autophagic elimination of intracellular microbes. LIR, LC3-interacting region (consensus sequence shown and key positions in red); aromatic pocket-filling W (or F/Y) and aliphatic pocket-filling L (or I/V) form an intermolecular parallel β sheet with LC3s or GABARAPs, at the interface between the amino-terminal α-helical domain and ubiquitin-like fold of LC3s/GABARAPs. CLIR, a LIR specific for LC3C, whereby aromatic residue is not present (X) to fill in the aromatic pocket and instead the interactions are stabilized by compensatory hydrophobic contacts provided by additional aliphatic residues located between the W and L position anchors. KIR, KAEP1-interacting region; NES, nuclear export signal; NLS, nuclear localization signal; PB1, protein-binding domain (homopolymerization of p62 hetero-oligomerization between p62 and NBR1, or interactions with other partners); TR, TRAF6-interacting region (also a multipartner binding region); ZZ, ZZ-type zinc finger (ZnF) domain; FW, four-Trp domain, also known as the NBR1 box; UBAN, a parallel coiled-coiled dimer UBD with specificity for linear ubiquitin chains; CC, coiled coil; UBA, a three-helix bundle UBD (ubiquitin-binding domain) with affinity for monoubiquitin and the more open conformation of K63 ubiquitin linkages; UBZ, a Zn finger ββα fold UBD binding mono- and polyubiquitin; SKICH, skeletal muscle and kidney enriched inositol phosphatase carboxyl homology domain; GIR, galectin-interacting region. Sequestosome 1/p62 has been shown to affect M. tuberculosis clearance (Ponpuak et al. 2010). NDP52 has been implicated in elimination of M. tuberculosis in murine macrophages (Watson et al. 2012); note, however, that NDP52 is severely truncated in this species. (Image modified from Deretic et al. 2013.)
LC3-ASSOCIATED PHAGOCYTOSIS
An example of a known exception to using all parts of the autophagy pathway is a process called LC3-associated autophagy (LAP), which is a merger between conventional phagocytosis and autophagy (Cemma and Brumell, 2012). LAP appears to be a modification of the conventional phagocytic pathway distinguished by the recruitment of LC3, which enhances phagosomal fusogenicity with lysosomes (Fig. 4). LAP does not require ULK1 but needs Beclin 1-hVPS34 and LC3-conjugation systems (Henault et al. 2012). LAP occurs following uptake of various extracellular targets: bacteria or particles coated with Toll-like receptor (TLR) agonists (Sanjuan et al. 2007; Xu et al. 2007; Lee et al. 2010), phagocytosed dead cells (Martinez et al. 2011), live epithelial cells engulfed via entotisis by neighboring cells (Florey et al. 2011), rod outer segments phagocytosed by retinal pigment epithelial cells (Kim et al. 2013), and FcγR-dependent uptake of immune complexes (Henault et al. 2012). M. tuberculosis and M. bovis BCG bacilli that reside inside the phagosome can be subjected to LAP on induction of autophagy by physiological, immunological, or pharmacological means (Gutierrez et al. 2004; Harris et al. 2007; Xu et al. 2007; Jagannath et al. 2009; Pilli et al. 2012). LAP enhances conventional phagosomes by conferring improved lytic properties, enhanced bactericidal functions (Gutierrez et al. 2004; Harris et al. 2007; Sanjuan et al. 2007; Cemma and Brumell 2012; Pilli et al. 2012), better antigen presentation (Jagannath et al. 2009; Lee et al. 2010), and enhanced intracellular trafficking of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) complexed with pattern-recognition receptors (PRRs) (Chaturvedi et al. 2008; Lee et al. 2010; Henault et al. 2012). The recapitulation of phagosomal/autophagosomal maturation steps in LAP is linked to PI3P generation, long known to be key for phagosomal maturation (Vergne et al. 2004), whereas the role of LC3 may be a manifestation of the tethering and fusogenic properties of LC3 (Nakatogawa et al. 2007; Weidberg et al. 2011).
Figure 4.
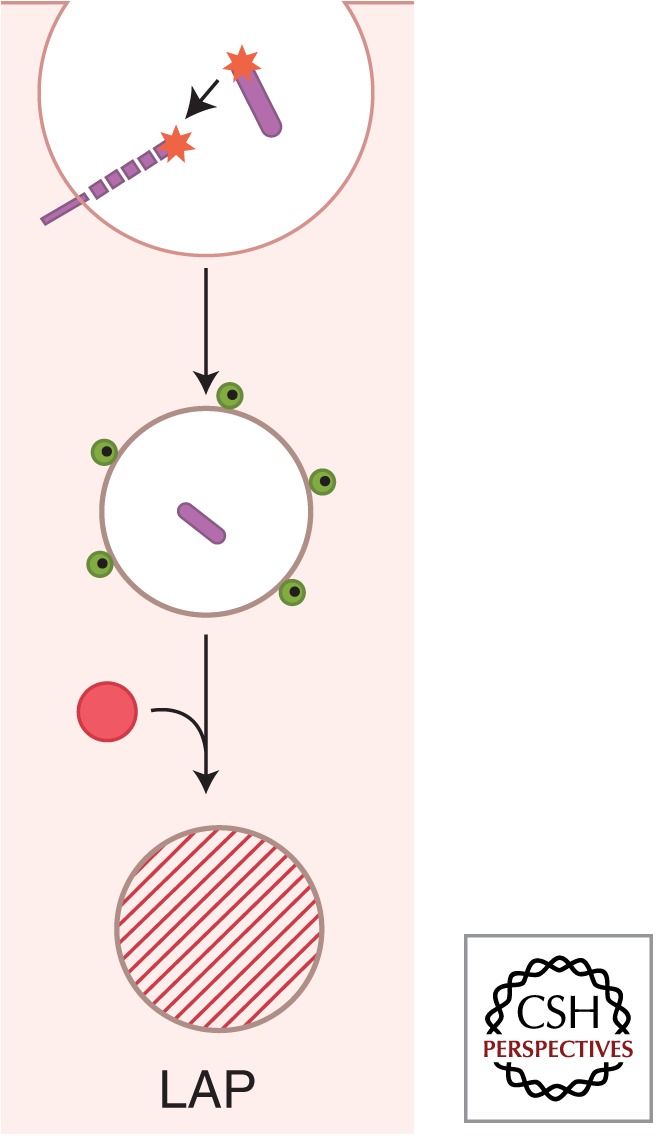
LC3-associated phagocytosis (LAP) as an intersection between autophagy and phagocytosis. Autophagy is frequently morphologically described as formation of double-membrane autophagosomes in the cytoplasm. This is requisite when an internal phagosome is derived from intracellular membranes such as the endoplasmic reticulum (ER). One important exception in the context of the role of autophagy during infection is the formation of conventional phagosomes that are also decorated with LC3 (green dots). Note that LC3 is only on the cytofacial side of the phagosomal membrane, and that this is a single membrane as in the case of conventional phagosomes. Depicted also is a Toll-like receptor molecule recognizing pathogen-associated molecular patterns (asterisks), involved in the induction of LAP and capable of concomitantly inducing conventional autophagy in the cell. The presence of LC3 on these phagosomes promotes maturation of the standard phagosome into autolysosomes. LAP depends on Beclin 1-hVPS34, LC3-conjugation systems, and other parts of autophagy pathway but is independent of ULK1, which is needed to generate double-membrane autophagosomes during starvation from internal ER membranes. The role of LC3 may be a manifestation of the tethering and fusogenic properties of LC3; furthermore, concomitantly generated autolysosomes are enriched in their bactericidal properties, whereas other immunologically active compartments such as those involved in antigen presentation and TLR signaling (e.g., TLR9) can enhance or exacerbate a variety of immune responses. (Image modified from Deretic et al. 2013.)
IMMUNE SIGNALING AND AUTOPHAGY AS AN ANTI-M. tuberculosis EFFECTOR
In addition to being controlled globally by starvation, autophagy responds to innate immunity signals and cytokine stimulation during immune responses (Deretic et al. 2013). Autophagy can be induced via PRRs. TLR ligands and TLRs activate autophagy (Xu et al. 2007; Delgado et al. 2008) via TRAF6 (Shi and Kehrl 2010), which activates or stabilizes Beclin 1 (Shi and Kehrl 2010) and ULK1 (Nazio et al. 2013). NOD2 can activate autophagy via RIPK2 and ULK1 (Lupfer et al. 2013). Second messenger 2′-5′ cyclic GMP-AMP (cGAMP) generated by mammalian cGAMP synthase in response to the presence of cyctosolic DNA (viral, mitochondrial, or bacterial, including mycobacterial) or secreted bacterial 3′-5′ cyclic-di-GMP or cyclic-di-AMP can stimulate autophagy and ULK1 (Watson et al. 2012; Konno et al. 2013). IL-1β stimulates autophagy (Pilli et al. 2012), which is of high relevance for M. tuberculosis control. MyD88 signaling is key to early protection against M. tuberculosis, although TLRs, which act via MyD88, appear not to be essential for protection (Fremond et al. 2004, 2007; Scanga et al. 2004; Holscher et al. 2008). In contrast, IL-1 receptor signaling, which also relies on MyD88 as an adapter, is key to early control of M. tuberculosis (Mayer-Barber et al. 2010, 2011; Guler et al. 2011). IL-1β eliminates M. tuberculosis through autophagy (Pilli et al. 2012). Th1 cytokines activate autophagy to kill intracellular M. tuberculosis (Harris et al. 2007). Stimulation of autophagy mediated through P2X purinergic receptors can eliminate intracellular M. tuberculosis (Biswas et al. 2008). Activation of anti-M. tuberculosis autophagy has also been reported via direct cell–cell contacts between specific T cells and M. tuberculosis–infected macrophages (Petruccioli et al. 2012).
MECHANISMS OF INTRACELLULAR KILLING OF M. tuberculosis BY AUTOPHAGY
Autophagy eliminates intracellular M. tuberculosis through lytic and antimicrobial properties unique to autolysosomes, which are much more robust antimicrobial compartments than conventional phagosomes (Ponpuak et al. 2010). A step-by-step analysis of autophagy factors, starting with an SLR, Sequestosome 1/p62, has established that the entire pathway is important for anti–M. tuberculosis action of autophagy (Ponpuak et al. 2010). Another SLR, NDP52, has been reported as being necessary for elimination of M. tuberculosis in mouse macrophages (Watson et al. 2012), but this cannot be the case, at least in murine cells, because Mus musculus lacks a functional NDP52 gene (Fig. 3) (Deretic et al. 2013). Induction of autophagy promotes maturation and acidification of M. tuberculosis phagosomes and their conversion into mycobactericidal organelles (Gutierrez et al. 2004; Harris et al. 2007; Fabri et al. 2011b) via processes akin to LAP (Cemma and Brumell 2012). Thus, induction of autophagy overcomes the classical M. tuberculosis virulence determinant known as inhibition of phagosome-lysosome (Hart et al. 1987; Vergne et al. 2004). Among the constituents delivered via these enhanced trafficking pathways are conventional anti–M. tuberculosis antimicrobial peptides including cathelicidin potentially through autophagy-stimulated fusion with lysosomes where cathelicidin is stored (Yuk et al. 2009; Fabri et al. 2011b). Moreover, autophagy captures cytosolic proteins and partially digests them. These autophagosomal contents, which have enhanced antimycobacterial properties (Ponpuak et al. 2010), are delivered to the M. tuberculosis phagosome in cells induced for autophagy. A specific manifestation of this is that induction of autophagy generates and delivers to M. tuberculosis a mixture of neo-antimicrobial peptides (known as cryptides [Ponpuak and Deretic 2011]) produced through autophagic proteolysis of otherwise innocuous cytosolic proteins such as ribosomal proteins (Ponpuak et al. 2010) and ubiquitin (Alonso et al. 2007). A certain fraction of intracellular M. tuberculosis bacilli escape from phagosomes into the cytoplasm or are in contact with the cytosol (van der Wel et al. 2007). The bacilli that are in contact with the cytosol represent a minor proportion of the total intracellular M. tuberculosis, but are nevertheless subject to a cleanup through selective autophagy (Watson et al. 2012).
An unexpected link between action of the frontline antituberculosis chemotherapeutics and autophagy has been reported (Kim et al. 2012a). The key antituberculosis drugs, isoniazid and pyrazinamide, express their full potency in cooperation with a functional autophagy (Kim et al. 2012a). During treatment of M. tuberculosis with isoniazid or pyrazinamide, the release of bacterial products induces autophagy in infected macrophages in association with a generation of reactive oxygen species (ROS) from mitochondrial and NADPH oxidase sources (Kim et al. 2012a). This is in keeping with induction of autophagy by ROS released by mitochondria and NADPH oxidase (Scherz-Shouval et al. 2007; Huang et al. 2009). The enhanced effectiveness of these antibiotics in synergy with autophagy may explain the sterilizing properties of pyrazinamide. Furthermore, several compounds that can induce autophagy have been shown to inhibit M. tuberculosis (Floto et al. 2007; Lam et al. 2012; Sundaramurthy et al. 2013), raising the possibility of host-based therapies based on autophagy-inducing drugs.
M. tuberculosis POSSESSES COUNTERMEASURES AGAINST AUTOPHAGY
Mycobacterial infections show evidence of mTOR activation (mTOR inhibits autophagy) or autophagy induction, correlating with the species or strain virulence (Zullo and Lee 2012). Furthermore, metabolic reprogramming of host macrophages by M. tuberculosis (Russell et al. 2009; Singh et al. 2012) leads to an increase in lipid droplets, the cytoplasmic stores of neutral lipids such as triglycerides and cholesterol esters. Lipid droplets are dynamic organelles (Fujimoto and Parton 2011) that are known to intersect with the autophagic pathway (Singh et al. 2009; Velikkakath et al. 2012). The host cell reprogramming by M. tuberculosis reduces autophagic capacity of the cell and protects intracellular M. tuberculosis from autophagic elimination (Singh et al. 2012).
A number of candidate antiautophagy factors encoded by M. tuberculosis have been reported. An M. tuberculosis protein termed Eis (enhanced intracellular survival) (Shin et al. 2010a; Ganaie et al. 2011; Kim et al. 2012b) may interact with specific autophagy factors or affect upstream signaling regulators. Eis is an ε-aminoacetyltransferase that acetylates Lys-55 in a JNK-specific phosphatase (Kim et al. 2012b). JNK action is important for activation of the key autophagy regulator Beclin 1, and thus Eis may modulate autophagy (Shin et al. 2010a; Ganaie et al. 2011; Kim et al. 2012b). A mycobacterial glycolipid lipoarabinomannan has also been reported as inhibiting autophagy (Shui et al. 2011). ESX-1, a type VII secretion system of M. tuberculosis, releases a 6-kDa protein ESAT-6 to block M. tuberculosis phagosomal maturation into degradative autolysosomal organelles (Romagnoli et al. 2012; Zhang et al. 2012). Pharmacological agonists of autophagy can overcome the ESAT-6-based block (Romagnoli et al. 2012), in keeping with the observations that physiological or immunological stimulation of autophagy (e.g., by starvation or IFN-γ) (Gutierrez et al. 2004; Harris et al. 2007) can kill M. tuberculosis overpowering antiautophagy mechanisms of M. tuberculosis.
IFN-γ, TH1 VERSUS TH2 CYTOKINES, AND VITAMIN D IN ANTI-M. tuberculosis AUTOPHAGY
Response of autophagy, as an antimicrobial effector, to Th1 and Th2 cytokines correlates with the general association of Th1 versus Th2 cytokines with their restriction versus permissiveness to intracellular pathogens. IFN-γ, a key Th1 cytokine, can turn on autophagy (Gutierrez et al. 2004; Harris et al. 2007). Th2 cytokines IL-4 and IL-13 inhibit IFN-γ-dependent autophagy in a dominant fashion; that is, they can trump IFN-γ effects during simultaneous exposure (Harris et al. 2007; Ghadimi et al. 2010). Th2 cytokines can also override other physiological inducers of autophagy, although the inhibitory signaling pathways differ in terms of interference with IFN-γ or, for example, starvation as inducers of autophagy (Harris et al. 2007). IL-4 and IL-13 inhibit IFN-γ-induced autophagy via a pathway dependent on STAT6, whereas they inhibit starvation-induced autophagy via Akt signaling. IL-10 can also be inhibitory to autophagy (Van Grol et al. 2010; Park et al. 2011).
There are important hormonal agonists of IFN-γ activation of autophagy, including sterol-derived vitamin D metabolite calcitriol, also known by various names as 1,25-dihydroxyvitamin D3, 1,25-dihydroxycholecalciferol, or 1,25(OH)D3. Calcitriol circulates at very low concentrations in the serum and is generated in the kidney. However, calcitriol can additionally be generated in human macrophages from its precursor calcidiol (25-hydroxy vitamin D3; present at much higher concentrations in the serum) by a specific 1-α hydroxylase induced in macrophages in response to TLR activation (Liu et al. 2006), stimulation with IFN-γ (Reichel et al. 1987), or exposure to other stimuli (Evans et al. 2006). Low levels of calcitriol and its precursor calcidiol in the serum have been associated with susceptibility to tuberculosis (Nnoaham and Clarke 2008). Of note, lower calcidiol and calcitriol levels also factor in HIV–M. tuberculosis coinfections (Campbell and Spector 2012). Recent studies have indicated a strong synergistic role between calcitriol with IFN-γ during induction of autophagy in human macrophages as a contributing mechanism to M. tuberculosis killing (Yuk et al. 2009; Shin et al. 2010b; Fabri et al. 2011a,b; Campbell and Spector 2012; Anandaiah et al. 2013; Klug-Micu et al. 2013). Calcitriol induces autophagy via Ca2+ and Ca2+/calmodulin-dependent kinase kinase-β (CaMKKβ) (Hoyer-Hansen et al. 2007), which in turn activates AMPK (AMP-activated protein kinase) (Mihaylova and Shaw 2011). Studies outside of the tuberculosis field have shown that CaMKKβ, which responds to calcitriol via Ca2+, promotes induction of autophagy in a manner sensitive to membrane-permeant Ca2+ chelators (Hoyer-Hansen et al. 2007) by activating a phosphorylation cascade involving ULK1 and then Beclin 1, as discussed above in the section Autophagy Pathway (Egan et al. 2011; Kim et al. 2011; Russell et al. 2013). The phosphorylation of AMPK by CaMKKβ occurs on the same critical Thr 172 residue as with other key kinases that control AMPK activation (Mihaylova and Shaw 2011). The aforementioned cathelicidin has also been reported to play a role in induction of autophagy by calcitriol (Yuk et al. 2009). However, the proposed activation of autophagy by cathelicidin has no defined mechanism in contrast to its documented effector functions of an antimicrobial peptide with enhanced delivery to intracellular M. tuberculosis through autophagy (Fabri et al. 2011b). Whereas in human macrophages calcitriol is required for IFN-γ-induced autophagy (Fabri et al. 2011b), it is not necessary in human macrophages when autophagy is induced by nutritional (starvation) or pharmacological (rapamycin) means (Fabri et al. 2011b). Calcidiol and calcitriol levels appear not to be a determinant of autophagy induction by IFN-γ in murine macrophages (Gutierrez et al. 2004; Singh et al. 2006; Harris et al. 2007). This is in keeping with the notion that expression of antimicrobial peptides (e.g., cathelcidin) in murine cells is not controlled by vitamin D response elements (Fabri et al. 2011b).
GENETIC PREDISPOSITIONS TO TUBERCULOSIS AND AUTOPHAGY
In keeping with the above role of calcitriol in autophagy, human genetic polymorphisms in the gene encoding vitamin D receptor are associated with susceptibility to tuberculosis when combined with low serum levels of calcidiol, a precursor to calcitriol (Wilkinson et al. 2000). There are further genetic links between autophagy and risks for active tuberculosis. A recent genome-wide association study (GWAS) shows a widespread overlap between genetic risks for inflammatory bowel disease (IBD) and tuberculosis (Jostins et al. 2012). As a subset of these overlaps, genetic polymorphisms in autophagy genes affect both IBD and tuberculosis. For example, polymorphisms in ATG16L1 and IRGM, have been initially identified through GWASs as autophagy risk loci for Crohn’s disease (Wellcome Trust Case Control Consortium 2007), a common form of IBD. Subsequent human population studies have shown that IRGM, encoding an autophagy-modulating factor (Singh et al. 2006, 2010), is also a risk factor for tuberculosis (Intemann et al. 2009; Che et al. 2010; King et al. 2011; Bahari et al. 2012).
AUTOPHAGY PROTECTS AGAINST M. tuberculosis INFECTION IN VIVO
Autophagy protects in vivo against bacillary burden, inflammation, lung pathology (Fig. 2B,C), and death in transgenic mouse models of M. tuberculosis infection (Castillo et al. 2012; Watson et al. 2012). This has been shown with virulent M. tuberculosis strains, H37Rv (Castillo et al. 2012) and Erdman (Watson et al. 2012), using Atg5fl/fl LysM-Cre transgenic mice with autophagy defective in myeloid cells. Atg7fl/fl LysM-Cre mice with a similar autophagy defect in myeloid cells show increased pathology when challenged with M. bovis BCG, whereas higher numbers of bacilli are taken up by Atg7fl/fl LysM-Cre+ macrophages ex vivo ascribed in part to changes in phagocytic receptors (Bonilla et al. 2013). Although the details of respiratory infection and strains used varied, all studies observed higher bacillary load, exacerbated lung involvement, and increased lung pathology relative to autophagy-competent mice (Castillo et al. 2012; Watson et al. 2012; Bonilla et al. 2013). The two studies with virulent M. tuberculosis strains showed increased mortality of Atg5fl/fl LysM-Cre+ mice (Castillo et al. 2012; Watson et al. 2012).
AUTOPHAGY PROTECTS AGAINST EXCESSIVE INFLAMMATION AND TUBERCULOSIS PATHOGENESIS
Most prominently, IL-1 has been found elevated in the infected Atg5fl/fl LysM-Cre+ mice relative to their Atg5fl/fl LysM-Cre− littermates (Castillo et al. 2012; Watson et al. 2012). In contrast, IFN-γ or IL-4 responses are equal in autophagy-proficient and autophagy-deficient mice (Castillo et al. 2012). Of IL-1s, IL-1α has turned out to be the dominant cytokine in the infected Atg5fl/fl LysM-Cre+ lung. IL-1β, albeit increased in autophagy-deficient mice, is present at low absolute levels in the lung challenged with disparate pathogens (Castillo et al. 2012; Lupfer et al. 2013). Although inflammasome is hyperactivated in autophagy-defective mice (Saitoh et al. 2008; Nakahira et al. 2011; Zhou et al. 2011), the lung environment may suppress excess IL-1β (Castillo et al. 2012; Lupfer et al. 2013). Nevertheless, the lung immune cells from mice with autophagic defect in myeloid cells show propensity towards extended Th17 polarization (Castillo et al. 2012). This is attributable to a cell-autonomous defect in autophagy-deficient macrophages, which secrete excess IL-1α (Castillo et al. 2012). IL-1α, just like IL-1β, promotes Th17 polarization leading to extended IL-17 response during M. tuberculosis infection of Atg5fl/fl LysM-Cre+ mice (Castillo et al. 2012). This scenario makes it impossible for IFN-γ, one of the functions of which is a suppression of excessive and pathology-inducing IL-1β responses by inhibiting inflammasome activation (Mishra et al. 2013), to overcome the alternative IL-1 responses via IL-1α. Unlike IL-1β, which depends on inflammasome activation, IL-1α can be activated independently of inflammasome via calpain (Castillo et al. 2012; Gross et al. 2012).
The above anti-inflammatory action of autophagy may not stop at direct control of intracellular M. tuberculosis and tempering of the IL-1 response (see Fig. 5 for an overall model). We propose a model that extends to potential effects on type I IFN responses. Type I interferon (IFN) is a key biosignature of active tuberculosis (Berry et al. 2010; Maertzdorf et al. 2012; Ottenhoff et al. 2012; Teles et al. 2013). M. tuberculosis–mediated induction of type I IFN requires the ESX-1 system (Stanley et al. 2007; Pandey et al. 2009; Novikov et al. 2011; Manzanillo et al. 2012), and this signaling has been shown to be induced by M. tuberculosis extracellular DNA through IRF3 (Manzanillo et al. 2012). Mice lacking IRF3 (or lacking IFNAR) survive M. tuberculosis infection longer than wild-type mice (Manca et al. 2005; Manzanillo et al. 2012). Because autophagy clears M. tuberculosis along with its extracellular DNA (Watson et al. 2012), it is possible that removal of this PAMP (bacterial extracellular DNA) prevents activation of now well-delineated pathways of induction of type I IFN via dicyclic nucleotide second messengers and STING (Ablasser et al. 2013; Civril et al. 2013; Wu et al. 2013). Specific molecular intersections between autophagy (e.g., ULK1) and these systems (e.g., STING) have been recently uncovered (Konno et al. 2013). It is possible that autophagic removal of other type I IFN-inducing products, such as the M. tuberculosis peptidoglycan N-glycolyl MDP (Pandey et al. 2009), may also act to prevent excess type I IFN production.
Figure 5.

Proposed model of how autophagy interferes with progression to active disease during M. tuberculosis infection. (A) Proposed processes contributing to progression into active disease pertinent to the role of autophagy: uncontrolled M. tuberculosis growth; endogenous sources of excessive inflammation (e.g., damaged organelles such as depolarized mitochondria, which are the source of reactive oxygen species [ROS] and mitochondrial DNA released into the cytosol) acting as damage-associated molecular patterns (DAMPs) that amplify inflammatory responses to the point of causing excessive tissue damage; M. tuberculosis pathogen-associated molecular patterns (PAMPs; e.g., mycobacterial N-glycolyl muramyl dipeptide or bacterial DNA released from or associated with the bacilli) inducing type I IFN, which is a biomarker of active disease and suppresses measured host responses that inhibit M. tuberculosis proliferation and thus curtails their protective effectiveness. (B) Autophagy (represented by crescents) eliminates the above promoters of active disease and thus acts as an antibacterial and tissue-sparing process.
CONCLUSIONS AND OUTLOOK
Autophagy is a cell-autonomous innate immunity defense that enables host cells to capture intracellular pathogens and kill them in lysosomal organelles that during autophagy acquire enhanced bactericidal properties. Equally or perhaps more importantly, autophagy can suppress excessive inflammatory responses that contribute to progression to active disease. This occurs via autophagic elimination of endogenous cellular agonists of inflammasome or calpain activation, thus preventing excess IL-1 response, and may also remove mycobacterial products eliciting other pathogenesis-associated responses such as type I IFN linked to active disease. When balanced, both IL-1 and type I IFN may have their role in protection but without autophagy to remove the contributing agonists, these important cytokine responses may be out of alignment and cause pathology and active disease. Consequently, pharmacological activation of autophagy may offer therapeutic host-targeted options for better control of M. tuberculosis infection.
ACKNOWLEDGMENTS
Work in V. Deretic's laboratory is supported by grants AI042999 and AI111935 from the National Institutes of Health and a Bill and Melinda Gates Grand Challenge Explorations grant.
Footnotes
Editors: Stefan H.E. Kaufmann, Eric J. Rubin, and Alimuddin Zumla
Additional Perspectives on Tuberculosis available at www.perspectivesinmedicine.org
REFERENCES
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498: 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso S, Pethe K, Russell DG, Purdy GE. 2007. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci 104: 6031–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandaiah A, Sinha S, Bole M, Sharma SK, Kumar N, Luthra K, Li X, Zhou X, Nelson B, Han X, et al. 2013. Vitamin D rescues impaired Mycobacterium tuberculosis–mediated tumor necrosis factor release in macrophages of HIV-seropositive individuals through an enhanced Toll-like receptor signaling pathway in vitro. Infect Immun 81: 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182: 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahari G, Hashemi M, Taheri M, Naderi M, Eskandari-Nasab E, Atabaki M. 2012. Association of IRGM polymorphisms and susceptibility to pulmonary tuberculosis in Zahedan, Southeast Iran. Scientific World Journal 2012: 950801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, et al. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466: 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgisdottir AB, Lamark T, Johansen T. 2013. The LIR motif—Crucial for selective autophagy. J Cell Sci 126: 3237–3247 [DOI] [PubMed] [Google Scholar]
- Biswas D, Qureshi OS, Lee WY, Croudace JE, Mura M, Lammas DA. 2008. ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol 9: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla DL, Bhattacharya A, Sha Y, Xu Y, Xiang Q, Kan A, Jagannath C, Komatsu M, Eissa NT. 2013. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 39: 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Spector SA. 2012. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog 8: e1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, et al. 2012. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci 109: E3168–E3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cemma M, Brumell JH. 2012. Interactions of pathogenic bacteria with autophagy systems. Curr Biol 22: R540–R545 [DOI] [PubMed] [Google Scholar]
- Chaturvedi A, Dorward D, Pierce SK. 2008. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 28: 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che N, Li S, Gao T, Zhang Z, Han Y, Zhang X, Sun Y, Liu Y, Sun Z, Zhang J, et al. 2010. Identification of a novel IRGM promoter single nucleotide polymorphism associated with tuberculosis. Clin Chim Acta 411: 1645–1649 [DOI] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP. 2013. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498: 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. 2008. Toll-like receptors control autophagy. EMBO J 27: 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. 2005. Autophagy in innate and adaptive immunity. Trends Immunol 26: 523–528 [DOI] [PubMed] [Google Scholar]
- Deretic V. 2010. Autophagy of intracellular microbes and mitochondria: Two sides of the same coin? F1000 Biol Rep 2: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V, Saitoh T, Akira S. 2013. Autophagy in infection, inflammation, and immunity. Nat Rev Immunol 13: 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deter RL, De Duve C. 1967. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 33: 437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deter RL, Baudhuin P, De Duve C. 1967. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol 35: C11–C16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CE, Mylchreest S, Mee AP, Berry JL, Andrew JG. 2006. Cyclic hydrostatic pressure and particles increase synthesis of 1,25-dihydroxyvitamin D3 by human macrophages in vitro. Int J Biochem Cell Biol 38: 1540–1546 [DOI] [PubMed] [Google Scholar]
- Fabri M, Realegeno SE, Jo EK, Modlin RL. 2011a. Role of autophagy in the host response to microbial infection and potential for therapy. Curr Opin Immunol 23: 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, et al. 2011b. Vitamin D is required for IFN-γ-mediated antimicrobial activity of human macrophages. Sci Transl Med 3: 104ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. 2011. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 13: 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floto RA, Sarkar S, Perlstein EO, Kampmann B, Schreiber SL, Rubinsztein DC. 2007. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy 3: 620–622 [DOI] [PubMed] [Google Scholar]
- Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. 2004. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest 114: 1790–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, Jacobs M, Ryffel B, Quesniaux VF. 2007. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol 179: 1178–1189 [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Parton RG. 2011. Not just fat: The structure and function of the lipid droplet. Cold Spring Harb Perspect Biol 3: a004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganaie AA, Lella RK, Solanki R, Sharma C. 2011. Thermostable hexameric form of Eis (Rv2416c) protein of M. tuberculosis plays an important role for enhanced intracellular survival within macrophages. PLoS ONE 6: e27590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadimi D, de Vrese M, Heller KJ, Schrezenmeir J. 2010. Lactic acid bacteria enhance autophagic ability of mononuclear phagocytes by increasing Th1 autophagy-promoting cytokine (IFN-γ) and nitric oxide (NO) levels and reducing Th2 autophagy-restraining cytokines (IL-4 and IL-13) in response to Mycobacterium tuberculosis antigen. Int Immunopharmacol 10: 694–706 [DOI] [PubMed] [Google Scholar]
- Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. 2012. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36: 388–400 [DOI] [PubMed] [Google Scholar]
- Guler R, Parihar SP, Spohn G, Johansen P, Brombacher F, Bachmann MF. 2011. Blocking IL-1α but not IL-1β increases susceptibility to chronic Mycobacterium tuberculosis infection in mice. Vaccine 29: 1339–1346 [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766 [DOI] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. 2013. Autophagosomes form at ER-mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. 2007. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27: 505–517 [DOI] [PubMed] [Google Scholar]
- Hart PD, Young MR, Gordon AH, Sullivan KH. 1987. Inhibition of phagosome-lysosome fusion in macrophages by certain mycobacteria can be explained by inhibition of lysosomal movements observed after phagocytosis. J Exp Med 166: 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, et al. 2012. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 37: 986–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, Lenz I, Sonntag T, Kroger S, Akira S, et al. 2008. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol 38: 680–694 [DOI] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. 2007. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol Cell 25: 193–205 [DOI] [PubMed] [Google Scholar]
- Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH. 2009. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci 106: 6226–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, Goldberg MB, Xavier RJ. 2012. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella typhimurium. Cell Host Microbe 12: 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intemann CD, Thye T, Niemann S, Browne EN, Amanua Chinbuah M, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, Helm S, et al. 2009. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5: e1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, Mizushima N. 2012. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269 [DOI] [PubMed] [Google Scholar]
- Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr, Eissa NT. 2009. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med 15: 267–276 [DOI] [PubMed] [Google Scholar]
- Jiang S, Dupont N, Castillo EF, Deretic V. 2013. Secretory versus degradative autophagy: Unconventional secretion of inflammatory mediators. J Innate Immun 5: 474–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. 2012. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez E, Carranza C, Hernandez-Sanchez F, Leon-Contreras JC, Hernandez-Pando R, Escobedo D, Torres M, Sada E. 2012. NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol 42: 880–889 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, Lee SH, Cha GH, Kim JM, Lee ZW, et al. 2012a. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 11: 457–468 [DOI] [PubMed] [Google Scholar]
- Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, Im HN, Kim J, Kim do J, Lee SJ, et al. 2012b. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci 109: 7729–7734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, et al. 2013. Noncanonical autophagy promotes the visual cycle. Cell 154: 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KY, Lew JD, Ha NP, Lin JS, Ma X, Graviss EA, Goodell MA. 2011. Polymorphic allele of human IRGM1 is associated with susceptibility to tuberculosis in African Americans. PLoS ONE 6: e16317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. 2009. A role for ubiquitin in selective autophagy. Mol Cell 34: 259–269 [DOI] [PubMed] [Google Scholar]
- Klug-Micu GM, Stenger S, Sommer A, Liu PT, Krutzik SR, Modlin RL, Fabri M. 2013. CD40 ligand and interferon-γ induce an antimicrobial response against Mycobacterium tuberculosis in human monocytes. Immunology 139: 121–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN. 2013. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155: 688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Nath L, Kamal MA, Varshney A, Jain A, Singh S, Rao KV. 2010. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 140: 731–743 [DOI] [PubMed] [Google Scholar]
- Lam KK, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, Anderson HJ, Andersen RJ, Av-Gay Y, Roberge M. 2012. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog 8: e1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A. 2010. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32: 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469: 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wandel MP, Li F, Liu Z, He C, Wu J, Shi Y, Randow F. 2013. Sterical hindrance promotes selectivity of the autophagy cargo receptor NDP52 for the danger receptor galectin-8 in antibacterial autophagy. Sci Signal 6: ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, et al. 2006. Toll-like receptor triggering of a vitamin D–mediated human antimicrobial response. Science 311: 1770–1773 [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. 2005. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120: 237–248 [DOI] [PubMed] [Google Scholar]
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, et al. 2013. Receptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14: 480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertzdorf J, Weiner J, 3rd, Mollenkopf HJ, Bauer T, Prasse A, Muller-Quernheim J, Kaufmann SH. 2012. Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci 109: 7853–7858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G. 2005. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res 25: 694–701 [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11: 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. 2013. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501: 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. 2011. Microtubule-associated protein 1 light chain 3 α (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci 108: 17396–17401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, et al. 2010. Caspase-1 independent IL-1β production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184: 3326–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. 2011. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35: 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. 2011. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM. 2013. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol 14: 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y. 2011. The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al. 2004. Autophagy defends cells against invading group A streptococcus. Science 306: 1037–1040 [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. 2011. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y. 2007. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130: 165–178 [DOI] [PubMed] [Google Scholar]
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. 2013. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 15: 406–416 [DOI] [PubMed] [Google Scholar]
- Nnoaham KE, Clarke A. 2008. Low serum vitamin D levels and tuberculosis: A systematic review and meta-analysis. Int J Epidemiol 37: 113–119 [DOI] [PubMed] [Google Scholar]
- Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, Myers TG, Rabin RL, Trinchieri G, Sher A, et al. 2011. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1β production in human macrophages. J Immunol 187: 2540–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenhoff TH, Dass RH, Yang N, Zhang MM, Wong HE, Sahiratmadja E, Khor CC, Alisjahbana B, van Crevel R, Marzuki S, et al. 2012. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS ONE 7: e45839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, Fitzgerald KA, Sassetti CM, Kelliher MA. 2009. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 5: e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, Park CG, Chun T. 2011. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol 48: 720–727 [DOI] [PubMed] [Google Scholar]
- Petruccioli E, Romagnoli A, Corazzari M, Coccia EM, Butera O, Delogu G, Piacentini M, Girardi E, Fimia GM, Goletti D. 2012. Specific T cells restore the autophagic flux inhibited by Mycobacterium tuberculosis in human primary macrophages. J Infect Dis 205: 1425–1435 [DOI] [PubMed] [Google Scholar]
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, et al. 2012. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponpuak M, Deretic V. 2011. Autophagy and p62/sequestosome 1 generate neo-antimicrobial peptides (cryptides) from cytosolic proteins. Autophagy 7: 336–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HWt, Kyei GB, Johansen T, Vergne I, et al. 2010. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32: 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel H, Koeffler HP, Norman AW. 1987. Synthesis in vitro of 1,25-dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 by interferon-γ-stimulated normal human bone marrow and alveolar macrophages. J Biol Chem 262: 10931–10937 [PubMed] [Google Scholar]
- Romagnoli A, Etna MP, Giacomini E, Pardini M, Remoli ME, Corazzari M, Falasca L, Goletti D, Gafa V, Simeone R, et al. 2012. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8: 1357–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. 2009. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol 10: 943–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. 2013. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. 2008. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456: 264–268 [DOI] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. 2007. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257 [DOI] [PubMed] [Google Scholar]
- Scanga CA, Bafica A, Feng CG, Cheever AW, Hieny S, Sher A. 2004. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect Immun 72: 2400–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. 2007. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26: 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A. 2013. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Kehrl JH. 2010. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 3: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, Lee SH, Lee ZW, Cho SN, Kim JM, et al. 2010a. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog 6: e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DM, Yuk JM, Lee HM, Lee SH, Son JW, Harding CV, Kim JM, Modlin RL, Jo EK. 2010b. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol 12: 1648–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui W, Petzold CJ, Redding A, Liu J, Pitcher A, Sheu L, Hsieh TY, Keasling JD, Bertozzi CR. 2011. Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macrophage phagosome maturation and autophagosome accumulation. J Proteome Res 10: 339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SB, Davis AS, Taylor GA, Deretic V. 2006. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313: 1438–1441 [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. 2009. Autophagy regulates lipid metabolism. Nature 458: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, et al. 2010. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 12: 1154–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. 2012. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 12: 669–681 [DOI] [PubMed] [Google Scholar]
- Stanley SA, Johndrow JE, Manzanillo P, Cox JS. 2007. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 178: 3143–3152 [DOI] [PubMed] [Google Scholar]
- Sundaramurthy V, Barsacchi R, Samusik N, Marsico G, Gilleron J, Kalaidzidis I, Meyenhofer F, Bickle M, Kalaidzidis Y, Zerial M. 2013. Integration of chemical and RNAi multiparametric profiles identifies triggers of intracellular mycobacterial killing. Cell Host Microbe 13: 129–142 [DOI] [PubMed] [Google Scholar]
- Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, et al. 2013. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339: 1448–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. 2012. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482: 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129: 1287–1298 [DOI] [PubMed] [Google Scholar]
- Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. 2010. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS ONE 5: e11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N. 2012. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell 23: 896–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergne I, Chua J, Singh SB, Deretic V. 2004. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol 20: 367–394 [DOI] [PubMed] [Google Scholar]
- Watson RO, Manzanillo PS, Cox JS. 2012. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150: 803–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. 2011. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 20: 444–454 [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson RJ, Llewelyn M, Toossi Z, Patel P, Pasvol G, Lalvani A, Wright D, Latif M, Davidson RN. 2000. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: A case-control study. Lancet 355: 618–621 [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. 2007. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27: 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. 2011. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, Lee ZW, Lee SH, Kim JM, Jo EK. 2009. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6: 231–243 [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang H, Zhao Y, Mao F, Wu J, Bai B, Xu Z, Jiang Y, Shi C. 2012. Effects of Mycobacterium tuberculosis ESAT-6/CFP-10 fusion protein on the autophagy function of mouse macrophages. DNA Cell Biol 31: 171–179 [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]
- Zullo AJ, Lee S. 2012. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J Biol Chem 287: 12668–12678 [DOI] [PMC free article] [PubMed] [Google Scholar]