

Abstract
Chloroquine was a cheap, extremely effective drug against Plasmodium falciparum until resistance arose. One approach to reversing resistance is the inhibition of chloroquine efflux from its site of action, the parasite digestive vacuole. Chloroquine accumulation studies have traditionally relied on radiolabelled chloroquine, which poses several challenges. There is a need for development of a safe and biologically relevant substitute. We report here a commercially-available green fluorescent chloroquine-BODIPY conjugate, LynxTag-CQGREEN, as a proxy for chloroquine accumulation. This compound localized to the digestive vacuole of the parasite as observed under confocal microscopy, and inhibited growth of chloroquine-sensitive strain 3D7 more extensively than in the resistant strains 7G8 and K1. Microplate reader measurements indicated suppression of LynxTag-CQGREEN efflux after pretreatment of parasites with known reversal agents. Microsomes carrying either sensitive- or resistant-type PfCRT were assayed for uptake; resistant-type PfCRT exhibited increased accumulation of LynxTag-CQGREEN, which was suppressed by pretreatment with known chemosensitizers. Eight laboratory strains and twelve clinical isolates were sequenced for PfCRT and Pgh1 haplotypes previously reported to contribute to drug resistance, and pfmdr1 copy number and chloroquine IC50s were determined. These data were compared with LynxTag-CQGREEN uptake/fluorescence by multiple linear regression to identify genetic correlates of uptake. Uptake of the compound correlated with the logIC50 of chloroquine and, more weakly, a mutation in Pgh1, F1226Y.
Introduction
Despite years of intense global effort to eradicate it, malaria is still one of the deadliest infectious diseases, killing more than 600 000 people in 2010 alone [1], [2]. The severest form of malaria is caused by the protozoan parasite Plasmodium falciparum. Chloroquine (CQ), once a spectacularly successful antimalarial drug, was first discovered by the German chemist Johann Andersag but was mistakenly thought to be too toxic for therapeutic purposes, an incident which became known as “the resochin error” (resochin being the name given to the compound by Andersag) [3], [4]. CQ was so effective that it inspired optimism for the eradication of malaria. However, resistance soon arose, first appearing along the Thai-Cambodian border in the 1950s. By the 1970s, CQ resistance had spread throughout the world [5], [6]. This resistance is generally attributed to mutations in the pfcrt (P. falciparum chloroquine resistance transporter) gene, which codes for a transporter situated on the membrane of the parasite digestive vacuole (DV).
During parasite development in the intraerythrocytic cycle, haemoglobin is digested in the DV and the toxic heme moiety is released, which the parasite crystalizes into non-toxic hemozoin [7]. CQ is generally thought to kill the parasite by inhibiting the formation of hemozoin and thus preventing the detoxification of free heme [8]–[10]. In wild-type parasites CQ diffuses through the DV membrane and is diprotonated in the acidic environment of the DV, acquiring a net positive charge which prevents it from escaping the DV; however, mutant PfCRT found in CQ-resistant parasites effluxes this charged CQ out of the DV, removing it from its site of action [11]. Although the current first line artemisinin-combination therapies are effective in clearing parasitaemia, resistance against artemisinins has emerged [12]–[17]. There is therefore an urgent need to develop novel antimalarial strategies. Several research groups, including our own, have tried different approaches to tackle the problem of CQ resistance by either reversing CQ resistance with a PfCRT inhibitor or synthesizing “reversed” CQ analogues that cannot be effluxed by PfCRT [18]–[24]. The ultimate goal is to reintroduce CQ as a viable treatment for malaria. Both development of PfCRT inhibitors and synthesis of “reversed” CQ analogues require a sensitive assay for CQ uptake which is typically performed by the use of radiolabelled CQ [22], [25]–[28]. Such methods are difficult to adopt in a high-throughput screen and may raise concerns of safety. To overcome this technical difficulty, fluorescent derivatives of chloroquine have recently been developed and used for this purpose; fluorophores used include 6-(N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)amino)hexanoic acid (NBD) [29], coumarin [21], [30], and 4, 4-difluoro-4-bora-3a, 4a-diaza-s-indacene (BODIPY) [31].
BODIPY derivatives typically exhibit strong fluorescence and are relatively inert in biological conditions [32]. Furthermore, their maximum emission wavelengths are in the green-red region [32], allowing them to be used with many DNA dyes that fluoresce blue, such as the DAPI and Hoechst stains. These properties make BODIPY a promising candidate as a marker for CQ uptake in P. falciparum. We therefore present here the characterization of a commercially-available BODIPY-CQ conjugate, LynxTag-CQGREEN, in several laboratory strains and clinical isolates.
Methods
Parasite culture and synchronization
P. falciparum laboratory strains 3D7 (MRA-102), K1 (MRA-159), 7G8 (MRA-154), HB3 (MRA-155), CS2 (MRA-96), T9-94 (MRA-153), and Dd2 (MRA-156) were obtained from MR4, ATCC Manassas Virginia. Strain T9/96 was obtained from The European Malaria Reagent Repository. A further twelve clinical isolates were collected from the Mae Sot district, Tak Province, in northwest Thailand at the Shoklo Malaria Research Unit; these isolates are prefixed ‘SMRU’. Parasites were continuously cultured in complete malaria culture media (MCM) consisting of RPMI 1640 (Life Technologies) supplemented with 0.5% (w/v) Albumax I (Invitrogen), 0.005% (w/v) hypoxanthine, 0.03% (w/v) L-glutamate, 0.25% (w/v) gentamycin, with human erythrocytes at 2.5% haematocrit. Cultures were gassed with a mixture of 3% CO2, 4% O2 and 93% N2 and incubated at 37°C. Synchronization of parasite cultures was performed by resuspending erythrocytes in 5% (w/v) D-sorbitol and incubating at 37°C for 10 min, after which the erythrocytes were washed twice, resuspended in MCM and returned to culture conditions. Thin Giemsa smears were made before each experiment to determine parasitemia and parasite stage.
Compound preparation
For work involving parasites, chlorpheniramine maleate salt, chlorpromazine hydrochloride, desipramine hydrochloride, promethazine hydrochloride, verapamil hydrochloride and CQ diphosphate (all from Sigma-Aldrich) were dissolved in PBS to a working concentration of 1 mM. LynxTag-CQGREEN (BioLynx Technologies, Singapore; hereafter abbreviated to ‘CQGREEN’) was dissolved in DMSO to the same concentration. All compounds were stored at −20°C and protected from light. For microsome uptake assays, methiothepin mesylate salt, metergoline, loperamide hydrochloride, octoclothepin maleate salt, mibefradil dihydrochloride hydrate, L703,606 oxalate salt hydrate, and chlorprothixene hydrochloride (all from Sigma-Aldrich) were dissolved in DMSO to 10 mM and stored at 4°C. Verapamil hydrochloride, adenosine triphosphate (ATP), and CQ diphosphate (all from Sigma-Aldrich) were dissolved in water to 7.5 mM, 50 mM and 0.1 M respectively and stored at −20°C. Tritiated CQ (3H-CQ; from Moravek Biochemicals and Radiochemicals) was diluted in water to 5.32 µM and stored at −20°C; specific activity was 4.7 Ci/mmol.
Reinvasion half-maximal inhibitory concentration (IC50)
Synchronized ring-stage cultures at 1–2% parasitemia, 1.25% haematocrit were incubated with either CQ or CQGREEN at a range of concentrations for 48 h in 96-well flat-bottomed plates at culture conditions. Following this, cells were stained with 1 µg/ml of Hoechst 33342 (Invitrogen) for 30 min at 37°C, washed twice and resuspended in PBS. Parasitemia was then assessed with the CyAn ADP flow cytometer (Beckman Coulter). IC50s were determined by plotting the measurements in Graphpad Prism 5 using a variable slope logistic curve.
Confocal imaging
200 µl cultures of 3D7 at 3% parasitemia, 1.25% haematocrit were incubated with CQGREEN for 2 h at 2 µM in a 96-well plate format. Erythrocytes were then washed twice and stained with Hoechst 33342 as previously. Wet mounts of stained parasites were visualized under ×100 magnification with the Fluoview FV1000 confocal microscope (Olympus). Hoechst and CQGREEN were excited at 405 nm and 488 nm with emissions captured at 430–470 nm and 505–525 nm respectively.
Parasite CQGREEN uptake assay
Synchronized trophozoite-stage cultures at 3–5% parasitemia were resuspended in 200 µl of MCM with 2 µM of CQGREEN to 2.5% haematocrit in a 96-well plate format. The parasites were then incubated for 2 h at culture conditions, after which they were washed twice and resuspended in PBS. Cells were allowed to settle in a Nunc F96 MicroWell black non-treated polystyrene plate (Thermo Scientific) for 1 h. Fluorescence was then measured with the Infinite M200 microplate reader (Tecan) with excitation and emission wavelengths of 488 nm and 520 nm respectively. K1 chemoreversal assays were performed by pretreatment with 10 µM of the reversal agents for 30 min prior to the addition of CQGREEN.
Preparation of microsomes carrying PfCRT
PfCRT originating from P. falciparum strains Dd2 or 3D7 were expressed in Pichia pastoris KM71 and microsomes harvested as described previously [33]. Microsomal levels of PfCRT were determined by western blot with standard curves generated from blots of purified PfCRT.
Uptake kinetics in microsomes
In order to assess the Michaelis-Menten kinetics of CQGREEN uptake by the microsomes, total or non-specific uptake was measured. Non-specific uptake was determined by pretreating microsomes with unlabelled CQ at 1000 times of the concentration of CQGREEN used, for 15 min at 37°C, before adding CQGREEN; total uptake was determined without the pretreatment. Reactions were carried out in accumulation buffer (0.25 M sucrose, 10 mM Tris–HCl, 5 mM MgCl2, and 3 mM ATP, pH 7.5). Following this, microsomes were washed twice in accumulation buffer, then lysed in lysis buffer (0.75 M HCl, 1% Triton X-100, 77.5% isopropanol) on ice for at least one hour. Unlabelled CQ was excluded from the washing step as the addition of CQ was observed to cause displacement of CQGREEN from the microsomes, possibly due to the higher affinity of CQ for PfCRT. Measurement of the fluorescence intensity was performed using the FLUOstar Galaxy microplate reader (BMG Labtech) with excitation and emission wavelengths of 480 nm and 520 nm respectively. For each experiment, measurements were made in triplicate and the mean calculated. Specific uptake was then calculated by subtracting non-specific uptake from total uptake. Non-linear regression analysis with the Michaelis-Menten model (GraphPad Prism 5) was then applied to obtain the Vmax and Km of specific uptake.
CQGREEN uptake in PfCRT microsomes
Unless stated otherwise, microsomes were incubated with 15 µM CQGREEN at 37°C for 15 min. For uptake inhibition assays, microsomes were pre-incubated with chemoreversal compounds [21] for 15 min at 37°C prior to the addition of CQGREEN. All data presented are specific uptake based on the calculations stated above.
3H-CQ uptake in PfCRT microsomes
3H-CQ uptake was measured as described previously [33], with some modifications. Non-specific uptake was determined by pre-incubation with 200-fold unlabelled CQ. Incubation was performed with 3H-CQ at 308 nM for 5 min, after which 200-fold unlabelled CQ was added to stop the reaction. Microsomes were then precipitated by the addition of polyethylene glycol (PEG) 8000 and washed twice with accumulation buffer containing 200-fold unlabelled CQ to remove excess 3H-CQ. Microsomes were then resuspended in scintillation buffer and agitated overnight. Radioactivity was measured using the LS 5600 Scintillation Counter (Beckman). All data presented are specific uptake.
Genotyping of strains and isolates
To assess pfmdr1 polymorphisms, parasite DNA from in vitro cultures was extracted with the QIAamp DNA Mini kit (Qiagen) as per the manufacturer's instructions. For pfcrt polymorphisms, total RNA was extracted with the RNeasy Mini Kit (Qiagen) and reverse transcription performed with SuperScript III (Invitrogen) as per manufacturers' instructions. Polymerase chain reaction (PCR) mixtures were made with 200 µM of each dNTP, 0.5 µM forward primer, 0.5 µM reverse primer, 0.02 U/µl Phusion DNA polymerase (Thermo Scientific), 6 µl of 5× Phusion HF buffer, and 1 µl of genomic DNA or cDNA to a total reaction volume of 30 µl. Thermocycler parameters were as follows: 98°C for 30 s, followed by 35 cycles of 98°C for 10 s, 60°C for 30 s, and 72°C for 1 min. Primers used for pfmdr1 sequencing were 5′- ATGGGTAAAGAGCAGAAAGA and 5′- TCCACAATAACTTGCAACAGT, or 5′- GTCAAGCGGAGTTTTTGC and 5′- TATTCTCTGTTTTTGTCCAC. Pfcrt-specific primers were 5′- GACGAGCGTTATAGAGAAT and 5′- CTTCGGAATCTTCATTTTCT. PCR products were purified with the QIAquick PCR purification kit as per manufacturer's instructions. Purified PCR products were sequenced by a commercial vendor (AIT Biotech, Singapore). Copy number of pfmdr1 was assessed by real-time PCR as previously reported [34]. Briefly, reaction mixtures were prepared with TaqMan universal PCR master mix (Applied Biosystems), 5.5 mM MgCl2, 300 nM dNTPs, 300 nM each of forward and reverse primers, and 100 nM of the probe. Thermocycler parameters were 95°C for 10 min, then 40 cycles of 95°C for 15 s and 60°C for 1 min. Forward and reverse primers used were 5′- TGCATCTATAAAACGATCAGACAAA and 5′- TCGTGTGTTCCATGTGACTGT respectively, and TaqMan probe was 5′- 6FAM-TTTAATAACCCTGATCGAAATGGAACCTTTG-TAMRA. A reference gene, β-tubulin, was also included; primers were 5′- TGATGTGCGCAAGTGATCC and 5′- TCCTTTGTGGACATTCTTCCTC, while the probe was 5′- VIC-TAGCACATGCCGTTAAATATCTTCCATGTCT-TAMRA. The threshold cycle (Ct) was analysed by the comparative Ct method, based on DNA amplification efficiencies of the pfmdr1 and β-tubulin genes. Pfmdr1 copy number was calculated according to the following formula: ΔCt = CtR - CtG, where CtR is the reference β-tubulin Ct, and CtG is that of pfmdr1. Each TaqMan run included three reference DNA samples from clones 3D7, K1, and Dd2 having pfmdr1 copy numbers of 1, 1, and 3 respectively.
Statistical analyses
All statistical analyses were performed with SPSS 21. Chemoreversal assays were assessed with Student's t test, 2-tailed. Multiple linear regression was performed with the stepwise method, using the log of IC50s and with dummy coded values for the respective amino acid residues.
Ethics statement
The blood collection protocol for in vitro malaria culture was approved by the Institutional Review Board (NUS-IRB Reference Code: 11–383, Approval Number: NUS-1475) of the National University of Singapore (NUS). All participants provided written informed consent. The clinical isolates used were obtained under ethical guidelines in the approved protocol: OXTREC Reference Number 29–09 (Center for Clinical Vaccinology and Tropical Medicine, University of Oxford, Oxford, United Kingdom). Use of field isolates in NUS was in accordance with NUS IRB (Reference Code: 12–369E).
Results and Discussion
Validation of CQGREEN localization and antimalarial activity
CQ is generally believed to accumulate in the DV as a result of ion-trapping [35], [36]. Confocal microscopy was therefore performed to ascertain the localization of CQGREEN in the parasite. Cultures of P. falciparum 3D7 were co-stained with Hoechst dye and CQGREEN, revealing a preferential accumulation of CQGREEN in the parasite DV (Fig. 1). Interestingly, CQGREEN fluorescence was also observed in the parasite cytosol but not in the erythrocyte cytosol. As CQGREEN is a CQ analog, the antimalarial potency of CQGREEN should be similar to that of CQ. To assess this, reinvasion IC50s of CQ and CQGREEN on the laboratory strains 3D7, 7G8 and K1 were determined. CQGREEN showed the same general trend of antimalarial activity as CQ, in that it is most potent against 3D7, followed by 7G8, then K1 (Fig. 2A, 2B).
Figure 1. CQGREEN localization in P. falciparum 3D7.
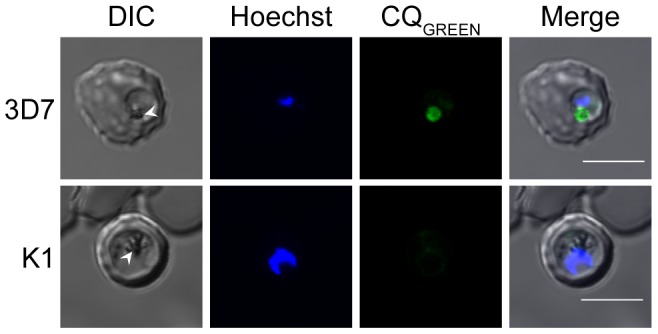
Parasites were stained with CQGREEN and Hoechst and visualized via confocal microscopy under a 100× objective. CQGREEN accumulates in the DV but also slightly stains parasite cytosol; erythrocyte cytosol is not stained. Arrowheads denote the DV. Scale bars represent 5 µm.
Figure 2. Validation of CQGREEN activity and uptake in laboratory strains.

(A, B) CQGREEN IC50s recapitulates CQ IC50s in three P. falciparum laboratory strains, 3D7, 7G8 and K1. 3D7 is a CQ-susceptible strain, 7G8 is moderately resistant, while K1 is highly CQ-resistant. Data shown are geometric means from at least 3 experiments. As the error from IC50s are not symmetrical, error bars indicate 95% confidence intervals instead of the standard error of the mean (S.E.M.). (C) CQGREEN uptake as measured by fluorescence is increased in CQ-resistant K1 when pretreated with chemoreversal agents. 3D7 exhibits highest uptake of CQGREEN. Data are means from at least 3 experiments; error bars are S.E.M. Veh: vehicle control; VPM: verapamil; CPZ: chlorpromazine; CPN: chlorpheniramine; DSP: desipramine; PMZ: promethazine. *: p<0.05.
CQGREEN fluorescence as a proxy for CQ uptake in parasites
Next, we determined if CQGREEN uptake by the highly CQ-resistant strain K1 can be increased by pre-treatment with chemosensitizers. Verapamil, chlorpromazine, chlorpheniramine, desipramine, and promethazine have previously been reported to reverse CQ resistance and increase CQ uptake in CQ-resistant strains [37]–[41]. CQ-sensitive 3D7 was included as a reference for complete reversal. All reversal agents except desipramine induced a significant increase in CQGREEN fluorescence (Fig. 2C). Desipramine is in fact a less potent reversal agent compared to verapamil when tested in the resistant strain Dd2, and two CQ-resistant field isolates [42]; this may explain why desipramine's effect on CQGREEN uptake did not achieve statistical significance. Taken together with the CQGREEN IC50 data, we believe that the reversibility of CQGREEN uptake by known chemoreversal agents suggests that CQGREEN shares similar structural properties with CQ.
Uptake of CQGREEN in microsomes bearing PfCRT
To test if CQGREEN can be transported by PfCRT, we have expressed PfCRT in Pichia pastoris and used the microsomes derived to study CQGREEN uptake. Figure 3 shows that CQGREEN uptake in Dd2 PfCRT-expressing microsomes is specific. At the highest concentration of CQGREEN used (200 µM), uptake was close to saturated. Michaelis-Menten approximation of CQGREEN uptake kinetics in Dd2 microsomes (Figure 3) yields a Vmax and Km of 938.5 nmol/mg PfCRT/min and 105.1 µM respectively, which are approximately 2000 and 500 times higher compared to when 3H-CQ was used [33]. Conjugation of CQ with the BODIPY fluorophore may have altered the affinity of PfCRT for the molecule. However, both the high (micromolar) Km and non-saturation of CQGREEN transport are consistent with a previous report in a Xenopus oocyte system using 3H-CQ [43]. We have also compared CQGREEN uptake in microsomes with PfCRT originating from either CQ-sensitive 3D7 or CQ-resistant Dd2. CQGREEN uptake in Dd2 PfCRT-expressing microsome was 96.07 nmol/mg PfCRT/min, which was about three times that of 3D7 PfCRT (31.64 nmol/mg PfCRT/min) (Figure 4). PfCRT-mediated transport of CQ is thought to be ATP-dependent [25], [44]. Here we demonstrated that removal of ATP completely abolished CQGREEN uptake in both Dd2 and 3D7 PfCRT microsomes (Figure 4). Verapamil, a known CQ resistance chemosensitizer which has no effect on CQ-sensitive strains, can reverse CQGREEN uptake in Dd2 PfCRT to almost that of 3D7 level but has almost no effect on 3D7 PfCRT (Figure 4). These results suggest that CQGREEN is similar to CQ in that (1) it is differentially recognized by CQ-resistant versus CQ-sensitive PfCRT, (2) its uptake by PfCRT is ATP-dependent, and (3) its uptake by CQ-resistant PfCRT is verapamil-reversible. Others have shown that extensive CQ side-chain modifications can render the CQ analogues not transportable by PfCRT and abolish their verapamil sensitivity [45], [46]. Our findings show that the additional BODIPY moiety in CQGREEN still allows CQGREEN to be differentially recognized by resistant versus sensitive PfCRT and these transport activities are still sensitive to verapamil. To demonstrate the usefulness of CQGREEN in screening for PfCRT inhibitors, we found that CQGREEN uptake by Dd2 PfCRT microsomes can be inhibited by mibefradil, a novel potent chemosensitizer [21], in a dose-dependent manner (Figure 5). Inhibition of CQGREEN uptake by a panel of known chemoreversal agents was compared to that using 3H-CQ (Figure 6). Mean uptake of CQGREEN was positively correlated with that of 3H-CQ (β of 60.24, p = 0.002), showing moderately good agreement between CQGREEN and 3H-CQ uptake (R 2 = 0.766). However, 3H-CQ uptake was roughly 100 times greater than that of CQGREEN.
Figure 3. CQGREEN uptake by Dd2 PfCRT microsomes.
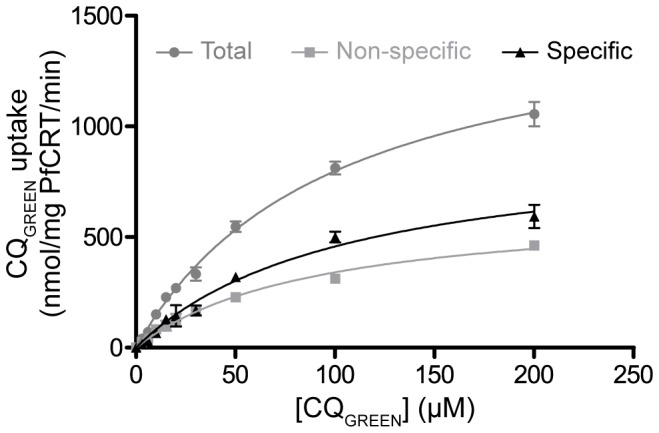
Total uptake of CQGREEN was measured at various CQGREEN concentrations in microsomes carrying Dd2 PfCRT. Non-specific uptake of CQGREEN was measured with pre-treatment of excess unlabelled CQ. Specific uptake was estimated as the difference between total and non-specific uptake. Vmax and Km of the specific uptake was 938.5 nmol/mg PfCRT/min and 105.1 µM respectively. Data are means ± S.E.M.; n≥3.
Figure 4. ATP-dependent, verapamil-sensitive uptake of CQGREEN in microsomes.

Yeast microsomes expressing CQ-sensitive or -resistant PfCRT (“PfCRT-3D7” and “PfCRT-Dd2” respectively), or microsomes from plasmid vector control (“No PfCRT”), were incubated with CQGREEN under different conditions. Preincubation with 150 µM verapamil abrogated CQGREEN uptake from PfCRT-Dd2 but did not affect uptake in PfCRT-3D7 microsomes. Removal of ATP from buffer abolished CQGREEN uptake entirely. **, ***: p<0.005 and p<0.001 respectively, against untreated control. ###: p<0.001. N.s.: not significant. Data presented are means ± S.E.M.; n≥3.
Figure 5. CQGREEN uptake by resistant-type PfCRT is inhibited by mibefradil in a dose-dependent manner.

Microsomes were preincubated with varying concentrations of the PfCRT inhibitor mibefradil prior to addition of CQGREEN. At the highest concentration of 10 µM, mibefradil drastically suppressed CQGREEN uptake in PfCRT-Dd2 microsomes but had no significant effect on uptake in PfCRT-3D7 microsomes. *, ***: p<0.05 and p<0.001 respectively, against no mibefradil control (Ctrl). Data presented are means ± S.E.M.; n≥3.
Figure 6. Accumulation of CQGREEN and 3H-CQ in PfCRT-Dd2 microsomes.

Microsomes were incubated with 10 µM of chemosensitizers before addition of CQGREEN or 3H-CQ. Ctrl: negative control; Ver: verapamil; Mtp: methiothepin; Mgl: metergoline; Lop: loperamide; Oct: octoclothepin; Mib: mibefradil; L703: L703,606; Chl: chlorprothixene. *: p<0.05, comparing CQGREEN uptake against control. ‡: p<0.05, comparing 3H-CQ uptake against control. Data presented are means ± S.E.M.; n≥3.
Polymorphisms and copy number variation in pfcrt and pfmdr1
Eight laboratory strains and twelve clinical isolates were sequenced for polymorphisms in PfCRT and Pgh1, the proteins encoded by the genes pfcrt and pfmdr1 respectively. Residues examined were 72, 74, 75, 76, 220, 271, 326, 356, and 371 for PfCRT, and 86, 184, 1034, 1042, 1226, and 1246 for Pgh1. These residues were chosen for analysis as they were previously implicated in modulating multidrug resistance as well as resistance against CQ [47], [48]. Copy number of pfmdr1 was also determined for each strain and isolate. All clinical isolates showed Dd2-type PfCRT mutations, whereas Pgh1 mutations and pfmdr1 copy numbers were more varied (Table 1).
Table 1. CQ IC50s, PfCRT and Pgh1 polymorphisms, and pfmdr1 copy number.
| PfCRT residue no. | Pgh1 residue no. | pfmdr1 copy number | |||||||||||||||
| Laboratory strains | CQ IC50 (nM) | 72 | 74 | 75 | 76 | 220 | 271 | 326 | 356 | 371 | 86 | 184 | 1034 | 1042 | 1226 | 1246 | |
| T9/96 | 24 | C | M | N | K | A | Q | N | I | R | N | Y | S | N | F | D | 1 |
| 3D7 | 31 | C | M | N | K | A | Q | N | I | R | N | Y | S | N | F | D | 1 |
| HB3 | 42 | C | M | N | K | A | Q | N | I | R | N | F | S | D | F | D | 1 |
| CS2 | 115 | C | I | E | T | S | E | S | I | I | Y | Y | S | N | F | D | 3 |
| T9-94 | 146 | C | I | E | T | S | E | S | I | I | Y | Y | S | N | F | D | 3 |
| 7G8 | 146 | S | M | N | T | S | Q | D | L | R | N | F | C | D | F | Y | 1 |
| Dd2 | 276 | C | I | E | T | S | E | S | T | I | Y | Y | S | N | F | D | 3 |
| K1 | 340 | C | I | E | T | S | E | S | I | I | Y | Y | S | N | F | D | 1 |
Bolded residues indicate deviation from 3D7 haplotype. CQ IC50s are geometric means of at least 3 measurements.
Genetic correlates of CQGREEN uptake
All strains and isolates were assayed for CQGREEN uptake in the trophozoite stage, and their CQ IC50s determined with the standard reinvasion assay. For the entire data set, CQGREEN uptake was inversely correlated to the log of CQ IC50, with an R 2 of 0.53 (Fig. 7A). However, multiple linear regression with sequencing and copy number data revealed that CQGREEN uptake was significantly correlated with not only CQ logIC50 but also a F1226Y substitution in Pgh1 (β of -587.32 and 178.70, p<0.001 and p = 0.024 respectively; adjusted R 2 of 0.615). None of the other mutations was significantly correlated with CQGREEN uptake. Separating the data set to two subpopulations improved the R 2, to 0.72 in the Pgh1 1226F group and 0.676 in the Pgh1 1226Y group (Fig. 7B). It is tempting to conclude that the Pgh1 F1226Y substitution plays a significant causal role in modulating CQGREEN uptake, given that it is also correlated with resistance to artemisinin, mefloquine and lumefantrine [48] and Pgh1's putative sequestration of cytosol-active drugs in the DV [49]. However, it must be kept in mind that the F1226Y mutation was only detected in the clinical isolates, and given the localized collection of these isolates within a small geographical region, F1226Y is likely to be strongly correlated with other undiscovered mutations. In fact, the PfCRT and Pgh1 haplotypes of the F1226Y mutants examined were identical, apart from SMRU0501 which had an additional Pgh1 Y184F substitution (Table 1). It is notable that for the F1226Y mutants, CQGREEN uptake could range as high as that of the CQ-susceptible strains (Fig. 7B) while still maintaining CQ resistance. One possible gene modulating CQGREEN uptake could be pfmrp, which has been proposed to efflux drugs across the parasite plasma membrane into the parasitophorous vacuolar lumen [49], which would still contribute to CQGREEN fluorescence but sequester the drug from its site of action. Knock-out mutants of this gene exhibit increased susceptibility to CQ, quinine, artemisinin, piperaquine, and primaquine [50]. Alternative mechanisms besides efflux of CQ could perhaps also contribute to CQ resistance, such as decreased susceptibility to CQ-induced apoptosis-like cell death [51].
Figure 7. CQGREEN uptake correlates with CQ resistance.

(A) CQGREEN fluorescence is inversely correlated with CQ logIC50. Total data set shows moderate R 2 of 0.53. (B) When split into subpopulations on the basis of Pgh1 residue 1226, R 2 is improved. Data shown are means of at least 3 experiments. Error bars represent S.E.M.
Conclusions
CQGREEN is a commercially available fluorescent CQ analog that interacts with the parasite in a similar fashion to CQ. CQGREEN presents some advantages over traditional radiolabelled CQ in uptake studies: it is safer to handle as it is not radioactive, and its fluorescence properties allows it to be monitored by common fluorescence equipment. Using a defined microsomal platform, we showed that CQGREEN interacts with PfCRT in a manner similar to CQ. Its use as a predictor of CQ susceptibility is enhanced if residue 1226 of Pgh1 is known. Unlike a typical reinvasion assay which may require 48 h or more, the use of CQGREEN allows for measurement of CQ susceptibility within several hours. We believe that CQGREEN could be a valuable tool in future drug discovery projects or used in the identification of factors involved in drug resistance.
Acknowledgments
We thank the patients and staff of Shoklo Malaria Research Unit (SMRU) for their contributions to this project. We would also like to extend our gratitude to the following for P. falciparum strains obtained through MR4's BEI Resources Repository, NIAID, NIH: DJ Carucci (3D7, MRA-102), TE Wellems (HB3, MRA-155; Dd2, MRA-156), SJ Rogerson (CS2, MRA-96), D Walliker (T9-94, MRA-153), and DE Kyle (7G8, MRA-154; K1, MRA-159). P. falciparum T9/96 was obtained from The European Malaria Reagent Repository (http://www.malariaresearch.eu).
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
As part of the Oxford Tropical Medicine Research Program of Wellcome Trust–Mahidol University, Shoklo Malaria Research Unit (SMRU) is funded by the Wellcome Trust of Great Britain. The authors further thank the National Research Foundation (NRF2009NRF-POC002–102), the National Medical Research Council (NMRC/1310/2011; NMRC/EDG/1038/2011), and the Agency for Science, Technology and Research (A*STAR, Singapore) for their generous support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, et al. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet 379: 413–431 10.1016/S0140-6736(12)60034-8 [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization (2012) World Malaria Report 2012. Available: http://www.who.int/malaria/publications/world_malaria_report_2012/en/index.html. Accessed 2013 Mar 11.
- 3. Krafts K, Hempelmann E, Skórska-Stania A (2012) From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy. Parasitol Res 111: 1–6 10.1007/s00436-012-2886-x [DOI] [PubMed] [Google Scholar]
- 4. Coatney GR (1963) Pitfalls in a Discovery: The Chronicle of Chloroquine. Am J Trop Med Hyg 12: 121–128. [DOI] [PubMed] [Google Scholar]
- 5. Butler A, Khan S, Ferguson E (2010) A brief history of malaria chemotherapy. J R Coll Physicians Edinb 40: 172–177 10.4997/JRCPE.2010.216 [DOI] [PubMed] [Google Scholar]
- 6. Wellems TE, Plowe CV (2001) Chloroquine-Resistant Malaria. J Infect Dis 184: 770–776 10.1086/322858 [DOI] [PubMed] [Google Scholar]
- 7. Sullivan DJ Jr, Gluzman IY, Goldberg DE (1996) Plasmodium hemozoin formation mediated by histidine-rich proteins. Science 271: 219–222. [DOI] [PubMed] [Google Scholar]
- 8. Hempelmann E (2007) Hemozoin Biocrystallization in Plasmodium falciparum and the antimalarial activity of crystallization inhibitors. Parasitol Res 100: 671–676 10.1007/s00436-006-0313-x [DOI] [PubMed] [Google Scholar]
- 9. Gildenhuys J, Roex T le, Egan TJ, de Villiers KA (2013) The Single Crystal X-ray Structure of β-Hematin DMSO Solvate Grown in the Presence of Chloroquine, a β-Hematin Growth-Rate Inhibitor. J Am Chem Soc 135: 1037–1047 10.1021/ja308741e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rathore D, Jani D, Nagarkatti R, Kumar S (2006) Heme detoxification and antimalarial drugs – Known mechanisms and future prospects. Drug Discov Today Ther Strateg 3: 153–158 10.1016/j.ddstr.2006.06.003 [DOI] [Google Scholar]
- 11. Summers RL, Martin RE (2010) Functional characteristics of the malaria parasite's “chloroquine resistance transporter”: implications for chemotherapy. Virulence 1: 304–308 10.4161/viru.1.4.12012 [DOI] [PubMed] [Google Scholar]
- 12. Pradines B, Bertaux L, Pomares C, Delaunay P, Marty P (2011) Reduced in vitro susceptibility to artemisinin derivatives associated with multi-resistance in a traveller returning from South-East Asia. Malar J 10: 268 10.1186/1475-2875-10-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miotto O, Almagro-Garcia J, Manske M, MacInnis B, Campino S, et al. (2013) Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat Genet 45: 648–655 10.1038/ng.2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, et al. (2013) Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci 110: 240–245 10.1073/pnas.1211205110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, et al. (2014) A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505: 50–55 10.1038/nature12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mok S, Imwong M, Mackinnon MJ, Sim J, Ramadoss R, et al. (2011) Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics 12: 391 10.1186/1471-2164-12-391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, et al. (2013) Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis. doi:10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed]
- 18. Peyton DH (2012) Reversed chloroquine molecules as a strategy to overcome resistance in malaria. Curr Top Med Chem 12: 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin SK, Oduola AMJ, Milhous WK (1987) Reversal of Chloroquine Resistance in Plasmodium falciparum by Verapamil. Science 235: 899–901. [DOI] [PubMed] [Google Scholar]
- 20. Burgess SJ, Selzer A, Kelly JX, Smilkstein MJ, Riscoe MK, et al. (2006) A Chloroquine-like Molecule Designed to Reverse Resistance in Plasmodium falciparum . J Med Chem 49: 5623–5625 10.1021/jm060399n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ch'ng J-H, Mok S, Bozdech Z, Lear MJ, Boudhar A, et al.. (2013) A Whole Cell Pathway Screen Reveals Seven Novel Chemosensitizers to Combat Chloroquine Resistant Malaria. Sci Rep 3. doi:10.1038/srep01734. [DOI] [PMC free article] [PubMed]
- 22. Martin RE, Butterworth AS, Gardiner DL, Kirk K, McCarthy JS, et al. (2012) Saquinavir Inhibits the Malaria Parasite's Chloroquine Resistance Transporter. Antimicrob Agents Chemother 56: 2283–2289 10.1128/AAC.00166-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adovelande J, Delèze J, Schrével J (1998) Synergy between two calcium channel blockers, verapamil and fantofarone (SR33557), in reversing chloroquine resistance in Plasmodium falciparum . Biochem Pharmacol 55: 433–440 10.1016/S0006-2952(97)00482-6 [DOI] [PubMed] [Google Scholar]
- 24. Kelly JX, Smilkstein MJ, Cooper RA, Lane KD, Johnson RA, et al. (2007) Design, Synthesis, and Evaluation of 10-N-Substituted Acridones as Novel Chemosensitizers in Plasmodium falciparum . Antimicrob Agents Chemother 51: 4133–4140 10.1128/AAC.00669-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanchez CP, Stein W, Lanzer M (2003) Trans Stimulation Provides Evidence for a Drug Efflux Carrier as the Mechanism of Chloroquine Resistance in Plasmodium falciparum . Biochemistry (Mosc) 42: 9383–9394 10.1021/bi034269h [DOI] [PubMed] [Google Scholar]
- 26. Sanchez CP, McLean JE, Stein W, Lanzer M (2004) Evidence for a Substrate Specific and Inhibitable Drug Efflux System in Chloroquine Resistant Plasmodium falciparum Strains. Biochemistry (Mosc) 43: 16365–16373 10.1021/bi048241x [DOI] [PubMed] [Google Scholar]
- 27. Martin RE, Marchetti RV, Cowan AI, Howitt SM, Broer S, et al. (2009) Chloroquine Transport via the Malaria Parasite's Chloroquine Resistance Transporter. Science 325: 1680–1682 10.1126/science.1175667 [DOI] [PubMed] [Google Scholar]
- 28. Zishiri VK, Hunter R, Smith PJ, Taylor D, Summers R, et al. (2011) A series of structurally simple chloroquine chemosensitizing dibemethin derivatives that inhibit chloroquine transport by PfCRT. Eur J Med Chem 46: 1729–1742 10.1016/j.ejmech.2011.02.026 [DOI] [PubMed] [Google Scholar]
- 29. Cabrera M, Natarajan J, Paguio MF, Wolf C, Urbach JS, et al. (2009) Chloroquine Transport in Plasmodium falciparum. 1. Influx and Efflux Kinetics for Live Trophozoite Parasites Using a Novel Fluorescent Chloroquine Probe. Biochemistry (Mosc) 48: 9471–9481 10.1021/bi901034r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ch'ng J-H, Kotturi SR, Chong AG-L, Lear MJ, Tan KS-W (2010) A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis 1: e26 10.1038/cddis.2010.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alcantara LM, Kim J, Moraes CB, Franco CH, Franzoi KD, et al. (2013) Chemosensitization potential of P-glycoprotein inhibitors in malaria parasites. Exp Parasitol 134: 235–243 10.1016/j.exppara.2013.03.022 [DOI] [PubMed] [Google Scholar]
- 32. Tram K, Yan H, Jenkins HA, Vassiliev S, Bruce D (2009) The synthesis and crystal structure of unsubstituted 4, 4-difluoro-4-bora-3a, 4a-diaza-s-indacene (BODIPY). Dyes Pigments 82: 392–395 10.1016/j.dyepig.2009.03.001 [DOI] [Google Scholar]
- 33. Tan W, Gou DM, Tai E, Zhao YZ, Chow LMC (2006) Functional reconstitution of purified chloroquine resistance membrane transporter expressed in yeast. Arch Biochem Biophys 452: 119–128 10.1016/j.abb.2006.06.017 [DOI] [PubMed] [Google Scholar]
- 34. Price RN, Uhlemann A-C, Brockman A, McGready R, Ashley E, et al. (2004) Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. The Lancet 364: 438–447 10.1016/S0140-6736(04)16767-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Homewood CA, Warhurst DC, Peters W, Baggaley VC (1972) Lysosomes, pH and the anti-malarial action of chloroquine. Nature 235: 50–52. [DOI] [PubMed] [Google Scholar]
- 36. Yayon A, Cabantchik ZI, Ginsburg H (1984) Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J 3: 2695–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Schalkwyk DA, Walden JC, Smith PJ (2001) Reversal of Chloroquine Resistance in Plasmodium falciparum Using Combinations of Chemosensitizers. Antimicrob Agents Chemother 45: 3171–3174 10.1128/AAC.45.11.31713174.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kyle DE, Oduola AMJ, Martin SK, Milhous WK (1990) Plasmodium falciparum: modulation by calcium antagonists of resistance to chloroquine, desethylchloroquine, quinine, and quinidine in vitro . Trans R Soc Trop Med Hyg 84: 474–478 10.1016/0035-9203(90)90004-X [DOI] [PubMed] [Google Scholar]
- 39. Martiney JA, Cerami A, Slater AFG (1995) Verapamil Reversal of Chloroquine Resistance in the Malaria Parasite Plasmodium falciparum Is Specific for Resistant Parasites and Independent of the Weak Base Effect. J Biol Chem 270: 22393–22398 10.1074/jbc.270.38.22393 [DOI] [PubMed] [Google Scholar]
- 40. Oduola AM, Sowunmi A, Milhous WK, Brewer TG, Kyle DE, et al. (1998) In vitro and in vivo reversal of chloroquine resistance in Plasmodium falciparum with promethazine. Am J Trop Med Hyg 58: 625–629. [DOI] [PubMed] [Google Scholar]
- 41. Egan T, Kaschula C (2007) Strategies to reverse drug resistance in malaria. Curr Opin Infect Dis 20: 598–604 10.1097/QCO.0b013e3282f1673a [DOI] [PubMed] [Google Scholar]
- 42. Bayoumi RAL, Babiker HA, Arnot DE (1994) Uptake and efflux of chloroquine by chloroquine-resistant Plasmodium falciparum clones recently isolated in Africa. Acta Trop 58: 141–149 10.1016/0001-706X(94)90053-1 [DOI] [PubMed] [Google Scholar]
- 43. Summers RL, Dave A, Dolstra TJ, Bellanca S, Marchetti RV, et al. (2014) Diverse mutational pathways converge on saturable chloroquine transport via the malaria parasite's chloroquine resistance transporter. Proc Natl Acad Sci 111: E1759–E1767 10.1073/pnas.1322965111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krogstad DJ, Gluzman IY, Herwaldt BL, Schlesinger PH, Wellems TE (1992) Energy dependence of chloroquine accumulation and chloroquine efflux in Plasmodium falciparum. Biochem Pharmacol 43: 57–62 10.1016/0006-2952(92)90661-2 [DOI] [PubMed] [Google Scholar]
- 45. De D, Krogstad FM, Cogswell FB, Krogstad DJ (1996) Aminoquinolines That Circumvent Resistance in Plasmodium falciparum in Vitro . Am J Trop Med Hyg 55: 579–583. [DOI] [PubMed] [Google Scholar]
- 46. Lakshmanan V, Bray PG, Verdier-Pinard D, Johnson DJ, Horrocks P, et al. (2005) A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. EMBO J 24: 2294–2305 10.1038/sj.emboj.7600681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valderramos SG, Valderramos J-C, Musset L, Purcell LA, Mercereau-Puijalon O, et al. (2010) Identification of a Mutant PfCRT-Mediated Chloroquine Tolerance Phenotype in Plasmodium falciparum . PLoS Pathog 6: e1000887 10.1371/journal.ppat.1000887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Veiga MI, Ferreira PE, Jörnhagen L, Malmberg M, Kone A, et al. (2011) Novel Polymorphisms in Plasmodium falciparum ABC Transporter Genes Are Associated with Major ACT Antimalarial Drug Resistance. PLoS ONE 6: e20212 10.1371/journal.pone.0020212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sanchez CP, Dave A, Stein WD, Lanzer M (2010) Transporters as mediators of drug resistance in Plasmodium falciparum . Int J Parasitol 40: 1109–1118 10.1016/j.ijpara.2010.04.001 [DOI] [PubMed] [Google Scholar]
- 50. Raj DK, Mu J, Jiang H, Kabat J, Singh S, et al. (2009) Disruption of a Plasmodium falciparum Multidrug Resistance-associated Protein (PfMRP) Alters Its Fitness and Transport of Antimalarial Drugs and Glutathione. J Biol Chem 284: 7687–7696 10.1074/jbc.M806944200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ch'ng J-H, Liew K, Goh AS-P, Sidhartha E, Tan KS-W (2011) Drug-induced permeabilization of parasite's digestive vacuole is a key trigger of programmed cell death in Plasmodium falciparum . Cell Death Dis 2: e216 10.1038/cddis.2011.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.