

Abstract
Bending of double-stranded DNA (dsDNA) is associated with many important biological processes such as DNA-protein recognition and DNA packaging into nucleosomes. Thermodynamics of dsDNA bending has been studied by a method called cyclization which relies on DNA ligase to covalently join short sticky ends of a dsDNA. However, ligation efficiency can be affected by many factors that are not related to dsDNA looping such as the DNA structure surrounding the joined sticky ends, and ligase can also affect the apparent looping rate through mechanisms such as nonspecific binding. Here, we show how to measure dsDNA looping kinetics without ligase by detecting transient DNA loop formation by FRET (Fluorescence Resonance Energy Transfer). dsDNA molecules are constructed using a simple PCR-based protocol with a FRET pair and a biotin linker. The looping probability density known as the J factor is extracted from the looping rate and the annealing rate between two disconnected sticky ends. By testing two dsDNAs with different intrinsic curvatures, we show that the J factor is sensitive to the intrinsic shape of the dsDNA.
Keywords: Molecular Biology, Issue 88, DNA looping, J factor, Single molecule, FRET, Gel mobility shift, DNA curvature, Worm-like chain
Introduction
Understanding the mechanical properties of dsDNA is of fundamental importance in basic sciences and engineering applications. The structure of dsDNA is more complicated than a straight helical ladder because roll, tilt, and twist angles between successive base pairs can vary with sequence. Thermal fluctuations can cause dsDNA to undergo diverse modes of conformational fluctuations such as bending, twisting and stretching. Transitions such as melting and kinking can also occur in extreme conditions.
Among these motions, dsDNA bending has the most noticeable biological impact1. dsDNA bending is associated with gene repression or activation by bringing two distant sites close to each other. It also plays an important role in DNA packaging inside the cell nucleus or a viral capsid. Bending deformation of dsDNA can be visualized experimentally by high-resolution microscopy (AFM2 and TEM3), and the thermodynamics and kinetics can be studied by looping assays, which chemically link juxtaposed sites of the dsDNA.
One such assay is ligase-dependent cyclization4. In this assay, dsDNA molecules with ‘sticky’ (cohesive) ends are circularized or dimerized by DNA ligase. By comparing the rates of circle and dimer formation, one can obtain an effective molar concentration of one end of the DNA in the vicinity of the other end, which is known as the J factor. This J factor is dimensionally equivalent to the probability density of finding one end of the DNA at a short distance from the other end, and thus reflects the flexibility of the DNA. Measuring the J factor as a function of DNA length reveals many characteristics about DNA mechanics including the persistence length4,5.
The worm-like chain (WLC) model has been widely regarded as the canonical polymer model for dsDNA mechanics based on its success in explaining the force-extension curves obtained in DNA pulling experiments6, and correctly predicting the J factors of dsDNAs longer than 200 bp7. However, using the cyclization assay on dsDNA molecules as short as 100 bp, Cloutier and Widom measured the J factors to be several orders of magnitude higher than the WLC model prediction8. A year later, Du et al. produced J factors in agreement with the WLC model using the cyclization assay with lower concentrations of ligase and attributed the anomalous result from the Widom group to high ligase concentrations used9. This controversy exemplifies the unavoidable influence of DNA ligase on cyclization kinetics when using the conventional assay9. Moreover, DNA ligase can also affect DNA structure and stiffness through nonspecific binding10,11.
To eliminate the technical concerns of protein-dependent looping assays, we recently demonstrated a protein-free looping assay based on Fluorescence Resonance Energy Transfer (FRET)12. In this method, looped conformations are detected by FRET between the donor and acceptor attached near the sticky ends of a DNA molecule. An objective-type total internal reflection fluorescence microscope (TIRFM) is used to record trajectories of reversible looping and unlooping events from surface-immobilized single DNA molecules for a prolonged period of time. This method features PCR-based assembly of DNA molecules to generate mismatch-free DNA molecules, which is a crucial improvement over a similar method by Vafabakhsh and Ha13. The single-molecule aspect of this protocol allows measurement of distributions in addition to ensemble averages while the FRET aspect allows one to measure DNA looping dynamics repeatedly from the same molecule, even in conditions that can impair ligase activity.
The TIRFM setup is shown in Figure 1. A custom-designed specimen stage is placed over an Olympus IX61 microscope body. 532 nm and 640 nm lasers are introduced from the side and are reflected by tiny elliptical mirrors14 into the high NA objective to achieve critical angle of incidence at the coverslip-water interface. We note that more widespread through-objective TIR using dichroic mirrors or prism-based TIR setups can also be used for this FRET application. The fluorescence image formed by the microscope is split into donor and acceptor images by a dichroic mirror. They are then re-imaged onto two halves of an EMCCD. Additional long-pass emission filters are used to reduce background signal.
Temperature control is essential for acquiring reproducible kinetic data. For temperature control, the objective is separated from the nosepiece of the microscope body to minimize heat transfer, and water from a temperature controlled chiller/heater is circulated through a brass collar that tightly fits around the interior metal beneath the objective jacket. This setup is able to achieve robust temperature control at the coverslip surface between 15 and 50 °C (Figure 2). In this work, the sample temperature was maintained at 24 °C.
The following protocol presents the step-by-step procedure for DNA construction, estimation of DNA shape, single-molecule experiment, and J factor determination.
Protocol
1. dsDNA Sample Preparation
Design globally curved DNAs by repeating a 10-mer sequence. For example, 5’-GTGCCAGCAACAGATAGC - (TTTATCATCCTTTATCATCCX)7 - TTTCATTCGAGCTCGTTGTTG-3’ is a 186-bp curved DNA where X is a random extra base and the sequence flanking the repeating 10-mer sequence are adapter sequences. NOTE: In this example two 10-mers with opposite preferences to nucleosome formation based on a large-scale nucleosome occupancy study by Kaplan et al.15 were chosen. Since the helical repeat of dsDNA is close to 10 bp, any net deflection of the helical axis of the 10-mer will accumulate to produce a shape like a circular arc (Figure 3A). Since the helical period is closer to 10.5 bp, an extra base is inserted after every two repeats to keep the curved structure as planar as possible. These sequences shorter than 200 bp can be ordered from companies that offer gene synthesis service. It is convenient to flank these sequences with common adapter sequences for subsequent steps. The procedure is schematized in Figure 3B.
Perform PCR with Primer 1 (GTGCCAGCAACAGATAGC) and Primer 2 (/5Cy3/TAAATTCCTACAACAACGAGCTCGAATG). NOTE: Primer 2 is labeled with the FRET donor Cy3 at the 5' end. A typical PCR recipe and cycling protocol are presented in Tables 1 and 2.
Perform PCR with Primer 3 (/5BioTEG/GAAACATAG/iCy5/GAATTTACCGTGCCAGCAACAGATAGC) and Primer 4 (CAACAACGAGCTCGAATG). NOTE: Primer 3 is labeled with the FRET acceptor Cy5 through the backbone and the biotin-linker at the 5' end. PCR recipe and cycling protocol are as above.
Purify the PCR products using a PCR cleanup kit.
Mix the Cy3-labeled product and the Cy5-labeled product in a buffer for strand exchange (100 mM NaCl, 10 mM Tris-HCl pH 7.0, 1 mM EDTA) at final concentrations of 0.4 µM and 0.1 µM, respectively. NOTE: The excess Cy3-DNA increases the concentration of the duplex carrying both Cy3 and Cy5 as a result of strand exchange.
Exchange strands by incubating at 98.5 °C for 2 min, gradually cooling to 5°C with ramp rate of 0.1 °C/sec, and incubating at 5 °C for 2 hr.
2. Gel Electrophoresis to Detect dsDNA Curvature
Pour the polyacrylamide gel16,17 by mixing acrylamide and bis-acrylamide solutions at 29.2:0.8 ratio, 5% (w/v) in 1X Tris/Borate/EDTA (TBE) buffer at pH 8.0. Note: 10 ml of gel solution contains: 1.217 ml 40% acrylamide, 0.667 ml 2% bis-acrylamide, 1 ml TBE 10X, 100 L ammonium persulfate (APS) 10%, 10 L TEMED, and the rest is dH2O. Full solidification of the gel takes about 30 min.
Load the polyacrylamide gel with DNA samples and the DNA ladder in 1X loading buffer (5% glycerol, 0.03% (w/v) bromophenol blue) and run the gel at 5-8 V/cm at 4 °C for 45 min or until the dye front approaches the end of the gel.
Stain the gel using 1X TBE buffer that contains 0.5 g/ml ethidium bromide for 30 min. Identify the DNA bands under UV illumination. Compare band positions with the size marker (100 bp DNA ladder) to calculate the apparent sizes of the DNA molecules. NOTE: Curved DNAs generally move slower than straight DNAs.
3. Flow Cell Preparation
Drill 6-7 pairs of holes along two opposite edges of a glass slide (3’’ x 1’’) using a drill press and diamond drill bits. After drilling, rub the slide in flowing water to remove visible glass powder. NOTE: The holes serve as perfusion inlets and outlets. During drilling, cooling the slide with water is important to prevent cracking.
Place the slides upright in a glass jar and fill it with water. Sonicate for 15 min and transfer them into another glass jar dedicated for acetone cleaning. Fill it with acetone and sonicate for 15 min. Rinse the slides with ethanol using a spray bottle and then with water. Place them in a polypropylene jar, fill it with 5 M potassium hydroxide, and sonicate for 15 min. Finally, sonicate the slides in water for 15 min. Clean the coverslips (no. 1, 24 x 40 mm) using the same protocol. NOTE: Cleaned slides and coverslips can be stored in dH2O for long term use.
Mix 1 mg of biotin-PEG-silane (MW 3400) with 80 mg of mPEG-silane (MW 2000) in 340 L of 0.1 M sodium bicarbonate solution. Mix well and centrifuge the mixture briefly to get rid of bubbles. NOTE: The functionalization of the surface with polyethylene glycol (PEG) helps reduce nonspecific binding of DNA to the surface.
Put 80 L of the PEG solution on each slide and gently lower a coverslip over it. Wait for 45 min. Separate the coverslip from the slide with tweezers, rinse them with copious amount of dH2O and let them dry in open air.
Place thin strips of double-stick tape across the slide to form channels. Align a coverslip over it and firmly press the coverslip against the slide to form liquid-tight channels. Use 5 minute epoxy to seal the edges of the channels.
4. Preparation of Trolox Solution
Put ~30 mg Trolox and 10 ml dH2O in a flask and use a magnet stir bar to stir the solution in open air for 18 hr.
Filter the solution using a 0.2 µm filter and adjust the pH to 7 by adding ~6 µl of 1 M Tris base (pH 11). Note: Trolox is an anti-blinking reagent which is commonly used in single-molecule studies18. The antifading action of Trolox comes from its oxidized derivative which is present in partially degraded Trolox solution19. Quickly dissolving Trolox in methanol or high pH Tris solution should be avoided because of inefficient oxidation.
5. Single-molecule Imaging
Inject 15 µl of NeutrAvidin solution (0.5 mg/ml) into the channel and wait for 2 min before rinsing with 100 µl of T50 buffer (10 mM Tris-HCl, 50 mM NaCl, pH 7.0).
Inject 50 µl of DNA sample (50-100 pM) into the channel. Wait for 5 min and rinse the unbound DNA away with 100 L of T50 buffer. NOTE: DNA molecules will specifically bind to the surface through the NeutrAvidin-biotin interaction.
Fill the channel with the imaging buffer that contains an oxygen scavenging system20 (100 mM PCD, 5 mM PCA, 1 mM Trolox, and 500 mM NaCl).
Set the EMCCD in frame-transfer mode to stream 2 x 2 binned images (256 x 256) to the computer at 25 frames per sec.
Put immersion oil on the microscope objective, and fix the flow cell on the microscope stage using specimen clips. Coarse-adjust the focus by looking at the laser reflection pattern on the wall. Fine-adjust the focus using the live view of fluorescence images on the monitor.
Begin data acquisition with the 532 nm laser on. Check the live view and adjust the focus if necessary. Stop data acquisition when most molecules have photobleached. NOTE: In this facility, a lab-written C program to control the microscope and display live images on the monitor as they are saved to the hard disk is used.
6. Image Processing and Data Analysis
NOTE: A time series of 256 x 256 images are processed by a MATLAB code to generate single-molecule time traces of Cy3 and Cy5 intensities. To pair pixels between the donor channel and the acceptor channel of the split-view image, 6-7 pairs of Cy3 and Cy5 spots, each pair from the same molecule evenly dispersed across the field of view, are manually picked, and an affine transformation is calculated using the coordinates of these spots as anchor points.
Using a MATLAB script, look through all single-molecule time traces that show multiple transitions between low and high FRET signals. Identify the looped and unlooped states. NOTE: The FRET signal is defined as the Cy5 intensity (Ia ) divided by the sum of Cy3 and Cy5 intensities ( Ia + Id ). Looped state has high FRET value while unlooped state has low FRET value. The histogram of FRET signals from a single molecule is bimodally distributed because of reversible looping and unlooping.
Find the threshold that separates the two distributions by determining the intersection between the two fitted Gaussian curves.
Calculate the FRET efficiency as
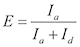 and assign the looped states with high FRET values and the unlooped states with low FRET values.
and assign the looped states with high FRET values and the unlooped states with low FRET values.Using a MATLAB script, analyze the cumulative number of molecules ( N(t) ) that looped (or reached the high FRET state) at different time lapses since the start of data acquisition. NOTE: Since a dsDNA molecule can start in any conformation of the unlooped state at the start of data acquisition, the rate of increase in the looped population reflects the mean looping rate averaged over initial conformations. Extract the looping rate kloop by fitting N(t) with an exponential function:
 If increases biphasically, it can be fitted with a double exponential function:
If increases biphasically, it can be fitted with a double exponential function: 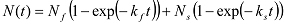 In this case, kloop is obtained from:
In this case, kloop is obtained from:  NOTE: Theoretically, the survival probability that a polymer has not looped at time t is not a single or double exponential function12. Exponential fitting is used as a practical means to extract the average looping time.
NOTE: Theoretically, the survival probability that a polymer has not looped at time t is not a single or double exponential function12. Exponential fitting is used as a practical means to extract the average looping time.
7. Determining the J Factor
NOTE: The J factor represents how concentrated one end of a dsDNA is around the other end. It can be determined by interpolating the concentration of one end segment of the DNA that would produce the same reaction rate with the other end segment as the measured looping rate. Experimentally, one end segment is immobilized on the surface, and the other end segment is introduced at a certain concentration c. If the measured annealing rate between the two ends is kanneal, then the J factor21 is given by ![]() . The annealing rate constant ( kanneal = kanneal / C ) is independent of the probe concentration.
. The annealing rate constant ( kanneal = kanneal / C ) is independent of the probe concentration.
Flow 20 µl of 30-50 pM biotin-Cy5 oligo (Primer 3) into a NeutrAvidin-coated channel. Rinse the channel with 100 µl T50 to wash away unbound oligos.
Prepare the imaging buffer as described in part 5 with the addition of Cy3 oligo (Primer 2) at a final concentration of 50 nM. Flow this imaging buffer into the channel.
While keeping the 532 nm laser on, briefly turn the 640 nm laser on to identify locations of surface-bound Cy5 oligos. Turn off the 640 nm laser and start monitoring the FRET signal. NOTE: Upon hybridization of a Cy3 oligo to a surface-bound Cy5 oligo, Cy5 fluorescence will arise from FRET.
Using a MATLAB script, analyze the number of molecules which start in the unbound state (low Cy5 intensity) but later turn into the annealed state (high Cy5 intensity) as a function of time from the Cy5 intensity traces.
Plot this number of annealed molecules vs. time. Fit this curve with a single exponential function (
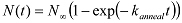 ) to obtain the annealing rate ( kanneal ).
) to obtain the annealing rate ( kanneal ).Repeat this experiment at different Cy3 oligo concentrations (60, 100, and 180 nM) to confirm linearity between the annealing rate and the reactant concentration. Extract the second-order annealing rate constant ( kanneal) from the slope.
Calculate the J factor from
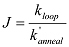 , where kloop is the looping rate measured in the same buffer condition.
, where kloop is the looping rate measured in the same buffer condition.
Representative Results
DNA molecules used for the looping study consist of a duplex region of variable sequence and length and single-stranded overhangs that are complementary to each other. The overhangs, which are 7-base long, can anneal to each other to capture the looped state. Each overhang contains either Cy3 or Cy5 that is linked in the DNA backbone through amidite chemistry. The Cy5-overhang is also linked with biotin-TEG (15-atom Tetra-Ethylene Glycol spacer) for surface immobilization (see Figure 4A). All these modifications can be incorporated into the dsDNA following a PCR-based protocol (Protocol 1 and Figure 3B). The chosen length and sequence of the overhangs work well to produce detectable and reversible FRET events in normal experimental conditions. The looping and the unlooping kinetics of the DNA loop are sensitive to the surrounding temperature, and therefore, a temperature controller is essential for reproducibility. Such a controller can be readily incorporated into the microscope (Figure 2).
Using TIRFM, anti-correlated fluctuations of Cy3 (donor) and Cy5 (acceptor) signals from surface-immobilized dsDNA molecules were observed (Figure 4B). In the looped state, the distance between two dyes is smaller than 5nm, which would result in high Cy5 signal and low Cy3 signal due to high FRET efficiency (Figures 4A and 4C). Several control experiments were performed to prove that the high FRET state represents the looped state. The lifetime of the high FRET state increased with increasing overhang length, which suggests that the high FRET state is stabilized by base pairing between the two overhangs. The lifetime of the high FRET state also decreased with decreasing DNA length, which is expected because the loop free energy increases as the loop becomes smaller12. Based on this evidence, the high and low FRET states can be assigned to the looped and unlooped states, respectively.
To test the effect of intrinsic curvature of dsDNA on looping, two 186 bp dsDNAs with different curvatures were constructed, which are named 'straight' (S-DNA) and 'curved' (C-DNA) based on their curvature. The repeating 10-mer sequence is TTTATCATCC for the C-DNA and TGACAGCAAC for the S-DNA. The overall curvature of each of these dsDNAs was checked by PAGE (polyacrylamide gel electrophoresis), which is the most widely used method to estimate dsDNA curvature16,22. Figure 5 shows that the S-DNA runs similarly to the DNA of the same size in the DNA ladder (compare lane 1 and lane 2 in Figure 5). On the other hand, the C-DNA shows a larger apparent size compared to its counterpart in the DNA ladder (~1.2 larger than its real size, see lane 1 and lane 3). This observation indicates that the C–DNA possesses a larger intrinsic curvature than the S-DNA.
Figure 6 shows a representative fluorescence time trace from the C-DNA (Figure 6A) and the corresponding FRET trace (Figure 6B). Compared to the S-DNA (Figures 4B and 4C), the C-DNA produces more high FRET events during the same period of time; the C-DNA loops 4-times more frequently than the S-DNA during the same acquisition time (Figure 7). This result demonstrates that intrinsic curvature of DNA can significantly affect looping dynamics.
To extract the J factor from the looping rate, one needs to measure the concentration-independent second-order rate constant for hybridization between two disconnected overhangs. A realization of such measurement is shown in Figure 8A. Hybridization of the Cy3 oligo to the immobilized Cy5 oligo produces Cy5 intensity bursts due to FRET. From multiple measurements with different Cy3-oligo concentrations (see Figure 8B), one can extract in a statistically robust way. In our experiment, the annealing rate constant between the two oligos in 500 mM NaCl was determined to be 0.45 ± 0.04 x 106 M-1 sec-1. The J factor was calculated by dividing the looping rate (kloop) by the second-order annealing rate constant (k'anneal). It was 61 ± 3 nM for the S-DNA and 265 ± 48 nM for the C-DNA. The J factor of the S-DNA (Figure 9) is in fair agreement with previous measurements at similar length7.
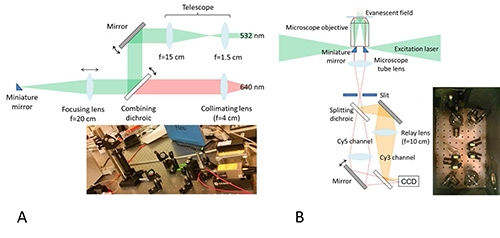
Figure 1. Objective-type TIRFM.A) Excitation optics. The 532 nm and 640 nm lasers are collimated separately to a similar diameter, merged into the same path by a dichroic mirror, and focused onto a tiny elliptical mirror placed underneath the objective back aperture. It is important to use an achromatic lens for focusing to minimize chromatic aberration. B) Microscope optics. The lateral position of the miniature mirror has to be finely adjusted so that it can reflect the laser beam through the objective while minimally blocking fluorescence emission. Therefore, it must be mounted on a small translation stage. Another mirror on the opposite side reflects the outgoing laser to prevent it from entering the detection optics. At the image plane outside the microscope body, an adjustable slit is placed to crop the image to half the size of the detection area of the CCD. The fluorescence emission is split by a dichroic mirror into two channels, and the relay lenses form the second image on the CCD with 1:1 magnification. One of the reflecting mirrors is slightly rotated to offset the donor and acceptor images. A bandpass and a longpass filters are used to reduce the background in the donor and acceptor channels, respectively. Click here to view larger image.
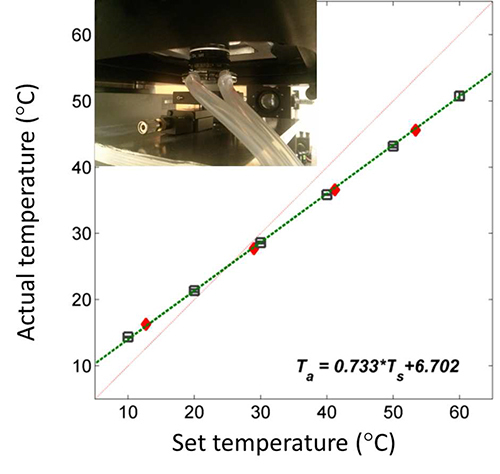 Figure 2. Temperature control for single-molecule experiments. The set temperature (Ts) is the reading on the water chiller/heater. The actual temperature (Ta) is measured by a thermocouple contacting the top surface of the coverslip on the microscope. The plot of the actual temperatures vs. six different set temperatures (black squares) shows excellent linearity (green dashed line). The robustness of the controller is shown by random measurements made at later times (red diamonds). The inset is the picture of the temperature controlling unit and the objective in their final assembly. The red line represents a slope of unity. Click here to view larger image.
Figure 2. Temperature control for single-molecule experiments. The set temperature (Ts) is the reading on the water chiller/heater. The actual temperature (Ta) is measured by a thermocouple contacting the top surface of the coverslip on the microscope. The plot of the actual temperatures vs. six different set temperatures (black squares) shows excellent linearity (green dashed line). The robustness of the controller is shown by random measurements made at later times (red diamonds). The inset is the picture of the temperature controlling unit and the objective in their final assembly. The red line represents a slope of unity. Click here to view larger image.
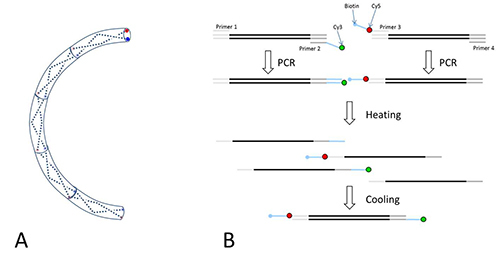 Figure 3. DNA Design.A) Curved dsDNA. A 10-mer dsDNA is represented as a curved tube segment, and the two single strands as dashed lines. The helical axis of any 10-mer DNA is not perfectly straight, and thus each concatenation of such a 10-mer will lead to incremental tilt of the helical axis in the same direction. This effect can be exploited to generate a planar, superhelical molecule. B) PCR-based preparation of dsDNA. Single-stranded DNAs are shown as a horizontal line. Their actual curvature is not depicted here. The central segment in black represents the unique part of the DNA fragment. Shown in light grey are the adapter sequences common to all DNAs used in this study. Primer 1 and 4 anneal to the front and back adapters, respectively. Primer 2 is labeled with Cy3 at the 5'-end. Primer 3 has a biotin tag at the 5'-end and an internally linked Cy5. The light blue regions of Primer 2 and 3 contain complementary sequences that function as sticky ends. Either a plasmid or genomic DNA that contains the sequence of interest is used as the template in two separate PCR reactions. Cy3-labeled dsDNA is produced using primer 1 and 2, and biotinylated Cy5-labeled DNA is produced using primer 3 and 4. These two PCR products are heated and cooled together for strand exchange. Four different dsDNAs are formed, only one of which contains Cy3, Cy5, and biotin. Click here to view larger image.
Figure 3. DNA Design.A) Curved dsDNA. A 10-mer dsDNA is represented as a curved tube segment, and the two single strands as dashed lines. The helical axis of any 10-mer DNA is not perfectly straight, and thus each concatenation of such a 10-mer will lead to incremental tilt of the helical axis in the same direction. This effect can be exploited to generate a planar, superhelical molecule. B) PCR-based preparation of dsDNA. Single-stranded DNAs are shown as a horizontal line. Their actual curvature is not depicted here. The central segment in black represents the unique part of the DNA fragment. Shown in light grey are the adapter sequences common to all DNAs used in this study. Primer 1 and 4 anneal to the front and back adapters, respectively. Primer 2 is labeled with Cy3 at the 5'-end. Primer 3 has a biotin tag at the 5'-end and an internally linked Cy5. The light blue regions of Primer 2 and 3 contain complementary sequences that function as sticky ends. Either a plasmid or genomic DNA that contains the sequence of interest is used as the template in two separate PCR reactions. Cy3-labeled dsDNA is produced using primer 1 and 2, and biotinylated Cy5-labeled DNA is produced using primer 3 and 4. These two PCR products are heated and cooled together for strand exchange. Four different dsDNAs are formed, only one of which contains Cy3, Cy5, and biotin. Click here to view larger image.
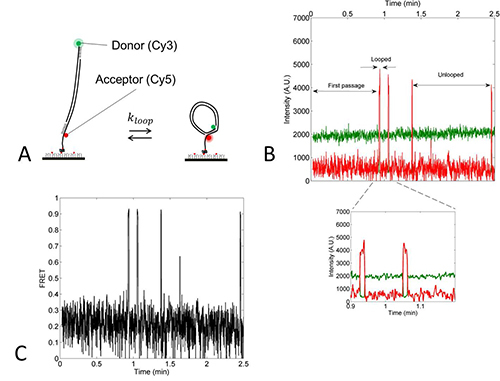 Figure 4. Experimental scheme.A) Single-molecule FRET experiment. dsDNA molecules labeled with Cy3 (green) on one side and Cy5 (red) on the other are immobilized on the PEG (polyethylene glycol)-coated surface. Reversible looping and unlooping of dsDNAs result in high FRET (looped) and low FRET (unlooped) states, respectively. B) A typical single-molecule time trace of donor (green) and acceptor (red) fluorescence intensities. The high and low FRET states are identified as the looped and unlooped states, respectively. Also indicated is the first looping time that is used to calculate the looping rate. (Inset) The zoom-in figure shows the anti-correlation of the Cy3 and Cy5 intensities. C) A time trace of FRET efficiency calculated from donor and acceptor intensity traces in B. Figure 4B and 4C were taken from the straight DNA (S-DNA) as described in the main text. The low FRET at 0.2 is due to direct excitation of Cy5 by the 532 nm laser, and confirms the presence of fluorescently active Cy5. Click here to view larger image.
Figure 4. Experimental scheme.A) Single-molecule FRET experiment. dsDNA molecules labeled with Cy3 (green) on one side and Cy5 (red) on the other are immobilized on the PEG (polyethylene glycol)-coated surface. Reversible looping and unlooping of dsDNAs result in high FRET (looped) and low FRET (unlooped) states, respectively. B) A typical single-molecule time trace of donor (green) and acceptor (red) fluorescence intensities. The high and low FRET states are identified as the looped and unlooped states, respectively. Also indicated is the first looping time that is used to calculate the looping rate. (Inset) The zoom-in figure shows the anti-correlation of the Cy3 and Cy5 intensities. C) A time trace of FRET efficiency calculated from donor and acceptor intensity traces in B. Figure 4B and 4C were taken from the straight DNA (S-DNA) as described in the main text. The low FRET at 0.2 is due to direct excitation of Cy5 by the 532 nm laser, and confirms the presence of fluorescently active Cy5. Click here to view larger image.
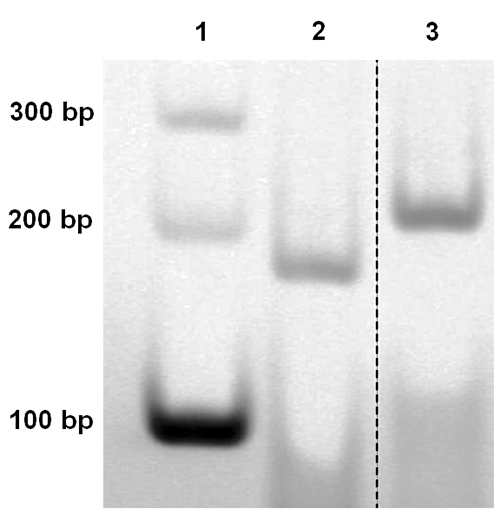 Figure 5. Polyacrylamide gel electrophoresis (29.2:0.8 acrylamide/bis-acrylamide, 5% in TBE buffer, pH 8.0) of synthetic DNAs. From left to right, the lanes contain 1 kb marker, the 186 bp Straight DNA and the 186 bp Curved DNA. This figure is partially adapted from a previous publication12 with permission. Click here to view larger image.
Figure 5. Polyacrylamide gel electrophoresis (29.2:0.8 acrylamide/bis-acrylamide, 5% in TBE buffer, pH 8.0) of synthetic DNAs. From left to right, the lanes contain 1 kb marker, the 186 bp Straight DNA and the 186 bp Curved DNA. This figure is partially adapted from a previous publication12 with permission. Click here to view larger image.
 Figure 6. Representative traces from the curved DNA (C-DNA) (A) and its corresponding FRET trace (B). Comparison of the intensity and FRET traces between S-DNA (Figures 4B and 4C) and C-DNA (Figures 6A and 6B) clearly shows that the curved DNA loops more frequently than the straight DNA in the same buffer condition. (Inset) The zoom-in figure shows the anti-correlation of the Cy3 and Cy5 intensities. Click here to view larger image.
Figure 6. Representative traces from the curved DNA (C-DNA) (A) and its corresponding FRET trace (B). Comparison of the intensity and FRET traces between S-DNA (Figures 4B and 4C) and C-DNA (Figures 6A and 6B) clearly shows that the curved DNA loops more frequently than the straight DNA in the same buffer condition. (Inset) The zoom-in figure shows the anti-correlation of the Cy3 and Cy5 intensities. Click here to view larger image.
 Figure 7. The mean first looping times of the straight and the curved DNAs. Each time was extracted from ~500 molecules in 5 different fields. The error bars represent the standard error of the mean (SEM) calculated from 3 independent experiments. Click here to view larger image.
Figure 7. The mean first looping times of the straight and the curved DNAs. Each time was extracted from ~500 molecules in 5 different fields. The error bars represent the standard error of the mean (SEM) calculated from 3 independent experiments. Click here to view larger image.
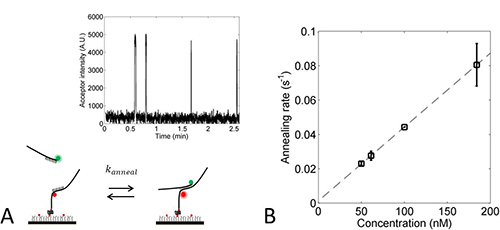 Figure 8. Determining the annealing rate constant from oligo hybridization. A) Schematic view of the annealing rate measurement. Cy5 primer is immobilized on the surface, and Cy3 primer is present at 50-180 nM. Binding and unbinding of the Cy3 primer at 50 nM occur reversibly and lead to detectable acceptor bursts, which are shown in the inset. B) The annealing rate depends linearly on the concentration of the free oligo. The slope of the line represents the second-order annealing rate constant (k'anneal). Click here to view larger image.
Figure 8. Determining the annealing rate constant from oligo hybridization. A) Schematic view of the annealing rate measurement. Cy5 primer is immobilized on the surface, and Cy3 primer is present at 50-180 nM. Binding and unbinding of the Cy3 primer at 50 nM occur reversibly and lead to detectable acceptor bursts, which are shown in the inset. B) The annealing rate depends linearly on the concentration of the free oligo. The slope of the line represents the second-order annealing rate constant (k'anneal). Click here to view larger image.
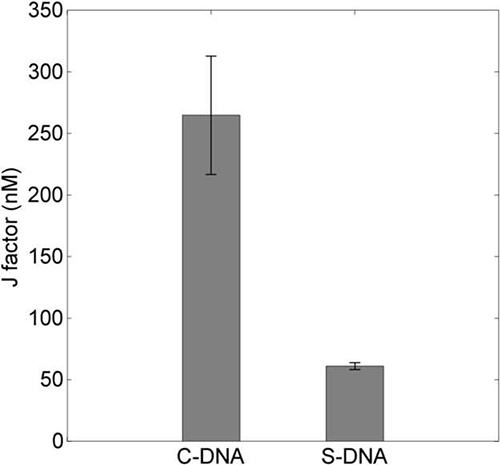 Figure 9. The J factors of the straight and curved DNAs calculated in nM. The J factor was determined based on two measurables: the looping rate of the dsDNA loop and the annealing rate constant of the free complementary overhangs. Click here to view larger image.
Figure 9. The J factors of the straight and curved DNAs calculated in nM. The J factor was determined based on two measurables: the looping rate of the dsDNA loop and the annealing rate constant of the free complementary overhangs. Click here to view larger image.
| Component | Volume/50 μl reaction | Final concentration |
| Water | 36 μl | |
| 5x phusion buffer | 10 μl | 1x |
| 10 mM dNTPs | 1 μl | 200 μM each |
| Forward primer (25 μM) | 1 μl | 0.5 μM |
| Reverse primer (25 μM) | 1 μl | 0.5 μM |
| Phusion hot start DNA polymerase (2 U/μl) | 0.5 μl | 0.02 U/μl |
| Template DNA | 0.5 μl | ~1 ng |
Table 1. PCR reaction mix. The protocol is optimized for the Phusion DNA polymerase. Items should be added in this order.
| Cycle step | Temperature | Time | Number of cycles |
| Initial denaturation | 95 °C | 30 sec | 1 |
| Denaturation | 95 °C | 5 sec | 30 |
| Annealing | 60 °C | 10 sec | |
| Extension | 72 °C | 15 sec/kb | |
| Final extension | 72 °C | 5 min | 1 |
| 4 °C | hold |
Table 2. PCR cycling instructions. The annealing temperature is determined based on the melting temperatures of the primers. The extension time is optimized for the Phusion DNA polymerase.
Discussion
A simple single-molecule assay based on FRET was used to study looping kinetics of DNAs of different intrinsic shapes. Curved DNAs can be prepared by repeating a 10-mer sequence in phase with the helical period of 10.5 bp, and their curvatures can be estimated using PAGE. These dsDNAs are designed with sticky ends to allow transient loop stabilization. We extracted the looping rate from the exponential rise in the number of looped molecules over time. The annealing rate constant between the disconnected sticky end segments is used to determine the concentration equivalent of the looping probability density, which is known as the J factor.
In a ligase-based DNA cyclization assay, the measurement of DNA closure probability is sensitive to the ligase concentration, and the J factor can be overestimated if ligase is excessive9. Nonspecific binding of DNA ligase to DNA can also affect the looping10. Furthermore, ligase activity depends on salt and decays over time. Thus it is difficult to study DNA looping in conditions suboptimal for ligase activity. In contrast, a FRET-based looping assay is free from all of these concerns.
Our experimental protocol is similar to that of Vafabakhsh and Ha13 except two important aspects which are worthy of discussion. Vafabakhsh and Ha13 constructed their dsDNA molecules by ligating several synthetic oligos. Although the error (insertion, deletion, or substitution) rate per nucleotide in oligo synthesis is negligible (f 0.181%)23, the fraction of unintended DNA sequences increases linearly with oligo length. Therefore, a 100-bp duplex sample prepared by this protocol can contain up to ~33% imperfect duplexes which may result in higher looping rate24. In comparison, our protocol uses polymerase chain reaction (PCR) to synthesize the dsDNA molecules, which has a much higher fidelity than DNA chemosynthesis25. According to the manufacturer's data, the high-fidelity polymerase we used would generate a correct 100-bp DNA molecule at 99.8% probability after 30 cycles of PCR amplification.
Another key difference between the two studies is the surface immobilization scheme. In our study, DNA molecules are attached to the surface through their ends, whereas in the protocol by Vafabakhsh and Ha13, they are attached through an internal base. It was predicted that based on the confinement effect alone, an internally pinned dsDNA can loop more frequently than a free dsDNA, up to a factor of five depending on the location of internal pinning26. Therefore, when measuring the J factor as a function of DNA contour length, internal pinning can introduce unexpected variability. It is also possible that the modified base with the linker has a weaker base pairing capability, which can result in a higher looping rate. However, despite these apparent differences, the two protocols produce similar J factors at 100 bp (~3 nM vs. ~2 nM).
This FRET-based looping assay holds promise for studying a wide range of phenomena related to dsDNA looping including the effect of mismatches, nicks or gaps on dsDNA looping27, the effect of DNA binding proteins on dsDNA looping, and temperature dependence of dsDNA flexibility. These topics would be difficult to address with the conventional ligase-based cyclization. The FRET-based looping assay can also be used to measure the J factor vs. the contour length, which is the standard relationship to test polymer models. However, the protocol we presented here is not well-suited to studying looping of dsDNA shorter than 100 bp. Below this length, dsDNA looping becomes extremely slow, and therefore the measured rate will be affected by other unintended processes such as photobleaching. One can accelerate the apparent looping kinetics by using high salt concentration ([Na+] > 500 mM), but physiological relevance of such measurement becomes questionable. Hence, investigation of dsDNA bending at length scales below 100 bp will benefit from an approach orthogonal to cyclization-based assays.
Disclosures
The authors declare no conflicts of interest.
Acknowledgments
We thank James Waters, Gable Wadsworth and Bo Broadwater for critically reading the manuscript. We also thank four anonymous reviewers for providing useful comments. We acknowledge financial support from Georgia Institute of Technology, the Burroughs Wellcome Fund Career Award at the Scientific Interface, and the student research network grant from NSF Physics of Living Systems.
References
- Garcia HG, et al. Biological consequences of tightly bent DNA: The other life of a macromolecular celebrity. Biopolymers. 2007;85:115–130. doi: 10.1002/bip.20627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins PA, et al. High flexibility of DNA on short length scales probed by atomic force microscopy. Nature Nanotechnology. 2006;1 doi: 10.1038/nnano.2006.63. [DOI] [PubMed] [Google Scholar]
- Lionberger TA, et al. Cooperative kinking at distant sites in mechanically stressed DNA. Nucleic Acids Research. 2011;41:6785–6792. doi: 10.1093/nar/gkr666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore D, et al. DNA flexibility studied by covalent closure of short fragments into circles. Proc Natl Acad Sci U S A. 1981;78:4833–4837. doi: 10.1073/pnas.78.8.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geggier S, Vologodskii A. Sequence dependence of DNA bending rigidity. Proc Nat Acad Sci U S A. 1992;107:15421–15426. doi: 10.1073/pnas.1004809107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, et al. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science. 1992;258:1122–1126. doi: 10.1126/science.1439819. [DOI] [PubMed] [Google Scholar]
- Peters JP, Maher LJ. DNA curvature and flexibility in vitro and in vivo. Quarterly Reviews of Biophysics. 2010;43:23–63. doi: 10.1017/S0033583510000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier TE, Widom J. Spontaneous sharp bending of double-stranded DNA. Molecular Cell. 2004;14:355–362. doi: 10.1016/s1097-2765(04)00210-2. [DOI] [PubMed] [Google Scholar]
- Du Q, et al. Cyclization of short DNA fragments and bending fluctuations of the double helix. Proc Natl Acad Sci U S A. 2005;102:5397–5402. doi: 10.1073/pnas.0500983102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, et al. T4 DNA ligase is more than an effective trap of cyclized dsDNA. Nucl. Acids Res. 2007;35:5294–5302. doi: 10.1093/nar/gkm582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo C, et al. The effect of nonspecific binding of lambda repressor on DNA looping dynamics. Biophysical Journal. 2012;103:1753–1761. doi: 10.1016/j.bpj.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Kim HD. Measuring shape-dependent looping probability of DNA. Biophys. J. 2013;104:2068–2076. doi: 10.1016/j.bpj.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafabakhsh R, Ha T. Extreme bendability of DNA less than 100 base pairs long revealed by single-molecule cyclization. Science. 2012;337:1097–1101. doi: 10.1126/science.1224139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman L, et al. Viewing dynamic assembly of molecular complexes by multi-wavelength single-molecule fluorescence. Biophysical Journal. 2006;91:1023–1031. doi: 10.1529/biophysj.106.084004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo HS, Crothers DM. Calibration of DNA curvature and a unified description of sequence-directed bending. Proc Nat Acad Sci U S A. 1988;85:1763–1767. doi: 10.1073/pnas.85.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosseda G, et al. A temperature-induced narrow DNA curvature range sustains the maximum activity of a bacterial promoter in vitro. Biochemistry. 2010;49:2778–2785. doi: 10.1021/bi902003g. [DOI] [PubMed] [Google Scholar]
- Rasnik I, et al. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- Cordes T, et al. On the mechanism of Trolox as antiblinking and antibleaching reagent. J. Am. Chem. Soc. 2009;131:5018–5019. doi: 10.1021/ja809117z. [DOI] [PubMed] [Google Scholar]
- Aitken CE, et al. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WH, Hagerman PJ. Application of the method of phage T4 DNA ligase-catalyzed ring-closure to the study of DNA structure: II. NaCl-dependence of DNA flexibility and helical repeat. Journal of Molecular Biology. 1990;212:363–376. doi: 10.1016/0022-2836(90)90131-5. [DOI] [PubMed] [Google Scholar]
- Bolshoy A, et al. Curved DNA without A-A: experimental estimation of all 16 DNA wedge angles. Proc Natl Acad Sci U S A. 1991;88:2312–2316. doi: 10.1073/pnas.88.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucl. Acids Res. 2009;37:6984–6990. doi: 10.1093/nar/gkp687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vologodskii A, et al. Bending of short DNA helices. Artif DNA PNA XNA. 2013;4 doi: 10.4161/adna.23892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucl. Acids Res. 2002;30 doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JT, Kim HD. Equilibrium Statistics of a Surface-Pinned Semiflexible Polymer. Macromolecules. 2013;46:6659–6666. [Google Scholar]
- Mills JB, et al. Electrophoretic evidence that single-stranded regions of 1 or more nucleotides dramatically increase the flexibility of DNA. Biochemistry. 1994;33:1797–1803. doi: 10.1021/bi00173a024. [DOI] [PubMed] [Google Scholar]