

Abstract
Type 1 diabetic adolescent children on insulin therapy suffer episodes of both hyper- and hypoglycemic episodes. Glucose transporter isoform GLUT1 expressed in blood-brain barrier (BBB) and red blood cells (RBC) compensates for perturbed circulating glucose towards protecting the supply to brain and RBCs. We hypothesized that RBC-GLUT1 concentration, as a surrogate for BBB-GLUT1, is altered in T1D children. To test this hypothesis, we measured RBC-GLUT1 by ELISA in T1D children (n=72; mean age 15.3 ± 0.2 y) and control children (CON; n=11; mean age 15.6 ± 0.9 y) after 12 hours of euglycemia and during a hyperinsulinemic-hypoglycemic clamp with a nadir blood glucose of ~3.3 mmol/l for 90 min (Clamp I) or ~3 mmol/l for 45 min (Clamp Π). Reduced baseline RBC-GLUT1 was observed in T1D (2.4 ± 0.17 ng/ng membrane protein); versus CON (4.2 ± 0.61 ng/ng protein) (P < 0.0001). Additionally, baseline RBC GLUT1 in T1D negatively correlated with HbA1C (R= −0.23, p<0.05) but not in CON (R=0.06, p<0.9). Acute decline in serum glucose to 3.3 mmol/l (90 min) or 3 mmol/l (45 min) did not change baseline RBC-GLUT1 in T1D or CON children. We conclude that reduced RBC-GLUT1 encountered in T1D, with no ability to compensate by increasing during acute hypoglycemia over the durations examined, may demonstrate a vulnerability of impaired RBC glucose transport (serving as a surrogate for BBB), especially in those with the worst control. We speculate this may contribute to the perturbed cognition seen in T1D adolescents.
Keywords: Blood-brain barrier, hyperinsulinemic hypoglycemic clamp, Type1 diabetes, RBC Glucose Transporter 1
INTRODUCTION
Glucose, an essential fuel for oxidative metabolism of the brain, is transported across the endothelial cells of the brain microvasculature and astrocytic glial cells, both essential components of the blood-brain barrier (BBB) [1]. Transport across the BBB is mediated by the GLUT1 protein [1–3]. GLUT1 is a facilitative glucose transporter isoform which is also expressed in human red blood cells (RBC) [4]. The BBB GLUT1 protein concentration is altered by circulating glucose concentrations and it mediates 90% of glucose transport to the brain. In animal experiments, chemically-induced uncontrolled diabetes associated with chronic hyperglycemia was observed to down-regulate BBB GLUT1 concentrations [5, 6]. In contrast, insulin-induced chronic hypoglycemia led to a compensatory increase in the BBB GLUT1 concentrations and is thought to be a mechanism that protects the brain from decreased glucose supply [7, 8].
The description of GLUT1 deficiency syndrome [9, 10] in children demonstrated that a haploinsufficiency of the GLUT1 gene led to a decrease in RBC GLUT1 protein concentrations, which translated into reduced transport of glucose into these cells [11]. In addition, these infants demonstrated hypoglycorrhachia in the presence of normal cerebro-spinal fluid (CSF) lactate levels and euglycemia, which was associated with developmental delays, seizures, and impaired cognition [9, 10]. This latter constellation of symptoms supported the probability of impaired BBB glucose transport secondary to reduced GLUT1 concentrations. Thus it appears that the RBC GLUT1 concentration may serve as a surrogate measure for the human BBB GLUT1 concentrations in conditions such as diabetes mellitus where perturbations of circulating glucose, rather than a reduction in gene dosage, could potentially alter these protein concentrations and affect BBB glucose transport surfacing as cognitive impairments.
The emphasis on tight glycemic control has been shown to be associated with frequent episodes of hypoglycemia in children with type 1 diabetes (T1D) which may also cause cognitive impairment [12, 13]. There is increasing evidence of T1D associated CNS damage seen by imaging studies and longitudinal neuro-cognitive testing displaying associations with both hyper- and hypoglycemic episodes. Additionally, even mild hypoglycemia encountered in T1D can cause transient cognitive impairment which is greater than that observed in control children [13]. The reason behind this effect is only partially understood. Impaired cognition appears to be associated with suppression of counter-regulatory hormone responses to hypoglycemia in adults and children [13] due to hypoglycemia-associated autonomic failure [14]. This autonomic failure may be a marker of recurrent hypoglycemia, while hemoglobin A1C (HbA1C) serves as an integrated marker of glycemic control, influenced mostly by the degree of hyperglycemia over the preceding few months.
We hypothesize that the greater the degree of hyperglycemia, the greater the suppression of RBC GLUT1 concentration, a marker for BBB GLUT1 concentration. Due to this suppression, normal up-regulation of GLUT transporters in response to acute hypoglycemia will be impaired, limiting glucose entry into the brain. To test this hypothesis, we prospectively examined the effect of T1D compared to control adolescent children on RBC GLUT1 concentrations during euglycemia and insulin-induced acute hypoglycemia.
Research Design and Methods
Study Population
Twelve to eighteen year old T1D and control subjects that met the inclusion criteria were enrolled from the diabetes center of Children's Hospital of Pittsburgh. These criteria were diabetes duration greater than 2 years, absence of clinical complications and psychological and learning disorders. Non-diabetic adolescent children in the same age range were either friends or islet cell antibody negative siblings of T1D subjects and served as the control (CON) group. Parental consent and child assent were obtained for all subjects prior to enrollment and after the nature of the procedures were explained. The study protocol was approved by the Human Rights Committee of the Children's Hospital of Pittsburgh (PI: D.B.) and the Human Subject Protection Committee at University of California, Los Angeles (P.I./co-P.I.: S.D. and M.G.).
Hyperinsulinemic Clamp Study
All enrolled subjects were admitted to the General Clinical Research Center at the Children's Hospital of Pittsburgh on the day before each hyperinsulinemic clamp study. Heart rate, blood pressure, and temperature were monitored throughout the study. To prevent overnight hypoglycemia, intermediate and long acting insulin were discontinued 24 hours prior to the study and only pre-meal regular or lispro insulin were administered. Then euglycemia was maintained overnight by an intravenous insulin infusion in adolescent children with diabetes while the controls received a saline infusion (Figure 4A). Blood glucose was monitored hourly by an indwelling IV catheter and maintained at 5.6 mmol/l (or 100 mg/dL). This established the baseline period of euglycemia. After the collection of baseline samples, the diabetic and CON children underwent a hyperinsulinemic hypoglycemic clamp. Regular insulin was infused at 0.1 units/kg/hr together with a variable infusion rate of 20% dextrose water via an ante-cubital vein. Arterialized blood was collected every 5 minutes from a hand or wrist vein with the hand heated by a heating pad. Serum glucose was measured every 5 minutes and a larger sample was obtained for measurements of GLUT1 transporter concentrations at five time points during each clamp. The serum glucose concentration was maintained at 3.3 mmol/l for 90 min by altering the glucose infusion rate and then returned to 5.5 mmol/L for 90 minutes. (Clamp I). A second hypoglycemic clamp study was performed only in children with T1D where the serum glucose concentration was allowed to reach a nadir of 3.0 mmol/l for 45 min after a 135 min period of euglycemia (Clamp Π). Thus two glycemic thresholds over differing durations ensued.
Figure 4. Hyperinsulinemic clamp studies in T1D and CON groups.

4A: The schematic diagram for clamp 1 and clamp 2 studies.
4B: Hyperinsulinemic clamp I study in CON children (n=8). Left y axis demonstrates the serum glucose concentration in mmol/l and right y-axis indicates the RBC GLUT1 concentrations in ng/ng protein. The x-axis shows time points B, baseline; EU, euglycemia; H45 and H90 min, after 45 and 90 minutes of hypoglycemia; and EU30, after 30 min of return to euglycemia.
4C: Hyperinsulinemic clamp I study in T1D children (n=8). The left y-axis demonstrates serum glucose in mmol/L and right y-axis indicates RBC GLUT1 in ng/ng. B, The x-axis shows time points B, baseline; EU, euglycemia; H45 and H90 min, after 45 and 90 minutes of hypoglycemia; and EU30, after 30 min of return to euglycemia
4D: Hyperinsulinemic clamp II study in T1D children (n=8). Left x-axis demonstrates the serum glucose concentration in mmol/L and right y-axis indicates RBC GLUT1 concentrations in ng/ng protein. The x-axis shows time points : B, baseline; EU1 and EU2, after 60 and 120 minutes of euglycemia; H, during hypoglycemia; and EU30, after 30 min of return to euglycemia., * p<0.0001 Serum glucose values compared to the baseline serum glucose values for each group. α p<0.001 RBC GLUT1 concentration in T1D group compared to the corresponding RBC GLUT1 concentration in the CON group.
Blood Samples
In Clamp1 the blood samples for measurements of RBC-GLUT1 concentrations were collected at 0 min (baseline, labeled B), 60 min (period of hyperinsulinemia with euglycemia, labeled EU), at two time points of 45 and 90 min of hypoglycemia (H45 and H90) and again after 30 min of euglycemia (labeled EU30) (Figure 4A). In Clamp Π baseline blood sample was collected at 0 min (labeled B) followed by 2 samples (obtained every 60 minutes, EU1 and EU2) during the 135 min hyperinsulinemic euglycemia, after 45 min of hypoglycemia (H45) and again 30 min after the recovery of euglycemia (labeled EU30) (Figure 4A). These blood samples were centrifuged at 1000 rpm at 4°C. The supernatant constituted the serum and the red blood cell pellets were harvested and snap frozen in acetone soaked dry ice and stored at −80°C until assayed.
Serum Glucose and HbA1C Measurements
Serum glucose was measured by the glucose oxidase method using the YSI analyzer (Yellow Springs, Ohio). HbA1C was measured by HPLC (Biorad, Hercules, CA), with a normal range of 4.2–6.1% (22.4–43.2 mmol/mol).
RBC Membrane Preparation
Frozen RBCs were re-suspended in 40 volumes of an ice cold hypotonic solution containing 5 mM Na2HPO4, pH 8 for 30 min and then centrifuged for 10 min at 22,000 g at 4°C [15]. The RBC membrane ghosts were pelleted and the supernatant was carefully aspirated. The centrifugation process was repeated three times and the RBC membrane ghosts were pooled and re-suspended in a Tyrode solution (137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.36 mM NaH2PO4, 12 mM NaHCO3, 2 mM CaCl2), sonicated on ice, and centrifuged at 14,000 rpm for 10 min at 4°C. The RBC membrane ghosts were subjected to a protein assay using the Bradford dye-binding method [16].
Enzyme-Linked Immunosorbent Assay (ELISA)
Initially the antigen was bound to Nunc maxisorb coated 96 well ELISA plates by suspending either the purified human GLUT1 C-terminus peptide (17 amino acids; 0–2 ng range) [17], which was synthesized by the UCLA peptide synthesizing core facility (standard curve), or the RBC membrane (10, 25, 50, and 100 ng) in 50 μl of a carbonate coupling buffer (0.3 mM Na2CO3, 0.7 mM NaHCO3 containing 0.1g of sodium azide [NaN3] in 500 ml at pH 9.6). The coated plates were dried at 37°C for 10 hr and stored overnight at 4°C [18]. After two washes with phosphate buffered saline with Tween (PBST; 138 mM NaCl, 2.7 mM KCl, 0.1% Tween at pH 7.4), the coated plates were incubated in a 5% milk powder solution in PBST for 90 min. After rinsing, primary antibody incubation was carried out for one hr at room temperature with a rabbit anti-GLUT1 C-terminus peptide antibody (1:500 dilution) that was generated by us and confirmed for isoform specificity. The plates were rinsed twice with PBST for 5 min each at room temperature, followed by incubation for one hr at room temperature with the secondary antibody (1:2000 dilution of goat anti-rabbit IgG conjugated to HRP; Chemicon International, Inc, Temecula, CA). Following two washes with PBST, the antigen-antibody complex was treated with o-phenylene diamine dihydrochloride in a total volume of 100 μL of 0.02 M citric acid in a 0.07 M solution of sodium phosphate dibasic buffer (pH 6.3), which contained 0.02% (v/v) H2O2 [19, 20]. The colorimetric reaction was terminated with 50 μL 1M HCl and the optical density assessed at a wavelength of 490 nm using an ELISA plate auto reader (Molecular Devices Corporation, Sunnyvale, CA). All standards and samples were run in triplicate.
GLUT1 ELISA Standardization
All assays included a standard curve. A representative standard curve from 6 curves is shown in figure 1. GLUT1 peptide concentrations ranging from 0 to 1 ng are depicted on the x-axis and the assessed absorbance on the y-axis. Linearity of the standard curve was confirmed and noted to have an r value of 0.99, P < 0.0001. Inter-assay variability was 7–8% (calculated from 6 standard curves above) and intra-assay variability was < 5% (calculated from multiple readings on the same plate).
Figure 1. Standard curve for GLUT1 ELISA.

The standard curve (n=6) for ELISA is shown (means ±SEM) ranging from 0.2 to 1 ng of the GLUT1 peptide plotted against the optical density at the 450 nm wave length. (r=0.99, p<0.0001)
Statistical Analysis
The data are expressed as a mean ± SEM. Two way ANOVA and Fisher PLSD test were employed for comparison of baseline values between control and T1D groups. When more than two values (60 min after hyperinsulinemic euglycemia, 30 or 90 min of hypoglycemia in Clamp I, 30 min after hypoglycemia in Clamp II, and 30 min after recovery) were simultaneously compared as in clamp studies, ANOVA models were used followed by the Fisher PLSD test to validate inter-group and inter-time comparisons. Correlations between an independent and dependent variable were accomplished by Spearman correlation coefficients.. P values < 0.05 were considered statistically significant.
Results
Demographics of the Patient Population
Baseline serum glucose (6.4±0.2mmol/L) in 72 T1D children age of 15.3 ± 0.2 years was significantly higher than in 11 age matched (15.6 ± 0.9 years) control children (5.4 ±0.3 mmol/L) (Figure 2A, p<0.0014). HbA1C in T1D children was 8.5 (69 mmol/mol) ± 0.2%, which was slightly higher than the clinic average for this population (~8.1%, 65mmol/mol) and was significantly higher compared to 4.9 (30 mmol/mol) ± 0.2% in the control children (Figure 2B, p<0.0001).
Figure 2. Serum glucose and RBC GLUT1 in T1D and CON adolescents.
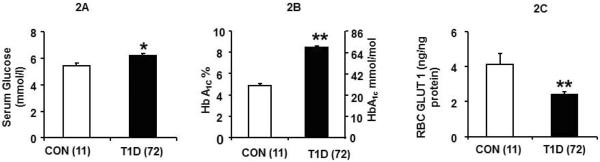
2A: Baseline serum glucose concentration (mmol/L) in CON (n=11) and T1D (n=72) groups, 2 B: HbA1C in CON and T1D groups, and, 2C: RBC GLUT1 (ng/ng protein) in CON and T1D, *p<0.0014, **p<0.0001 versus CON.
Baseline RBC GLUT1
Baseline RBC GLUT1 concentrations in the T1D group were half that of the CON group (2.4 ± 0.17 ng/ng membrane protein versus 4.2 ± 0.61 ng/ng, P < 0.0001) (Figure 2C). The baseline GLUT1 concentration in the T1D group showed an inverse relationship with HbA1C (Figure 3D, R = −0.23, p<0.05). In contrast, the RBC GLUT1 concentrations in the CON group, revealed no such correlation with HbA1C levels. (Figure 3C, R=0.0.06, p<0.85). T1D children with better diabetic control (HbA1C < 8% or <64 mmol/mol) demonstrated the highest RBC GLUT1 concentrations (Figure 3D). In both cases where HbA1C was either >8 or <8% (64 mmol/mol) in T1D adolescents, RBC GLUT1 concentrations were significantly lower and circulating glucose concentrations were significantly higher than that of the CON group (Figure 3A and3B).
Figure 3. Serum Glucose and RBC GLUT1 in CON and T1D comparing HbA1C <8% (64 mmol/mol) and HbA1C >8(64 mmol/mol).

3A:Baseline serum glucose (mmol/L) in CON (n=11), T1D with HbA1C <8% (<64mmol/mol, n=28)) and HbA1C >8% (>64 mmol/mol, n=44)), 3B: RBC GLUT1 (ng/ng protein) in CON (n=11), T1D with HbA1C <8 (<64 mmol/mol, n=28) and HbA1C >8% (>64 mmol/mol, n=44), 3C: Scatter plot between RBC GLUT1 and HbA1C in CON (n=11) showing no correlation and 3D: Scatter plot between RBC GLUT1 and HbA1C in T1D (n=72) showing lower serum HbA1C levels resulting in higher baseline RBC GLUT1 concentrations in T1D adolescents (n=72) (R=0.23, p<0.05).
Serum Glucose and RBC GLUT 1 during Hyperinsulinemic Euglycemia
Sixty minutes following the initiation of the hyperinsulinemic clamp with euglycemia, serum glucose concentrations were no different from the zero time. During this hyperinsulinemic euglycemic period the RBC GLUT1 concentrations in both the T1D and CON groups were similar to their respective baseline values (EU, Figure 4B, 4C and 4D). In the T1D group, the mean RBC GLUT1 concentration remained lower than the CON group (Fig. 4C at time point labeled EU and Fig. 4D at time points labeled EU1 and EU, p<0.001).
In control children, sixty minutes following the initiation of the hyperinsulinemic clamp with maintained euglycemia the plasma glucose concentrations were no different from the baseline (Figure 4B, time point labeled EU) and neither were the corresponding, RBC GLUT1 concentrations (Figure 4B). RBC GLUT1 concentration at point EU in CON (Figure 4B) remained higher than the corresponding point EU in the T1D group clamp I and EU1 and EU2 in Clamp II (Figure 4C and 4D, p<0.001).
Serum Glucose and RBC GLUT1 during Hyperinsulinemic Hypoglycemia
During hyperinsulinemic hypoglycemia (Clamp I), the mean serum glucose concentrations decreased from a baseline value of 5.5 ± 0.3 mmol/l to a nadir of 3.4 ± 0.1 mmol/l (P < 0.0001) in the CON group and from 7.1 ± 0.3 to 3.3 ± 0.1 mmol/l (P < 0.0001) in the T1D group, with the 90 min nadir being no different (Figures 4B and 4C, time point labeled H90). In order to assess the effect of more severe hypoglycemia, during Clamp II in T1D subjects, the serum glucose levels decreased from 6.4± 0.3 to a 45 minute nadir of 3.0 ± 0.1 mmol/l (P < 0.0001) (Figure 4D, Time point H45). Thereafter euglycemia was restored in all groups for 30 min at the time of blood sampling (time point labeled EU30).
In the T1D groups, RBC GLUT1 concentrations did not demonstrate any change during the hypoglycemia clamp I or clamp II studies (4C and 4D), the values remained lower than that of the CON group (p<0.001). In the CON group although a transient trend towards recovery of the RBC GLUT1 concentrations at H45 after the initial decrease during hyperinsulinemic euglycemia was evident, no significant change was observed, (Fig. 4B).
Discussion
We have shown that T1D adolescent children demonstrate a baseline reduction in RBC GLUT1 concentrations despite 12 hr imposition of serum glucose concentrations close to euglycemia. This decrease in RBC GLUT1 is most severe in those patients with a higher HbA1C concentration >8% (64 mmol/mol) supporting the hypothesis that, as seen in animal models, the more severe the long term hyperglycemia, the greater the suppression of RBC GLUT1 concentrations. This reduction under conditions of T1D-associated hyperglycemia may serve to protect against increased glucose transport into RBCs and perhaps other GLUT1 expressing cells including the BBB. As we could not measure BBB GLUT1 or CSF glucose concentrations, we used the RBC GLUT1 concentration as a surrogate for BBB GLUT1 concentrations. The hyperglycemia associated decrease in GLUT1 may play a role in maintaining near normal or decreased glucose transport across the RBC membrane and the BBB, thereby protecting against cellular glucose toxicity. In animal studies, chronic hyperglycemia caused a decline in brain GLUT1 concentrations [5, 6] occurring at the post-transcriptional level and involving GLUT1 mRNA stability, which relates to corresponding protein concentrations [20].
The functional significance of this reduction in RBC GLUT1 observed in T1D children is unknown from our present study. Previous studies undertaken in adults with T2D have demonstrated a significant reduction in RBC glucose transport [21, 22] despite the variable presence of a change in GLUT1 concentrations. This latter observation may perhaps be related to the chronic elevation in insulin concentrations in adults related to the obese state with increased body mass index [21, 22], as opposed to that encountered at baseline in children with T1D. In contrast, this same insulin effect on RBC GLUT1 was lacking in T1D children perhaps due to the already reduced baseline concentrations of RBC GLUT1 encountered. Further, prior adult studies in T2D and control subjects demonstrated the presence of GLUT1 on T lymphocytes attesting to the fact that GLUT1 is not unique to RBCs. T2D adults when compared to controls displayed reduced lymphocyte glucose uptake without changes in GLUT1 expression suggesting perturbations in the intrinsic activity of the transporter [23]. In a separate investigation, control subjects expressed a reduction in lymphocyte GLUT1 concentrations when maintained in vitro under both hyperglycemic and hypoglycemic conditions [24]. Our present study is the first in T1D children examining the impact of T1D on RBC GLUT1 concentrations. A previous study in adults with T1D complements our study as they observed a 21% decrease in glucose uptake in type 1 diabetics compared to the control subjects with only a 9% decrease in cerebral blood flow [25]. However, the plasma glucose and insulin concentrations were higher in diabetic patients versus controls and these observations were made from brain positron emission tomography (PET) as opposed to GLUT1 measurement in our study. Our findings of decreased RBC GLUT1 in T1D adolescents attest to the fact that chronic hyperglycemia as encountered in adult T2D may have very different consequences to that encountered with T1D in children on RBC GLUT1 concentrations. Thus, it is quite possible that the BBB GLUT1 concentrations are also decreased following a similar pattern as seen in RBCs of adolescent children in the present study.
In our present study, while in-vivo measurement of BBB GLUT1 in adolescent children is not possible, PET scan assessment of brain glucose uptake was not performed as it does not only reflect GLUT1 mediated glucose transport across the BBB. PET scans reflect total brain glucose uptake mediated by various brain GLUT isoforms and fails to distinguish between glucose transport and phosphorylation. A limitation of our study is that we could not determine whether reduced RBC GLUT1 concentrations in T1D reflected reduced GLUT1 mediated glucose transport across the BBB. Studies by others have determined changes in cognition and brain imaging in T1D individuals[26–30] Reduced BBB GLUT1 in T1D, if it exists as reflected by reduced RBC GLUT1, may mediate some of these brain functional changes.
Acute hypoglycemia failed to alter the RBC GLUT1 concentrations at both hypoglycemic levels whether over 45 min or 90 min in the T1D group. The small increase in RBC GLUT1 concentration in the CON group failed to reach significance, possibly because of the small numbers in this group. This absence of a hypoglycemia associated compensatory increase in GLUT1, if extrapolated to the BBB, may result in relatively reduced brain glucose transport when compared to the euglycemic state, and in turn can impair cognition. Thus, frequent short-term hypoglycemic episodes may have detrimental effects on cognition [12, 13, 31, 32], especially in those with very poor control, unless other compensatory events occur. Examples include an increase in brain blood flow [33, 34] or an epinephrine surge [34] which in turn increases the intrinsic activity of the GLUT1 protein [35]. It is also possible that chronic hypoglycemia is required to reproduce increases in BBB GLUT1 concentrations as seen in animals [5, 6], and acute episodes fail to have a significant effect because of inadequate time for a compensatory increase in GLUT1 synthesis or a decrease in GLUT1 degradation, which collectively would contribute towards an increased amount of GLUT1 protein. However, other proteins that were not measured in this study, such as stomatin, can influence the intrinsic activity of GLUT1 by suppressing it and affecting the glucose transporting function [4]. On the other hand, RBC's are entirely different from endothelial cells since the mature RBC does not have intracellular organelles that are necessary for synthesis, degradation, and recycling of membrane proteins. Therefore, a change in T1D GLUT1 concentrations may be secondary to a chronic hyperglycemia-associated down-regulation of GLUT1 in nascent RBCs that continue to dilute and finally dominate the total RBC pool over time, something that cannot be achieved over the duration of 90 min alone. In fetal sheep studies, a short time of 60 to 90 min proved inadequate for circulating glucose to alter brain GLUT1 concentrations as well despite the presence of intracellular organelles in these cells [36, 37].
While the T1D and CON children appear to have similar responses to an acute episode of hypoglycemia, namely no significant change in RBC GLUT1 concentrations, the combination of substrate deficiency and significantly lower amounts of GLUT1 in the T1D children at baseline could have a major detrimental effect on brain function should a similar circumstance exist at the BBB. Therefore, suppression by almost half the amount of GLUT1, may have a greater negative impact on BBB glucose transport during an episode of even mild hypoglycemia. This in turn may set the stage for a cumulative vulnerability in developing impaired cognition. Recently, recurrent hypoglycemia in children with T1D were associated with structural changes detected by diffusion tensor imaging as well as early cognitive decline [38]. The cellular changes evoked in central nervous system by recurrent hypoglycemia are not well known. A recent diffusion tensor imaging study by MRI illustrated lower values of white matter (WM) structures, specifically in the internal capsule, body of corpus callosum, right thalamus, temporal gyrus and posterior parietal lobe of young children with T1D, compared to normal controls [38]. These WM structural differences correlated with their HbA1C values while a higher time weighted HbA1C correlated with lower intellectual functioning and intelligence quotient [38]. One could postulate that long-term severe hyperglycemia could have a direct damaging effect or make the brain more susceptible to damage from frequent episodes of hypoglycemia.
We conclude that T1D in adolescent children causes a decline in RBC GLUT1 concentrations to almost half the control values. Acute episodes of hypoglycemia failed to increase these baseline RBC GLUT1 concentrations in both the control and T1D children. We speculate that the very low concentrations of RBC GLUT1 and absence of a compensatory increase may parallel that of the BBB GLUT1 concentrations, thereby setting the stage for more severe effects of acute hypoglycemia in T1D children especially those with the most poorly controlled diabetes.
Acknowledgements
We acknowledge the assistance of Lisa Rogers and Shanthie Thamotharan with setting up of ELISAs, and Hejing Wang for her biostatistical assistance. The study was funded by HD29487 PI Becker and the Children's Hospital of Pittsburgh GCRC MO1 RR 00084. SUD is supported by HD 33997, HD 25024, HD 41230, HD46979 and the National Center for Advancing Translational Sciences through UCLA CTSI Grant UL1TR000124.
Grant Support: This work was supported by grants from NIH – HD29487 (to DB) and NIH supported GCRCs at Children's Hospital of Pittsburgh (5MO1 RR 00084 to DB) and UCLA (MO1-RR00865 to MG) and NIH HD 33997 (to SUD) and National Center for Advancing Translational Sciences through UCLA CTSI Grant UL1TR000124 (to SUD).
Footnotes
Disclosure- None
References
- 1.Morgello S, et al. The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia. 1995;14(1):43–54. doi: 10.1002/glia.440140107. [DOI] [PubMed] [Google Scholar]
- 2.Devaskar S, et al. The neonatal rabbit brain glucose transporter. Brain Res Dev Brain Res. 1992;67(1):95–103. doi: 10.1016/0165-3806(92)90029-v. [DOI] [PubMed] [Google Scholar]
- 3.Mantych GJ, Sotelo-Avila C, Devaskar SU. The blood-brain barrier glucose transporter is conserved in preterm and term newborn infants. J Clin Endocrinol Metab. 1993;77(1):46–9. doi: 10.1210/jcem.77.1.8325958. [DOI] [PubMed] [Google Scholar]
- 4.Zhang JZ, Ismail-Beigi F. Activation of Glut1 glucose transporter in human erythrocytes. Arch Biochem Biophys. 1998;356(1):86–92. doi: 10.1006/abbi.1998.0760. [DOI] [PubMed] [Google Scholar]
- 5.Cornford EM, et al. Down-regulation of blood-brain glucose transport in the hyperglycemic nonobese diabetic mouse, in Neurochem Res. 1995. pp. 869–73. [DOI] [PubMed] [Google Scholar]
- 6.Maher F, Simpson IA, Vannucci SJ. Alterations in brain glucose transporter proteins, GLUT1 and GLUT3, in streptozotocin diabetic rats. Adv Exp Med Biol. 1993;331:9–12. doi: 10.1007/978-1-4615-2920-0_2. [DOI] [PubMed] [Google Scholar]
- 7.Lee DH, et al. Changes of glucose transporters in the cerebral adaptation to hypoglycemia. Diabetes Res Clin Pract. 2000;47(1):15–23. doi: 10.1016/s0168-8227(99)00107-2. [DOI] [PubMed] [Google Scholar]
- 8.Boado RJ, Pardridge WM. Glucose deprivation and hypoxia increase the expression of the GLUT1 glucose transporter via a specific mRNA cis-acting regulatory element. J Neurochem. 2002;80(3):552–4. doi: 10.1046/j.0022-3042.2001.00756.x. [DOI] [PubMed] [Google Scholar]
- 9.Klepper J, Voit T. Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: impaired glucose transport into brain-- a review. Eur J Pediatr. 2002;161(6):295–304. doi: 10.1007/s00431-002-0939-3. [DOI] [PubMed] [Google Scholar]
- 10.De Vivo DC, et al. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991;325(10):703–9. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- 11.Seidner G, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet. 1998;18(2):188–91. doi: 10.1038/ng0298-188. [DOI] [PubMed] [Google Scholar]
- 12.Ryan C, Gurtunca N, Becker D. Hypoglycemia: a complication of diabetes therapy in children. Pediatr Clin North Am. 2005;52(6):1705–33. doi: 10.1016/j.pcl.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Ryan CM, et al. Mild hypoglycemia associated with deterioration of mental efficiency in children with insulin-dependent diabetes mellitus. J Pediatr. 1990;117(1 Pt 1):32–8. doi: 10.1016/s0022-3476(05)82440-0. [DOI] [PubMed] [Google Scholar]
- 14.Cryer PE. Mechanisms of hypoglycemia-associated autonomic failure and its component syndromes in diabetes. Diabetes. 2005;54(12):3592–601. doi: 10.2337/diabetes.54.12.3592. [DOI] [PubMed] [Google Scholar]
- 15.Cloherty EK, et al. Regulation of GLUT1-mediated sugar transport by an antiport/uniport switch mechanism. Biochemistry. 1996;35(40):13231–9. doi: 10.1021/bi961208t. [DOI] [PubMed] [Google Scholar]
- 16.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 17.Mueckler M, et al. Sequence and structure of a human glucose transporter. Science. 1985;229(4717):941–5. doi: 10.1126/science.3839598. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt R, et al. An ELISA technique for quantification of surfactant apoprotein (SP)-C in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 2002;165(4):470–4. doi: 10.1164/ajrccm.165.4.2102080. [DOI] [PubMed] [Google Scholar]
- 19.Takakura Y, et al. Hexose uptake in primary cultures of bovine brain microvessel endothelial cells. I. Basic characteristics and effects of D-glucose and insulin. Biochim Biophys Acta. 1991;1070(1):1–10. doi: 10.1016/0005-2736(91)90139-y. [DOI] [PubMed] [Google Scholar]
- 20.Boado RJ. Brain-derived peptides increase blood-brain barrier GLUT1 glucose transporter gene expression via mRNA stabilization. Neurosci Lett. 1998;255(3):147–50. doi: 10.1016/s0304-3940(98)00731-9. [DOI] [PubMed] [Google Scholar]
- 21.Porter-Turner MM, et al. Relationship between erythrocyte GLUT1 function and membrane glycation in type 2 diabetes. Br J Biomed Sci. 68(4):203–7. doi: 10.1080/09674845.2011.11730351. [DOI] [PubMed] [Google Scholar]
- 22.Hu XJ, et al. The abnormality of glucose transporter in the erythrocyte membrane of Chinese type 2 diabetic patients. Biochim Biophys Acta. 2000;1466(1–2):306–14. doi: 10.1016/s0005-2736(00)00175-9. [DOI] [PubMed] [Google Scholar]
- 23.Piatkiewicz P, Czech A, Taton J. Glucose transport in human peripheral blood lymphocytes influenced by type 2 diabetes mellitus. Arch Immunol Ther Exp (Warsz) 2007;55(2):119–26. doi: 10.1007/s00005-007-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oleszczak B, Szablewski L, Pliszka M. The effect of hyperglycemia and hypoglycemia on glucose transport and expression of glucose transporters in human lymphocytes B and T: an in vitro study. Diabetes Res Clin Pract. 96(2):170–8. doi: 10.1016/j.diabres.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 25.van Golen LW, et al. Positron emission tomography measured cerebral blood flow and glucose metabolism are decreased in human type 1 diabetes. Diabetes. 2013 doi: 10.2337/db12-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Northam EA, Lin A. Hypoglycaemia in childhood onset type 1 diabetes--part villain, but not the only one. Pediatr Diabetes. 2010;11(2):134–41. doi: 10.1111/j.1399-5448.2009.00545.x. [DOI] [PubMed] [Google Scholar]
- 27.Northam EA, et al. Central nervous system function in youth with type 1 diabetes 12 years after disease onset. Diabetes Care. 2009;32(3):445–50. doi: 10.2337/dc08-1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Northam EA, Cameron FJ. Understanding the diabetic brain: new technologies but old challenges. Diabetes. 2013;62(2):341–2. doi: 10.2337/db12-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marzelli MJ, et al. Neuroanatomical correlates of dysglycemia in young children with type 1 diabetes mellitus. Diabetes. 2013 doi: 10.2337/db13-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arbelaez AM, Semenkovich K, Hershey T. Glycemic extremes in youth with T1DM: effects on the developing brain's structural and functional integrity. Pediatr Diabetes. 2013 doi: 10.1111/pedi.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan CM, Becker DJ. Hypoglycemia in children with type 1 diabetes mellitus. Risk factors, cognitive function, and management. Endocrinol Metab Clin North Am. 1999;28(4):883–900. doi: 10.1016/s0889-8529(05)70107-9. [DOI] [PubMed] [Google Scholar]
- 32.Jones TW, Davis EA. Hypoglycemia in children with type 1 diabetes: current issues and controversies. Pediatr Diabetes. 2003;4(3):143–50. doi: 10.1034/j.1399-5448.2003.00025.x. [DOI] [PubMed] [Google Scholar]
- 33.MacLeod KM, et al. The effects of acute hypoglycemia on relative cerebral blood flow distribution in patients with type I (insulin-dependent) diabetes and impaired hypoglycemia awareness. Metabolism. 1996;45(8):974–80. doi: 10.1016/s0026-0495(96)90266-8. [DOI] [PubMed] [Google Scholar]
- 34.Ryan CM, et al. Detection of symptoms by adolescents and young adults with type 1 diabetes during experimental induction of mild hypoglycemia: role of hormonal and psychological variables. Diabetes Care. 2002;25(5):852–8. doi: 10.2337/diacare.25.5.852. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu Y, et al. Effects of noradrenaline on the cell-surface glucose transporters in cultured brown adipocytes: novel mechanism for selective activation of GLUT1 glucose transporters. Biochem J. 1998;330(Pt 1):397–403. doi: 10.1042/bj3300397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das UG, et al. Time-dependent and tissue-specific effects of circulating glucose on fetal ovine glucose transporters. Am J Physiol. 1999;276(3 Pt 2):R809–17. doi: 10.1152/ajpregu.1999.276.3.R809. [DOI] [PubMed] [Google Scholar]
- 37.Anderson MS, et al. Glucose transporter protein responses to selective hyperglycemia or hyperinsulinemia in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;281(5):R1545–52. doi: 10.1152/ajpregu.2001.281.5.R1545. [DOI] [PubMed] [Google Scholar]
- 38.Aye T, et al. White matter structural differences in young children with type 1 diabetes: a diffusion tensor imaging study. Diabetes Care. 35(11):2167–73. doi: 10.2337/dc12-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]