

Abstract
DNA methylation and microRNAs (miRNAs) play crucial roles in maturation of postnatal mouse neurons. Aberrant DNA methylation and/or altered miRNA expression cause postnatal neurodevelopmental disorders. In general, DNA methylation in the 5'-flanking region suppresses gene expression through recruitment of methyl-CpG binding domain proteins (MBPs) to the cytosine residues of CpG dinucleotides. X-linked MeCP2 (methyl-CpG binding protein 2), a member of MBPs, is a methylation-associated transcriptional repressor with other functions in the central nervous system (CNS). miRNAs negatively regulate gene expression by targeting the 3'-untranslated region (3'UTR). Some miRNA genes harboring or being embedded in CpG islands undergo methylation-mediated silencing. One such miRNA is miR-7b which is differentially expressed through stages of neurodevelopment. In our present study, we focused on a canonical CpG island located in the 5'-flanking region of the murine miR-7b gene. Hypermethylation of this CpG island down-regulates miR-7b while recruiting MeCP2 to the methylated CpG dinucleotides. Meanwhile, Mecp2, a target of miR-7b, was up-regulated due to lack of restrain exerted by miR-7b during maturation of postnatal (PN) mouse neurons between PN3 and PN14. Our results indicate that miR-7b is a direct downstream gene transcriptional target while also being a negative post-transcriptional regulator of Mecp2 expression. We speculate that this bidirectional feed-back autoregulatory function of miR-7b and Mecp2 while linking DNA methylation and miRNA action maintains the homeostatic control of gene expression necessary during postnatal maturation of mammalian neurons.
Keywords: Neurodevelopment, epigenetic regulation, transcriptional control, miRNA biogenesis, DNA methylation, histone deacetylase
INTRODUCTION
Epigenetic mechanisms, including DNA methylation, histone modifications and regulatory non-coding RNAs play critical roles in neural development and maturation (Feng and Fan, 2009; Hsieh and Eisch, 2010). DNA methylation in the postnatal brain is of particular importance as brain-specific deletion of DNA methylation related machineries results in postnatal neurodevelopmental abnormalities and premature death in mice (Chahrour and Zoghbi, 2007; Nguyen et al., 2007). Generally, DNA methylation is intimately connected with histone modifications through recruitment of methyl-CpG binding domain proteins (MBPs) that suppress expression of methylation-dependent genes (Lewis et al., 1992). Once DNA sequences are methylated, they can directly repress transcription by blocking the binding of transcriptional activators to recognition DNA sequences (Watt and Molloy, 1988). Since not all transcription factor recognition DNA sequences contain CpG dinucleotides, an alternate model (Cedar and Bergman, 2009) consists of recruiting methyl-CpG binding domain proteins that associate with co-repressors, such as HDACs (histone deacetylases). This leads to forming chromatin remodeled co-repressor complexes (Buschhausen et al., 1987), which in turn stably maintain the repressed state by changing the surrounding chromatin structure (Nan et al., 1997).
X-linked MeCP2 (methyl-CpG binding protein 2), a member of MBPs, was originally identified as a transcriptional repressor in the central nervous system (CNS). MeCP2 is highly expressed in neurons and increases from early postnatal to adult stages of development (Shahbazian et al., 2002; Luikenhuis et al., 2004). The timing of Mecp2 expression with maturation of the CNS, and the delay in phenotypic effects due to developmental loss of MeCP2 suggests that MeCP2 plays a functional role in early postnatal rather than embryonic development (Chen et al., 2001). Genetic mouse models that compromise MeCP2 function demonstrate that MeCP2 is critical for normal neurobehavior (Chahrour et al., 2008). Accumulating evidence shows that loss-of-function mutations cause Rett syndrome (RTT) and duplications of the Mecp2 gene cause neurological disorders but not classic Rett syndrome (RTT) (Amir et al., 1999; Van Esch et al., 2005).
MicroRNAs (miRNAs) are a class of non-coding RNA transcripts that regulate gene expression at the post-transcriptional level. miRNAs control gene expression by binding to complementary sequences (miRNA response elements; MREs) in the 3'-untranslated region (3′-UTR) of target mRNA transcripts to facilitate their degradation and/or inhibit translation (Bartel, 2004). Although the specific mechanisms underlying miRNA regulation of neuronal development are not fully uncovered, current experimental evidence suggests that miRNAs can play a functional role during all stages of neuronal development and maturation (Fiore et al., 2008). This is necessary to provide a highly orchestrated program of gene expression critical for appropriate neuronal development and function (Smith et al., 2010). miRNA malfunction has been linked to certain neurological disorders such as Parkinson's disease (Kim et al., 2007), Huntington's disease (Johnson et al., 2008), Alzheimer's disease (Hebert et al., 2008), and Tourette's syndrome (Abelson et al., 2005).
Mature miRNAs are transcribed from corresponding miRNA genes by RNA polymerase II (Lee et al., 2004). Hence, expression of miRNAs shares the same genetic and epigenetic regulatory mechanisms including DNA methylation (Lujambio and Esteller, 2007). Although only subsets of miRNA genes either harbor CpG islands in their promoter regions or are themselves embedded within CpG islands, DNA methylation has been identified as a crucial step in microRNA biogenesis (Yu et al., 2005). Conversely, reports of microRNAs targeting DNA methyltransferases (Dnmts) 3a, 3b (Fabbri et al., 2007), Dnmt1 (Garzon et al., 2009) and Mecp2 can alter DNA methylation and expression of downstream genes (Urdinguio et al., 2010; Wu et al., 2010). Such investigations support interactions between miRNA biogenesis/function and the process of DNA methylation (Iorio et al., 2010). Further, Mecp2 expression is known to be regulated by a cluster that contains the CREB-induced miR-132 and miR-122, both miRs are known to regulate synaptic structure and activity (Klein et al., 2007; Hansen et al., 2010; Magill et al., 2010; Wanet et al., 2012). To date unidirectional influence of miRNAs on mRNAs/proteins that mediate the process of DNA methylation or miRNA(s) expression modified by promoter DNA methylation has been reported. We hypothesized that during postnatal neuronal maturation, certain miRNAs may negatively regulate gene expression required to mediate or complement while being regulated by DNA methylation themselves.
To test this hypothesis, we focused on miR-7b and Mecp2 during postnatal mouse neuronal development. We chose miR-7b which is located on chromosome 17, because it is expressed in various regions of the adult murine brain such as the hippocampus, deep layers of the neocortex, habenula, paraventricular and suprachiasmatic nuclei (Lee et al., 2006; Bak et al., 2008; Yuan et al., 2010; Herzer et al., 2012; Hansen et al., 2013), inhibits neuronal Fos translation which is a gene that reflects neuronal activity (Lee et al., 2006), and bears CpG islands in its promoter region capable of being methylated. However, not much is known about miR-7b in the postnatal brain. Additionally the role of neuronal miR-7b in post-transcriptional regulation of Mecp2 expression or the transcriptional influence of MeCP2 on miR-7b expression during postnatal brain development is unknown. We demonstrated that miR-7b is a negative regulator of Mecp2 through targeting the 3'-UTR of the mRNA. Conversely, miR-7b is repressed by MeCP2 enrichment at the methylated 5'-flanking region associated CpG island (mCpG). This dual interaction between DNA methylation and miRNA plays an important autoregulatory role during postnatal neuronal maturation between postnatal day (PN)3 and PN14.
MATERIALS AND METHODS
Brain tissues and neuronal cells
The National Institutes of Health guidelines for the care and use of mice were followed as approved by the Animal Research Committee of the University of California, Los Angeles. Brains were obtained after craniotomy from 3- (PN3) and 14-day (PN14) old C57/BL6 mice followed by careful elimination of meninges in chilled sterile Hank's Buffered Salt Solution (HBSS, pH 7.3). Single cell suspensions were prepared using MACS® Neural Tissue Dissociation Kit (Miltenyi Biotec, Auburn, CA) according to the recommended procedure as reported previously (Yu et al., 2004). Single cell suspensions were incubated with FITC-conjugated rat anti-mouse-CD90.2 antibody (eBioscience, San Diego, CA) for 15 min at 4°C followed by magnetically labeled goat anti-rat IgG microbeads (Miltenyi Biotec, Auburn, CA) for 15 min at 4°C and then sorted using MiniMACS separation (Miltenyi Biotec, Auburn, CA). To determine the purity of magnetically sorted cells, 5 × 106 stained cells were re-suspended in 4% paraformaldehyde in PBS and applied to the fluorescence activated cell sorting (FACS) Calibur flow-cytometer (Becton Dickinson, San Jose, CA) before and after sorting. Cells incubated with FITC-conjugated goat anti-rat IgG (H+L) (eBioscience, San Diego, CA) were used to set the background fluorescence. The purity of these cells and the validity of this isolation procedure have previously been shown (Chen et al., 2013). The CD90+ cells (MAP2 positive neurons) were separated from the CD90- cells (GFAP and S100β positive astrocytes) and employed in experiments as neurons.
Murine hippocampal neuron-like cell line and epigenetic reagents
Mouse hippocampal neuron-like cell line HT22 (Li et al., 1997) was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS (Invitrogen, Carlsbad, CA) (Suo et al., 2003). For study of epigenetic regulation, HT22 cells were treated with 0.5 μM 5-Aza-dC for 5 days and 200 nM of trichostatin A (TSA; Sigma-Aldrich, St. Louis, Mo) for 24 hrs which was added on the 5th day of incubation. Alternatively, the HT22 cells were treated first with 200 nM of TSA for 24 hrs alone or supplemented with 0.5 μM 5-Aza-dC during the last 12 hr of this 24 hr period.
Taqman primary or mature microRNA assay
RNAs were extracted using mirVana™ miRNA Isolation Kit (Applied Biosystems, Foster City, CA). To quantify primary or mature miR-7b, reverse transcription and qPCR were performed according to the TaqMan pri-miRNA or microRNA assay protocols (Applied Biosystems, Foster City, CA). U6 (RUN6B) was also assessed serving as the endogenous control. PCRs were conducted in three independent triplicates for each sample. Expression of pri-miR-7b or miR-7b was normalized to that of U6. The relative expression was quantified by the comparative CT method (Livak and Schmittgen, 2001).
DNA methylation analysis
Genomic DNA was isolated from mouse neuronal or HT22 cells using the DNeasy® Blood & Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer's recommendations. DNA methylation was detected by first modifying 1.5 μg of genomic DNA with sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen, Valencia, CA). CG Genome Universal Methylated and Unmethylated DNAs (MDNA, UDNA, Millipore, Billerica, MA) were also modified to serve as positive and negative controls. 100ng of the converted DNA was amplified in MethySYBR methylation specific PCR (MSP) with primers pre-confirmed to be specific for fully methylated and unmethylated miR-7b gene 5'-flanking region (MmiR-7b and UmiR-7b), which were designed using the MethPrimer software. Primers for converted (ActB) and unconverted (ActG) ß-actin special sequence (table 1) that contained no CpG sites as reported previously (Hattermann et al., 2008; Lo et al., 2009) were used as controls to correct the CT values. MethySYBR qMSPs were conducted in triplicate for each sample. CT values of MmiR-7b (methylated) and UmiR-7b (unmethylated) were normalized using corresponding standard curves and then corrected to the ActB values. Percent of fully methylated (PMR) and unmethylated (PUR) miR-7b amounts were calculated assuming MmiR-7b + UmiR-7b = 100%.
Table 1.
Primers used in this study
| Target | Sequence |
|---|---|
| MSP | |
| Methylated | 5'-GGTAGAAGTTGGGAGAGTGTTTTC-3' |
| 5'-CCGAAATCCTAATACTAAAACCGA-3' | |
| Unmethylated | 5'-AGGTAGAAGTTGGGAGAGTGTTTTT-3' |
| 5'-CCAAAATCCTAATACTAAAACCAAA-3' | |
| BSP | 5'-TATTTTGGTAGAAGGTAGAAGTTGG-3' |
| 5'-AAAAATTCCTTCCCTATACACTCTC-3' | |
| ChIP | 5'-ACGAAGAACAGAAACAAGGAGCGG-3' |
| 5'-TGCTGAAGCCTAGTCTCTTCGTGA-3' | |
| pGL2-7b-DNA Construction | |
| pGL2-7b-(L) | 5'- CGATGGTACCTGTTTAGGAAGGGCAGAGAA -3' |
| 5'- TCTAGCTAGCTTCCACATAGCACTGGCTCA -3' | |
| pGL2-7b-(S) | 5'-ATATGGTACCGGAAAGCCAGCCTCAATCCA-3' |
| 5'- TCTAGCTAGCTTCCACATAGCACTGGCTCA -3' | |
| MeCP2 MUT-3'UTR | |
| 1st | 5'-GGGAAGAAAGGAAAATATCGGCTGAATTACTTTCCAG-3' |
| 5'-CTGGAAAGTAATTCAGCCGATATTTTCCTTTCTTCCC-3' | |
| 2st | 5'-CCCCCAAAAAAAAGTCCAGAACCTTGTCGGCTGAAA |
| GCAGAGAGCATTATAATCAGGGCC-3' | |
| 5'-GGCCCTGATTATAATGCTCTCTGCTTTCAGCCGAC | |
| AAGGTTCTGGACTTTTTTTTGGGGG-3' | |
| ActB-F | 5'-TGGTGATGGAGGAGGTTTAGTAAGT-3' |
| ActB-R | 5'-AACCAATAAAACCTACTCCTCCCTTAA-3' |
| ActG- F | 5'-TGGTGATGGAGGAGGCTCAGCAAGT-3' |
| ActG- R | 5'-AGCCAATGGGACCTGCTCCTCCCTTGA-3' |
| MeCP2 RT-PCR | 5'-AGTCTTCCATACGGTCTGTGCA-3' |
| 5'-CTTCCCGCTTTTCTCACCAAG-3' | |
Bisulfite sequencing
Primers specific for modified DNA that did not contain any CpG sites in their sequence, thereby enabling both methylated and unmethylated DNA to be amplified by the same primer set, were used for bisulfite genomic sequencing PCR (BSP). The PCR products were cloned into pCR 2.1 TOPO vector (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Plasmid DNA was isolated from at least 6 clones (QIAprep spin miniprep, Qiagen, Valencia, CA), confirmed by EcoR I digestion (New England Biolab, Beverly, MA), sequenced using M13 forward primer and the Big Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and finally analyzed on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA).
SYBR Green real time qPCR assay
First-strand cDNA was synthesized from 1 μg of deoxyribonuclease treated total RNA using Superscript III First-strand synthesis system for RT-PCR (Invitrogen Life Technologies, Carlsbad, CA) as previously described (Shin et al., 2012; Chen et al., 2013). Mecp2 mRNAs were quantified using power SYBR Green-qPCR assay (Invitrogen Life Technologies) according to the manufacturer's recommendations. PCRs were conducted in independent triplicates for each sample. The gene expression amounts were standardized using an endogenous control gapdh gene as previously described (Shin et al., 2012; Chen et al., 2013).
Western blot analysis
Fifty μg of cellular proteins were analyzed by western blot analysis using the primary antibody against MeCP2 (1:500 dilution, size ~75 kDa, Sigma-Aldrich, St. Louis, Mo), MBD1 (1:1000, ~61 kDa, Millipore, Billerica, MA) and MBD2 (1:1000, ~50 kDa, Millipore, Billerica, MA). Vinculin (1:5000, Sigma-Aldrich, St. Louis, Mo) served as the internal loading control.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP was performed using the ChIP-IT™ Express and control kits (Active Motif, Carlsbad, CA) according to the manufacturer's instructions using anti-mouse MeCP2 antibody (Millipore, Billerica, MA). RNA Pol II and rat IgG (Active Motif, Carlsbad, CA) were employed as positive and negative controls respectively. ChIP-qPCR reactions were performed using SYBR® GreenER™ Assay (Invitrogen, Carlsbad, CA) with murine neuronal or HT22 cellular genomic DNA. Protein enrichment was expressed as the ratio of immunoprecipitated (IP)-DNA/input (target) to IP-DNA/input (RNA PoI II) and expressed as a -fold change in PN14 over PN3 (Chen et al., 2001).
DNA constructs used in in-vitro transfection assays
The 5'-flanking region sequences either harboring or deleting the CpG island of the miR-7b gene were inserted 5'- to the luciferase gene in the pGL2-basic plasmid using primers listed in table 1 to construct the miR-7b-luciferase reporter genes and were designated as pGL2-miR7b-(L) and pGL2-miR7b-(S) following a previously described approach (Chen et al., 2004). Precursor miR-7b clones (MmiR3270, Genecopoeia, Rockville, MD) and scrambled negative control clones (CmiR0001-MR01, Genecopoeia, Rockville, MD), wild type Mecp2-3'-UTR containing target sequences 1 and 2 of miR-7b, pEZX-Mecp2-3'UTR (Gene Accession: AF158181), and scrambled negative control were also constructed, with these 3'-UTR sequences inserted 3'- to the luciferase reporter gene (GeneCopoeia, Rockville, MD) using primers listed in table 1. The potential binding sequences of miR-7b on the Mecp2-3′-UTR were mutated using QuikChange ® XL Site-Directed Mutagenesis Kit with primers listed in table 1 (Stratagene, La Jolla, CA). Silencer® Select siRNA for Mecp2 and the scrambled negative control (siRNA) were purchased from Applied Biosystems (Foster City, CA; Target RefSeq No. = AF15818). These recombinants containing the desired DNA constructs were confirmed by full-length sequencing.
In-vitro DNA methylation
pGL2-miR7b DNA constructs were methylated in-vitro using M. Sss I (4 U/μg DNA) (New England, Beverly, MA) in the presence of 160 μM S-adenosylmethionine (SAM) for 16 hrs. The methylated DNA redesigned as pGL2-miR7b was digested with restriction enzymes either Sal I which is blocked by M. Sss I or BamH I which is not blocked by M. Sss I. Sensitivity to BamHI with resistance to Sal I indicates efficient DNA methylation.
Luciferase reporter assay
pGL2-M-Mir7b (methylated) or pGL2-U-Mir7b (unmethylated) was transiently transfected into wild type HT22 cells. pRL-TK was co-transfected as the transfection efficiency control. Methylated and unmethylated pGL2-basic was used as the respective negative controls. In the case of 3'-UTR Luciferase reporter assay, vectors containing Mecp2 WT-3'-UTR or MUT-3'-UTR were transfected. Seventy-two hours after transfection, Luciferase activities were quantified using a Dual-Luciferase® Reporter Assay System (Promega, Madison, WI) following the manufacturer's recommendations.
siRNA-mediated knockdown experiments
To attain RNAi, 5×106 freshly seeded HT22 cells were transfected with targeting Mecp2-siRNA, miR-7b inhibitor (Thermo Fisher Scientific, Lafayette, Co.) or the scrambled negative control at a final concentration of 200 nM in the case of Mecp2-siRNA or 25 nM for miR-7b inhibitor, using HiPerfect Transfection Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Cells were harvested at 48 hrs (for RNA extraction) and 72 hrs (for protein studies) after transfection.
Stable overexpression of miR-7b in HT22 cells
Pre-miR-7b or pre-miR-neg precursors were transfected into HT22 cells in 35-mm dishes using Lipofectamine™ LTX and Plus Reagent (Invitrogen. Carlsbad, CA). Twenty-four hours post-transfection, cells were reseeded into 100-mm dishes at various cell densities in DMEM containing 2.0 μg/mL of puromycin for 7 days followed by maintenance with 1.0 μg/mL of puromycin. The stable cell lines established were designated as HT22-7b and HT22-7b-neg. These cells were transiently transfected with a green fluorescent protein (GFP) reporter construct with the 3'-UTR containing the seed region complementary to miR-7b. This was accomplished using the multi-cloning site (MCS) containing miRNA SelectTM pMIR-GFP Reporter System (Cell BioLabs Inc., San Diego, CA) and following the manufacturer's experimental protocol.
Statistical analysis
Data analysis was performed using SigmaStat 3.5 (Chicago, USA). Student's t-test when two groups were compared and ANOVA followed by post-hoc Fisher's PLSD test when more than two groups were compared were employed. In addition, Pearson correlations were performed to determine correlation coefficients. P values of <0.05 were assigned significance.
RESULTS
Decreased miR-7b expression was associated with hypermethylation of the 5'-flanking region associated CpG island in postnatal mouse neurons
To assess miR-7b expression during maturation of postnatal mouse neurons, isolated mouse neurons at PN3 and PN14 (n=6 each) were used and miR-7b expression was measured using the Taqman miRNA assay with normalization to U6. MiR-7b mRNA decreased 7.8-fold in PN14 versus PN3 neurons (p<0.01) (Fig. 1A).
Figure 1. Decrease of miR-7b during maturation of postnatal mouse neurons and methylation of the CpG island within the 5'-flanking region and its correlation with miR-7b gene expression in postnatal murine neurons.

Neurons were isolated using MACS® technology with CD90.2 antibody from 3-day and 14-day old C57/BL6 mice (PN3 vs PN14, n=6 each). Enrichment of neurons was assessed by fluorescence activated cell sorting (FACS). Expression of miR-7b was measured using Taqman microRNA assay in triplicate for each sample with U6 serving as an internal control. (A) Top panel, representative gel photograph of miR-7b seen in PN3 and PN14 mouse neurons. Bottom panel, miR-7b concentrations were calculated and expressed in arbitrary units to display the age-related difference in postnatal murine neurons. Data are depicted as mean ± S.E.M from three independent experiments, each run in triplicate for each sample, **p<0.01. (B) A schematic representation of the 5′-flanking region of the miR-7b gene including the predicted proximal CpG island that contains 9 CpG sites and predicted transcription start site (TSS) located in chromosome 17. (C) DNA methylation detected by MSP. Lanes labeled M and U denote products amplified by primers recognizing methylated and unmethylated sequences. (D) Percent of fully methylated reference (PMR) values of miR-7b 5'-flanking region's CpG island assessed by MethySYBR qMSP. Data are depicted as mean ± SEM of three independent experiments, **p<0.01 when compared to PN3. (E) Representative bisulfite sequencing of the miR-7b gene CpG island within the 5'-flanking region from postnatal neurons. CpG dinucleotides are indicated by red (methylation) and green (unmethylated) arrow heads. (F) Average methylation frequency of a total of 9 CpG dinucleotides in PN3 and PN14 neurons was calculated from six clones of each sample, *p<0.05. (G) A distinct decrease in miR-7b expression is consistent with hypermethylation of this CpG island within PN14 murine neurons when compared to PN3. (H) The miR-7b expression and PMR were directly related in PN14 neurons using 2ΔΔCt methodology. Inverse correlation of miR-7b expression and the 5'-flanking region-associated CpG island methylation confirmed by Pearson's correlation analysis (n=6, correlation coefficient=−0.882, p<0.01) in postnatal murine neurons.
To investigate whether DNA methylation played a role in reduced miR-7b expression in PN14 neurons, we first attempted to detect the presence of CpG islands in the 5'-flanking region of the miR-7b gene. This was accomplished with a database by screening the chromosome locus (chr17:56242988-56243098) where the miR-7b gene is located (http://genome.ucsc.edu/). The promoter and transcriptional start site (TSS) were predicted using the Neural Network Promoter Prediction tool (http://www.fruitfly.org/seq_tools/promoter.html), and CpG island(s) were analyzed with the Methprimer program (http://www.urogene.org/methprimer/). This search resulted in discovering two canonical CpG islands spanning −1120 to −1003 bp (~118 bp) and −696 to −571 bp (~126 bp) regions located in the 5′-flanking region of the miR-7b gene. We primarily focused on the proximal CpG island that contained 9 CpG sites (Fig. 1B).
We next examined the methylation status of this miR-7b gene associated CpG island employing methylation specific PCR (MSP), MethySYBR PCR and bisulfite sequencing. Using MSP, both methylated and unmethylated amplification product bands were detected for each sample (Fig. 1C). Average values of PMR (percent fully methylated) derived from the MethySYBR PCR experiments were 24.7% in PN3 and 58.8% in PN14 neurons (as a percent of the GC reference standard, Fig 1D, p < 0.01). Furthermore, average methylation frequency of individual CpG dinucleotides calculated from the bisulfite sequencing experiments undertaken in PN3 and PN14 neurons displayed 32.3% and 57.9%, respectively (Fig. 1E & F, p < 0.05). Collectively our studies support progressive increase in methylation of the 5'-flanking region associated CpG island of the miR-7b gene during postnatal mouse neuronal maturation.
To examine the association between CpG methylation status and miR-7b gene expression, the 2−ΔΔCt values of miR-7b expression obtained in the Taqman microRNA assay (Fig. 1G) and MethySYBR based calculations of the CpG island PMR (Fig. 1G) were subjected to the Pearson's correlation analysis. An inverse correlation was found between decreased miR-7b expression and increased methylation of the CpG (mCpG) island during postnatal neuronal maturation (n=6+6, correlation coefficient = −0.882, p<0.01 Fig. 1H).
Methylation of the 5'-flanking region associated CpG island behaves as a cis-element functionally suppressing miR-7b expression
To determine the functional significance of the inverse association between mCpG and miR-7b expression during postnatal neuronal maturation, we transfected the two DNA constructs, pGL2-7b-(L) that contained and pGL2-7b-(S) that did not contain the canonical CpG island, and the pGL2-basic negative control (Fig. 2A) into HT22 neuron-like cells. Luciferase reporter activity as a read out of gene transcription was assessed in the case of both these miR-7b 5'-flanking region-luciferase DNA constructs in the native unmethylated state. Relative luciferase activity decreased dramatically in the methylated pGL2-miR7b-(L) that contained the CpG island versus the unmethylated counterpart (Fig. 2B, p < 0.01). However, no such change was observed upon methylation of the pGL2-miR7b-(S) versus the unmethylated counterpart (Fig. 2B, p>0.05). These results support a cis-acting functional role for the CpG island within the 5'-flanking region of the miR-7b gene. Methylation of this CpG island regulates miR-7b gene expression in the neuron-like HT22 cells.
Figure 2. Regulation of miR-7b expression by DNA methylation of the CpG island in the 5'-flanking region of the gene.

(A) A schematic representation of the truncated form of the miR-7b 5'-flanking region, TSS=transcriptional start site. The 5'-flanking region fragment of the miR-7b gene harboring [pGL2-7b-(L)] or deleting [pGL2-7b-(S)] the region containing the CpG island (black box) was cloned into pGL2-basic plasmid containing the luciferase (Luci) reporter gene and were used in transient transfection experiments. (B) miR-7b gene-luciferase reporter assay. Both miR-7b-(L) and miR-7b-(S) constructs drove the expression of the reporter gene (#p<0.01 when compared to the pGL2-control (con) vector that did not contain mir-7b sequences). After the constructs were methylated invitro by methylase M. Sss I in the presence of SAM, the relative luciferase activity decreased dramatically in pGL2-m7b-(L) (**p<0.01) with little change in pGL2-m7b-(S) (p>0.05) when compared to the respective wild type unmethylated counterparts. The relative luciferase activity was normalized to pRL-TK activity which served as a transfection control followed by correction using the pGL2-basic control. Data are represented as mean ± S.E.M of the six independent experiments with each sample performed in triplicate.
Recruitment of MeCP2 enriching the 5'-flanking region associated CpG island of the miR-7b gene occurs with postnatal mouse neuronal maturation
Similar to previous reports in whole brain (Skene et al., 2010), we detected increased Mecp2 expression in isolated and purified PN14 when compared to PN3 mouse neurons (Chen et al., 2013). Mecp2 mRNA measured by SYBR Green qPCR (11.2-fold, p<0.01, Fig. 3A) and MeCP2 protein (3-fold, p<0.05, Fig. 3B) assessed by Western blotting revealed an increase at PN14 versus PN3.
Figure 3. The up-regulation and recruitment of MeCP2 to the CpG island in the 5'-flanking region of the miR-7b gene during postnatal maturation of murine neurons.

Mecp2 mRNA expression in postnatal murine neurons was detected by SYBR green - qPCR assay for mRNA (A) and Western blot analysis for protein (B), the inset demonstrates representative blots with MeCP2 (above) and vinculin (internal control; below). (C & D) Representative gels showing the ChIP assay which was employed for detecting MeCP2 enrichment of the CpG island in the 5'-flanking region of the miR-7b gene with RNA Pol II serving as an unrelated antibody control and rat IgG (Active Motif, Carlsbad, CA) serving as the negative control. Input=positive PCR control (C). ChIP-qPCR was performed using SYBR® GreenER™ Assay (Invitrogen) to detect genomic DNA. MeCP2 enrichment was expressed as a ratio of immunoprecipitated (IP)-DNA/input (target) to IP-DNA/input (RNA PoI II) at PN14 in comparison to that at PN3 to calculate the change expressed in arbitrary units (D). In A, B and D, data are depicted as the mean ± S.E.M from 3 independent experiments, each sample run in triplicate. *p< 0.05 and **p< 0.01 when compared to PN3. (E) Recruitment of MeCP2 to the CpG island in the 5'-flanking region is positively correlated with miR-7b DNA methylation at the same region (Pearson's correlation analysis, n=12, correlation coefficient = 0.998, p<0.01). (F) Recruitment of MeCP2 is negatively correlated with miR-7b expression (Pearson's correlation analysis, n=12, correlation coefficient = −0.889, p<0.01).
To investigate whether increased expression was associated with increased recruitment of MeCP2 to mCpG of miR-7b gene, ChIP analysis was undertaken which revealed 7-9-fold increased MeCP2 associated with mCpG of miR-7b in PN14 versus PN3 neuronal cells (p<0.01, Fig. 3C & D). Pearson's correlation analysis demonstrated a direct correlation between recruitment of MeCP2 at the CpG island and methylation of this region in the miR-7b gene (n=6+6, correlation coefficient = 0.998, p<0.01; Fig. 3E) and a negative correlation between MeCP2 recruitment to mCpG and miR-7b expression (n=6+6, correlation coefficient = −0.889, p<0.01; Fig. 3F) during postnatal murine neuronal development. These correlational findings are consistent with the possibility that MeCP2 recruitment to the CpG island contained in the 5'-flanking region of the gene regulates miR-7b expression.
Recruitment of MeCP2 to the methylated CpG island suppresses miR-7b expression in HT22 neuronal cells
The findings consistent with inhibition of miR-7b expression related to hypermethylation and enrichment of MeCP2 at the 5'-flanking region of the gene during postnatal neuronal maturation was functionally validated employing HT22 neuron-like cells. When HT22 cells were treated with 5-Aza-dC (demethylating agent) for 5 days alone or in combination with TSA (histone deacetylases inhibitor) over 24 hrs, recruitment of MeCP2 to the CpG island in the miR-7b gene was completely abrogated (p<0.01, Fig. 4A). Consequently, the expression of mature miR-7b was enhanced markedly by 3.4-fold and 3.7-fold respectively with pretreatment of HT22 cells with 5-Aza-dC either by itself or in combination with TSA added subsequently (p < 0.05 each, Fig. 4B). This increase in miR-7b was concomitant with diminished Mecp2 expression at both the mRNA and protein level when HT22 cells were treated with either 5-Aza-dC alone or in combination with TSA (Fig. 4C, p<0.01).
Figure 4. Recruitment of MeCP2 at the methylated CpG island in the 5'-flanking region and its effect on miR-7b expression in HT22 cells.

(A) MeCP2 recruitment assessed by ChIP assay was depleted at the CpG island of the miR-7b gene in HT22 cells pre-treated with epigenetic regulating reagents, 5-Aza-dC (5-Aza) and 5-Aza + trichostatin A (TSA). Inp = Input without any antibody, IgG = non-specific IgG negative control and RNA pol II an unrelated antibody served as another control (B) Mature miR-7b expression measured by Taqman microRNA assay in HT22 cells treated with epigenetic regulating reagents. HT22 cells were treated with 5-Aza-dC (0.5 μM) for 120 hrs alone or supplemented with TSA (200nM) during the last 24 hrs. The inset demonstrates a representative gel showing mature miR-7b with U6 (internal control) bands with 25 bp markers in the first lane. Data in the graph below are depicted as the mean ± S.E.M of three independent experiments, *p<0.05 compared to the untreated HT22 cells (CON). (C)Mecp2 mRNA and protein were decreased in HT22 cells pre-treated with epigenetic regulation reagents. HT22 cells were treated with 5-Aza-dC (0.5 μM) for 120 hrs alone or supplemented with TSA (200 nM) during the last 24 hrs. The inset demonstrates western blots showing the MeCP2 protein (top panel) and vinculin (bottom panel) the internal loading control. Mecp2 mRNA measured by SYBR green qPCR assay is depicted as mean ± S.E.M of three independent experiments shown in the bar graph below, **p<0.01 compared to the wild type untreated HT22 cells (CON). (D) Demethylation of the CpG island in the 5'-flanking region of the miR-7b gene in response to 5-Aza (0.5 μM over 48 h) pre-treatment when compared to untreated CON measured by MSP, n=6 in each group, *p<0.0001 in the case of methylated (M) or #p<0.0001 in the case of unmethylated (U) PCR product versus CON (E) MeCP2 protein in arbitrary units (graph) and protein (inset: top panel shows MeCP2 protein and bottom panel vinculin, the internal loading control) concentrations diminished in response to 5-Aza (0.5 μM over 48 h), n=6 in each group, *p<0.0001 compared to the untreated CON cells. (F) and (G) Primary (Pri-) miR-7b (F) and mature miR-7b (G) expression increased (2.5- and 4-fold respectively) in HT22 cells pre-treated with TSA (200 nM for 24 h) alone or supplemented with 5-Aza-dC (0.5 μM) during the last 12 hr of the 24 hr period (3.5- and 10-fold respectively). Data are depicted as the mean ± S.E.M of 5 to 6 independent experiments. *p<0.01 versus the wild type-HT22 cells (CON). (H) and (I)Mecp2 mRNA (H) and MeCP2 protein (I) concentrations, both in arbitrary units (inset: top panel shows MeCP2 protein bands and bottom panel the vinculin protein bands as the internal loading control) are shown in response to TSA (200 nM for 24 h) or TSA+5-Aza (0.5 μM added during the last 12 h of the 24 h treatment period), n=6 in each group, *p<0.0001 versus the untreated control (CON) HT22 cells.
Next we assessed whether 5-Aza-dC treatment actually demethylated the CpG island within the 5'-flanking region of miR-7b gene by MSP using primers specific for methylated and unmethylated sequences (Table 1). While untreated control HT22 cells demonstrated the unmethylated (U) and methylated (M) DNA fragments containing the CpG island, 5-Aza-dC treatment increased the unmethylated (p<0.0001) while decreasing the methylated (p<0.0001) fractions when compared to the corresponding counterparts in untreated control HT22 cells (Fig. 4D). Along with this decrease in methylation of the CpG island within miR-7b gene, a drastic reduction in MeCP2 protein expression was observed (p<0.0001, Fig. 4E). Additionally, demethylation of the CpG island within the 5'-flanking region of miR-7b gene was associated with a reduction in MeCP2 protein concentrations, suggesting a relationship. These observations collectively support a functional role for recruitment of MeCP2 to mCpG island in transcriptionally regulating miR-7b.
Besides DNA methylation and recruitment of MeCP2, histone deacetylases are also known to transcriptionally suppress gene expression by modifying chromatin structure. We therefore examined whether initial inhibition of histone de-acetylases with TSA independent of demethylation or with subsequent demethylation by 5-Aza-dC would contribute towards primary and mature miR-7b expression in these neuron-like HT22 cells. We observed a 4-fold trend towards an increase with TSA (p=0.37) and a 10-fold increase with TSA+5-Aza-dC in primary miR-7b (Fig. 4F, p<0.008) and a 3-fold increase in mature miR-7b expression under both circumstances (Fig. 4G, p<0.01 each) compared to control cells. This increase in miR-7b expression was associated with a reciprocal decrease in Mecp2 mRNA (p<0.0001, Fig. 4H) and MeCP2 protein (p<0.0001, Fig. 4I) concentrations. These results support a heightened role for HDACs with methylation of the CpG island in suppressing transcription of primary miR-7b and expression of mature miR-7b, which in turn enhances Mecp2 expression and MeCP2 protein concentrations.
This effect of MeCP2 on transcriptional control of miR-7b was further investigated by specifically knocking down its expression by a siRNA approach. Following transfection of HT22 cells with a siRNA targeting Mecp2 for 72 hours, the recruitment of MeCP2 to the CpG island of the miR-7b gene was eliminated by 72.7% (p <0.01, Fig. 5A) while a 6-fold increase in miR-7b expression was observed in comparison to the wild type and siRNA-negative control (Fig. 5B). In contrast, when an inhibitor targeting miR-7b was added to HT22 cells, in the presence of an achieved 80% reduction in endogenous miR-7b expression (p<0.0001, Fig. 5C), a 40% increase in Mecp2 mRNA (p<0.05, Fig. 5D) and protein (p<0.02, Fig. 5E) concentrations was observed. These findings collectively illustrate an important functional trans-role for MeCP2 in DNA methylation-mediated repression of miR-7b transcription and expression.
Figure 5. Mecp2 knockdown and miR-7b inhibition – effect on Mecp2 and miR-7b expression.

(A) ChIP assay (inset demonstrates representative gels, bar graph the qPCR results in arbitrary units) demonstrates siRNA-mediated (Mecp2-siR) knockdown of MeCP2 abrogating its recruitment to the CpG island of the miR-7b gene compared to CON (untransfected cells) and scrambled sequences (siRNA-neg). Input = without any antibody, IgG = negative control in the presence of a non-specific antibody, and RNA pol II = in the presence of another unrelated antibody. Data shown as mean±SEM, n=3 each, **p<0.01 versus the CON and siRNA-neg. (B) Expression of miR-7b increased in HT22 cells pre-treated with Mecp2 specific targeted siRNA (Mecp2-siR) versus CON (untransfected) and scrambled sequences (siR-neg). n=3 each, *p<0.01. (C–E) miR-7b inhibitor (25 nM for 72 h) transfections diminished endogenous mature miR-7b concentrations shown in arbitrary units, n=6 each, *p<0.0001 versus CON cells (C), but increased Mecp2 mRNA, n=6 each, *p<0.05 versus CON cells (D), and MeCP2 protein (inset: representative western blots, top panel shows MeCP2 and bottom panel vinculin) quantified and depicted as arbitrary units and shown in the graph below, n=6 each, *p<0.02 versus CON cells (E), (F) 3'-UTR luciferase reporter assay. Relative luciferase activities were reduced in HT22 cells transfected with Mecp2-3'-UTRs, i.e. 1st and 2nd constructs separately or combined together with pre-miR-7b construct. After the conserved targeted 3'-UTR sequences of the miR-7b gene were mutated (Mecp2-3'-UTR-mut), the relative luciferase activity of the reporter gene was restored. Relative luciferase activities (in light units) are normalized to transfection efficiency by calculating ratios of firefly to renilla (pRL TK) luciferase activities.
Mecp2-3'UTR is a functional target of miR-7b
Mecp2 was predicted as one of the high-scoring candidate genes for a miR-7b target by four algorithms (TargetScan, PicTar, miRanda, miRbase Target). As shown in table 2, the Mecp2-encoded mRNA contains two 3'-UTR elements spanning dinucleotides +899 to +905 and +6371 to +6377 bp which are stringently complementary to the seed sequence of miR-7b. This information is indicative of a high probability of Mecp2-3'-UTR being the target sequence of miR-7b. To investigate whether miR-7b functionally recognizes Mecp2-3'UTR, expression clones of Mecp2-3'UTR containing 1st (+899 to +905 bp) and 2nd (+6371 to +6377 bp) targeting sequences were separately constructed 3'- to the luciferase reporter gene and transfected into HT22 cells along with pre-miR-7b. Pre-miR-7b co-transfection led to a 21.94% and 38.64% reduction in luciferase activity related to the sequences of the Mecp2-3′-UTR recombinants compared to corresponding negative control (MOCK) without the Mecp2-UTR (Fig. 5F, p < 0.05). Furthermore, co-transfection of the two Mecp2-3'-UTR sequences together with pre-miR-7b into HT22 cells reduced luciferase activity by 34.3% (p < 0.05). In contrast, the relative luciferase activity was restored following mutation of the conserved targeting regions of miR-7b binding in the Mecp2-3'-UTR (Fig. 5F). These experiments supported Mecp2-3'-UTR as a functional target of miR-7b, and indicated the 2nd targeted sequences of Mecp2-3′UTR to play a more pronounced role than that of the 1st target.
Table 2.
Bioinformaticprediction of miR-7b complementary to two 3' UTR elementsin MeCP2-encoded mRNA

|
Exogenous miR-7b reduced Mecp2 expression in HT22 neuronal cells
To determine whether miR-7b affects endogenous Mecp2 expression, we transfected miR-7b precursor or scrambled negative control into HT22 cells. Since inhibition of gene expression by miRNAs may be mediated by target mRNA degradation or translational suppression, Mecp2 mRNA and protein were examined at 48 hrs and 72 hrs after transfection, respectively. Fig. 6A shows that exogenous pre-miR-7b transfection dramatically reduced Mecp2 mRNA (2.7-fold) and MeCP2 protein concentrations versus the wild type HT22 cells and those cells transfected by the scrambled negative control. This observation indicates that reduction of Mecp2 mRNA levels by miR-7b occurs due to mRNA degradation and suppression of translation, and that miR-7b is a negative regulator of post-transcriptional processes regulating Mecp2 expression.
Figure 6. Mecp2 expression in transiently overexpressed and miR-7b expression and function in stably transfected HT22 cells.

(A) Mecp2 mRNA quantified by SYBR green-qPCR assay (in arbitrary units in the graph below) and protein by Western blots (inset: top panel shows MeCP2 and bottom panel vinculin) is shown in HT22 wild type (CON), transiently transfected with scrambled pre-miR-7b (miR-neg; negative control) and pre-miR-7b (miR-7b). n=6 per group and *p<0.02 versus wild type and pre-miR-7b negative control. (B) miR-7b expression assessed by Taqman microRNA assay in HT22 cells (CON = untransfected cells, (−) = miR-7b negative contains scrambled miR transfected cells, and (+) = miR-7b stably transfected cells) are shown along with U6 mRNA (internal control). (C) Quantification of Taqman microRNA assay of miR-7b is shown in HT22 untransfected control (CON), miR-7b negative (neg) contains scrambled miR transfected cells (−) and miR-7b stable transfectant (+) cells. N=6 per each cell type examined with each experiment performed in triplicate. **p<0.01 versus CON and (−) cells. (D) miR-7b-negative (neg) containing scrambled miR stably transfected cells (right panel) and miR-7b (+) stable transfectants (left panel) were transiently transfected with GFP reporter construct with the 3'-UTR containing seed region complementary to miR-7b. A diminution of GFP is seen in the presence of miR-7b (left panel) versus scrambled SiRNA negative control (right panel, arrows show the GFP positive HT22 cells).
We next examined the stable transfected HT22 mouse neuron-like cell lines created by us employing puromycin resistance to sub-select cellular clones containing miR-7b transfectants. Assessment of miR-7b expression by Taqman microRNA assay revealed an 8.2-fold increase over WT or the scrambled miRNA-7b (miR-7b-neg; negative control) transfected cell line (Fig. 6B & C, p < 0.01). To establish functionality of miRNA-7b, we transfected this cell line with a GFP (green fluorescent protein) construct containing a 3'-UTR region that seeds miR-7b, and noted a decline of GFP in this cell line (Fig. 6D). Since the alignment score between the 3'-UTR of Mecp2 mRNA and miR-7b is 168 with a PhastCons score of 0.575 (Table 2), we next examined Mecp2 mRNA expression (134 bp) in this HT22-miR-7b cell line and observed a decline versus that in CON or the scrambled miR-7b negative control cell line (miR-7b-neg) (Fig. 7 A & B; p < 0.024). This decline was 2.6-fold when assessed by the SYBR green qPCR assay (Fig. 7C, p < 0.05). Western blot analysis revealed a parallel decrease in Mecp2 protein concentrations in miR-7b stably transfected cell line (Fig. 7D).
Figure 7. miR-7b inhibits Mecp2 expression in miR-7b over-expressing stably transfected HT22 cells.

(A) Mecp2 and gapdh (internal control) mRNAs are shown in a representative gel from HT22 cells that are untransfected cells (CON), miR-7b negative (neg) contains the scrambled miR stably transfected cells and miR-7b stable transfectants. (B) Quantification of Mecp2 mRNA assessed by semi-quantification of the reverse transcription-PCR amplification product by densitometry and normalized to gapdh is shown in these three cell types, n=6 each with each experiment performed in triplicates; *p<0.024. (C) Quantification of Mecp2 mRNA assessed by SYBR green – qPCR assay is shown. A 2.6 fold decrease in miR-7b stable transfected versus the miR-7b-neg containing the scrambled miR transfected (miR-7b-neg) HT22 cells is seen. N=6 per group; *p<0.05 versus the mir-7b-neg cells. (D) Representative Western blots demonstrating MeCP2 (top panel) and vinculin (vin; internal control, bottom panel) in HT22 untransfected cells (CON), miR-7b negative (neg) contains the scrambled miR stable transfected cells and miR-7b stable transfected cells.
DISCUSSION
We have for the first time demonstrated auto-regulation between Mecp2 and a specific miRNA-7b during postnatal murine neuronal maturation. This observation is novel implying that tight regulation is necessary during this critically important process of neurodevelopment. Any disruption of this well orchestrated molecular process may have the potential of causing neurodevelopmental disorders. miRNAs in general and MeCP2 have basic roles in vertebrate organogenesis, disruption of either may underlie pathogenesis of disease. The biological importance of neuronal miRNAs is supported by the observed disruptive effects in mice after genetic inactivation of Dicer, a key enzyme in miRNA biogenesis (Kawase-Koga et al., 2009). The biological role of MeCP2 within mouse neurons is validated by loss-of-function mutations or duplications (Amir et al., 1999; Chen et al., 2001; Van Esch et al., 2005), both extremes causing aberrant neurobehavior comparable to Rett's syndrome, a form of autism spectrum disorder (Amir et al., 1999; Van Esch et al., 2005; Tarquinio et al., 2012). Various mutations of MeCP2 have also been described in girls who present with Rett's syndrome (Amir et al., 1999; Bartholdi et al., 2006). However, while miRNAs such as the CREB-induced miR-132 and miR-212 have been shown to unidirectionally and post-transcriptionally regulate Mecp2 expression (Klein et al., 2007; Hansen et al., 2010; Magill et al., 2010; Wanet et al., 2012), there is little information related to bidirectional autoregulation where miRNAs dictate the expression and thereby the function of MeCP2, which in turn affects miRNA biogenesis.
In addition to MeCP2, there is evidence from other systems that histone deacetylases (HDACs) either by themselves or synergistically with DNA methyltransferases can also block microRNA expression (Ghoshal et al., 2002; Wendtner, 2012). In our present study we observed that prior demethylation of miR-7b (over 5 days) was not a requisite for enticing additional HDAC induced suppression of miR-7b expression. In contrast, initial inhibition of HDACs alone enhanced pri- and mature forms of miR-7b expression, although the pri-miR-7b expression was further enhanced by subsequent DNA demethylation. These results suggest that histone modifications affecting conformation of chromatin along with DNA methylation regulate miR-7b biogenesis. We speculate that these two processes may not be inter-dependent but individually important in suppressing miR-7b expression.
In our present study, we primarily focused on miR-7b, which is predominantly expressed in the central nervous system and is one of the miRNAs regarding which limited information is available (Aboobaker et al., 2005; Bak et al., 2008). While mir-7b is expressed in the adult murine cortex, hippocampus and hypothalamus (Zhou et al., 2009; Herzer et al., 2012), and is induced in the latter location by chronic hyperosmolar stimulation (Lee et al., 2006; Smith et al., 2010), not much is known during postnatal brain development. Subsequently, differential expression of multiple miRNAs including miR-7b was noted in neurodegenerative disorders (Lehmann et al., 2012), implying a possible connection between miR-7b expression and the process of neurodegeneration.
However little is known about the downstream mediators of miR-7b. More recently a physical association was observed between miR-7b containing ribonucleoprotein complexes (miRNPs) in a human neuronal cell line as the mechanism by which miRs inhibit protein translation from the mRNAs (Nelson et al., 2004). In addition to these studies, miR-7b emerged as a player in regulating neural stem cell proliferation and differentiation. Overexpression of miR-7b reduced neural stem cell proliferation but increased neural differentiation. These studies though limited demonstrate that miR-7b plays a role in inhibiting neural stem cell proliferation while promoting differentiation through different stages of development. These two processes are of great importance during neurodevelopment encountered in the postnatal stage. These studies provided an impetus for our current study of miR-7b in the postnatal brain.
Based on this information, we embarked on the present investigation to determine first if miR-7b expression was affected by DNA methylation during postnatal neuronal maturation. We chose PN3 and PN14 since murine neuronal maturation (differentiation) which includes synaptogenesis and pruning occurs during the first three weeks of postnatal life, with peak maturation noted between PN10 and PN14 (Viberg et al., 2008). These two stages, namely PN3 depicted a pre-maturational phase and PN14 a peak neuronal maturational phase, allowing us to compare these two developmental stage specific differences between miR-7b expression and its regulatory role of Mecp2 expression. Initially, we observed a reduction in neuronal miR-7b expression with advancing postnatal age. Whether this postnatal decrease in miR-7b expression perpetuates neuronal cell proliferation versus differentiation, the former process being necessary for the process of synaptogenesis that is active at PN14 is unknown and requires future investigation.
The reciprocal association between decreasing miR-7b and increasing MeCP2 between PN3 and PN14 postnatal mouse neurons prompted us to explore the underlying molecular mechanism behind this observation. Beginning with in silico predictions and followed by the use of various experimental approaches, we have shown that expression of miR-7b is regulated by methylation of one canonical CpG island within the 5'-flanking region of the miR-7b gene. Increasing MeCP2 recruitment to these methylated CpG dinucleotides inhibits miR-7b gene expression. MeCP2 on the other hand was observed to be negatively regulated by miR-7b via targeting the Mecp2 mRNA 3'-UTR. This reciprocal interaction between miR-7b and MeCP2 perhaps caters to the homeostatic balance needed during maturation of postnatal mouse neurons. The age-dependent increasing methylation of a CpG island and recruitment with enrichment of MeCP2 restrains miR-7b expression, which in turn reduces the post-transcriptional inhibition of Mecp2 expression. As a result Mecp2 expression is enhanced, that then inhibits miR-7b gene expression, thereby constituting auto-regulation. Our present study demonstrates miR-7b, behaving as a direct downstream transcriptional target and at the same time functioning as a negative regulator of Mecp2 gene expression during developmental maturation of murine neurons (Fig. 8).
Figure 8. Schematic representation showing miR-7b behaving as a downstream direct transcriptional target while serving as a negative regulator of Mecp2 through a feedback loop involving CpG methylation.
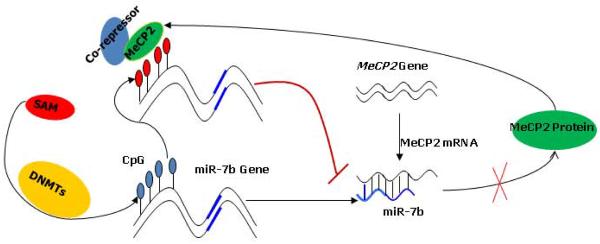
On the one hand, expression of miR-7b was inhibited by methylation of the 5'-flanking region containing CpG island and MeCP2 recruited; on other hand, Mecp2 was suppressed by miR-7b targeting the mRNA-3'-UTR of Mecp2. This interaction between miR-7b and Mecp2 may provide the homeostatic balance needed during postnatal murine neuronal maturation. Due to increasing methylation of the CpG island and MeCP2 recruitment, function of miR-7b is restrained thereby losing its inhibitory regulation of the Mecp2 gene expression. This resulted in enhanced Mecp2 expression which in turn inhibits the transcription of miR-7b gene and its expression.
Our present study required the precise identification of the gene promoter region and CpG islands. Complete sequence and function of the miR-7b transcript (i.e. exons) have been described previously (Farh et al., 2005; Lee et al., 2006), however no information regarding the miR-7b gene (non-coding regions) was available. Hence, we predicted the promoter and presence of a CpG island by computer analysis and authenticated their respective functions by employing the 5'-flanking region that contained the promoter with or without the CpG island in a gene-luciferase reporter assay. The bio-informatic prediction of the promoter DNA fragment demonstrated the ability to drive expression of the reporter gene. Hence, this DNA region was considered the canonical promoter region that contained the transcription start site and site recognition for RNA polymerase II, both being necessary for catalyzing transcription of DNA towards synthesizing mRNA precursors. Since our focus was on the CpG island of the miR-7b gene, further detailed characterization of other cis-elements including other distal predicted transcription start sites far beyond the CpG islands were not pursued in the present study.
In this study, we observed heterogeneous methylation of the miR-7b gene in murine neurons seen by the presence of methylated and unmethylated DNA fragments detected by methylated and unmethylated primers respectively in the methylation specific PCR experiments. This suggests that during maturation of neurons gene methylation may not occur in an “all-or-none” fashion, as seen with pre-implantation embryos (Haycock, 2009). Alternatively since our neuron-enriched fraction was 88.8% pure reflecting the CD90.2 marker, the possible presence of other non-neuronal cell types may have contributed to this heterogeneity in DNA methylation, although unlikely (since 100% of these cells were MAP2 positive). Methylation specific PCR is highly sensitive in detecting methylated cytosines, as it can reliably detect 1 methylated allele within a total of 2000 unmethylated alleles (Herman et al., 1996). Further the possibility exists that allele-specific DNA methylation (ASM) of the miR-7b gene with the other allele being unmethylated could generate a similar picture. Such a scenario has been described in a small number of metabolism-related genes, certain parentally imprinted genes and with X-inactivation (Herman et al., 2003; Kim and Ogura, 2009). Future studies may examine such possibilities in the case of miR-7b in greater detail.
The functional significance of our novel observations needs subsequent in depth investigation. However, speculatively one can surmise that miR-7b regulation of MeCP2 is critical in ensuring tight control of Mecp2 gene expression. Previous investigations using various MeCP2 mutant and transgenic mice have demonstrated that too little or too much of MeCP2 results in abnormal neurodevelopment and neurobehavior (Collins et al., 2004; Kishi and Macklis, 2004). Hence we have always known that Mecp2 expression within neurons has to be just right to ensure normal neurodevelopment. Thus, if miR-7b expression is altered during normal neurodevelopment, akin to the Mecp2 mutant or transgenic mice, it is quite possible that neurodevelopment and neurobehavior may be disrupted. Whether aberrant miR-7b expression in neurons results in a Rett-like syndrome remains to be examined in the future by creating miR-7b mutant and transgenic mice to validate functional importance of this microRNA. Further whether miR-7b mutations or duplications exist in human conditions related to autism spectrum disorders of an unknown etiology may be worthy of pursuit once murine models establish an association between disrupted miR-7b expression and dysfunction as related to a neuropsychiatric phenotype of neurodevelopmental disorders.
In summary, we have illustrated a novel autoregulatory mechanism responsible for homeostatic regulation of neuronal MeCP2 and miR-7b. This molecular feed-back mechanism may be innate and necessary for orchestrating normal postnatal neuronal maturation. While our present study also provides clues about interactions between miRNA biogenesis and DNA methylation with MeCP2 recruitment, more importantly disruption of this finely tuned molecular interaction between miR-7b and MeCP2 may form the basis of neurodevelopmental disorders presenting with neuropsychiatric dysfunction. Such functional importance can only be established in future in-vivo experiments employing gene manipulations in murine models, in lieu of discovering existing mutations or duplications in children with neurodevelopmental disorders.
ACKNOWLEDGMENTS
This work is supported by grants from the NIH HD33997 and HD25024 (to SUD). We are grateful to Dr. David Schubert at the Salk Institute, La Jolla, CA, for sharing the HT22 murine hippocampal neuron-like cell line with us.
ABBREVIATIONS
- 5-Aza-dC
5-aza-2′-deoxycytidine
- 3′-UTR
three prime untranslated region
- ChIP
chromatin immunoprecipitation
- HDACs
histone deacetylases
- TSA
trichostatin A
- MBPs
methyl-CpG binding domain proteins
- MeCP2
methyl-CpG binding protein 2
- miRNA
microRNA
- MSP
methylation specific polymerase chain reaction
- SAM
S-adenosylmethionine
Footnotes
None of the authors have any conflicts of interest.
REFERENCES
- Abelson JF, Kwan KY, O'Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, Davis NR, Ercan-Sencicek AG, Guez DH, Spertus JA, Leckman JF, Dure LSt, Kurlan R, Singer HS, Gilbert DL, Farhi A, Louvi A, Lifton RP, Sestan N, State MW. Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- Aboobaker AA, Tomancak P, Patel N, Rubin GM, Lai EC. Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. Proc Natl Acad Sci U S A. 2005;102:18017–18022. doi: 10.1073/pnas.0508823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Bak M, Silahtaroglu A, Moller M, Christensen M, Rath MF, Skryabin B, Tommerup N, Kauppinen S. MicroRNA expression in the adult mouse central nervous system. RNA. 2008;14:432–444. doi: 10.1261/rna.783108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartholdi D, Klein A, Weissert M, Koenig N, Baumer A, Boltshauser E, Schinzel A, Berger W, Matyas G. Clinical profiles of four patients with Rett syndrome carrying a novel exon 1 mutation or genomic rearrangement in the MECP2 gene. Clin Genet. 2006;69:319–326. doi: 10.1111/j.1399-0004.2006.00604.x. [DOI] [PubMed] [Google Scholar]
- Buschhausen G, Wittig B, Graessmann M, Graessmann A. Chromatin structure is required to block transcription of the methylated herpes simplex virus thymidine kinase gene. Proc Natl Acad Sci U S A. 1987;84:1177–1181. doi: 10.1073/pnas.84.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen Y, Shin BC, Thamotharan S, Devaskar SU. Creb1-Mecp2-(m)CpG complex transactivates postnatal murine neuronal glucose transporter isoform 3 expression. Endocrinology. 2013;154:1598–1611. doi: 10.1210/en.2012-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- Fiore R, Siegel G, Schratt G. MicroRNA function in neuronal development, plasticity and disease. Biochim Biophys Acta. 2008;1779:471–478. doi: 10.1016/j.bbagrm.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, Havelange V, Volinia S, Blum W, Rush LJ, Perrotti D, Andreeff M, Bloomfield CD, Byrd JC, Chan K, Wu LC, Croce CM, Marcucci G. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal K, Datta J, Majumder S, Bai S, Dong X, Parthun M, Jacob ST. Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol Cell Biol. 2002;22:8302–8319. doi: 10.1128/MCB.22.23.8302-8319.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KF, Sakamoto K, Wayman GA, Impey S, Obrietan K. Transgenic miR132 alters neuronal spine density and impairs novel object recognition memory. PLoS One. 2010;5:e15497. doi: 10.1371/journal.pone.0015497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- Hattermann K, Mehdorn HM, Mentlein R, Schultka S, Held-Feindt J. A methylation-specific and SYBR-green-based quantitative polymerase chain reaction technique for O6-methylguanine DNA methyltransferase promoter methylation analysis. Anal Biochem. 2008;377:62–71. doi: 10.1016/j.ab.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Haycock PC. Fetal alcohol spectrum disorders: the epigenetic perspective. Biol Reprod. 2009;81:607–617. doi: 10.1095/biolreprod.108.074690. [DOI] [PubMed] [Google Scholar]
- Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman H, Lu M, Anggraini M, Sikora A, Chang Y, Yoon BJ, Soloway PD. Trans allele methylation and paramutation-like effects in mice. Nat Genet. 2003;34:199–202. doi: 10.1038/ng1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzer S, Silahtaroglu A, Meister B. Locked nucleic acid-based in situ hybridisation reveals miR-7a as a hypothalamus-enriched microRNA with a distinct expression pattern. J Neuroendocrinol. 2012;24:1492–1504. doi: 10.1111/j.1365-2826.2012.02358.x. [DOI] [PubMed] [Google Scholar]
- Herzer S, Silahtaroglu A, Meister B. Locked Nucleic Acid-Based In Situ Hybridization Reveals miR-7a as a Hypothalamus-Enriched MicroRNA with a Distinct Expression Pattern. J Neuroendocrinol. 2012 doi: 10.1111/j.1365-2826.2012.02358.x. [DOI] [PubMed] [Google Scholar]
- Hsieh J, Eisch AJ. Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol Dis. 2010;39:73–84. doi: 10.1016/j.nbd.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta. 2010;1799:694–701. doi: 10.1016/j.bbagrm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Johnson R, Zuccato C, Belyaev ND, Guest DJ, Cattaneo E, Buckley NJ. A microRNA-based gene dysregulation pathway in Huntington's disease. Neurobiol Dis. 2008;29:438–445. doi: 10.1016/j.nbd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Kawase-Koga Y, Otaegi G, Sun T. Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev Dyn. 2009;238:2800–2812. doi: 10.1002/dvdy.22109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Ogura A. Changes in allele-specific association of histone modifications at the imprinting control regions during mouse preimplantation development. Genesis. 2009;47:611–616. doi: 10.1002/dvg.20541. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007;10:1513–1514. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Palkovits M, Young WS., 3rd miR-7b, a microRNA up-regulated in the hypothalamus after chronic hyperosmolar stimulation, inhibits Fos translation. Proc Natl Acad Sci U S A. 2006;103:15669–15674. doi: 10.1073/pnas.0605781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, Habbel P, Kalin R, Franzoni E, Rybak A, Nguyen D, Veh R, Ninnemann O, Peters O, Nitsch R, Heppner FL, Golenbock D, Schott E, Ploegh HL, Wulczyn FG, Lehnardt S. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 2012;15:827–835. doi: 10.1038/nn.3113. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lo PK, Watanabe H, Cheng PC, Teo WW, Liang X, Argani P, Lee JS, Sukumar S. MethySYBR, a novel quantitative PCR assay for the dual analysis of DNA methylation and CpG methylation density. J Mol Diagn. 2009;11:400–414. doi: 10.2353/jmoldx.2009.080126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A, Esteller M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle. 2007;6:1455–1459. [PubMed] [Google Scholar]
- Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, Mandel G, Goodman RH. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci U S A. 2010;107:20382–20387. doi: 10.1073/pnas.1015691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Hatzigeorgiou AG, Mourelatos Z. miRNP:mRNA association in polyribosomes in a human neuronal cell line. RNA. 2004;10:387–394. doi: 10.1261/rna.5181104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S, Meletis K, Fu D, Jhaveri S, Jaenisch R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. 2007;236:1663–1676. doi: 10.1002/dvdy.21176. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Shin BC, Dai Y, Thamotharan M, Gibson LC, Devaskar SU. Pre- and postnatal calorie restriction perturbs early hypothalamic neuropeptide and energy balance. J Neurosci Res. 2012;90:1169–1182. doi: 10.1002/jnr.23013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Treadwell J, Zhang D, Ly D, McKinnell I, Walker PR, Sikorska M. Large-scale expression analysis reveals distinct microRNA profiles at different stages of human neurodevelopment. PLoS One. 2010;5:e11109. doi: 10.1371/journal.pone.0011109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo Z, Wu M, Citron BA, Palazzo RE, Festoff BW. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J Biol Chem. 2003;278:37681–37689. doi: 10.1074/jbc.M301406200. [DOI] [PubMed] [Google Scholar]
- Tarquinio DC, Motil KJ, Hou W, Lee HS, Glaze DG, Skinner SA, Neul JL, Annese F, McNair L, Barrish JO, Geerts SP, Lane JB, Percy AK. Growth failure and outcome in Rett syndrome: Specific growth references. Neurology. 2012;79:1653–1661. doi: 10.1212/WNL.0b013e31826e9a70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdinguio RG, Fernandez AF, Lopez-Nieva P, Rossi S, Huertas D, Kulis M, Liu CG, Croce CM, Calin GA, Esteller M. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics. 2010;5:656–663. doi: 10.4161/epi.5.7.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, Moraine C, Marynen P, Fryns JP, Froyen G. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viberg H, Mundy W, Eriksson P. Neonatal exposure to decabrominated diphenyl ether (PBDE 209) results in changes in BDNF, CaMKII and GAP-43, biochemical substrates of neuronal survival, growth, and synaptogenesis. Neurotoxicology. 2008;29:152–159. doi: 10.1016/j.neuro.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Wanet A, Tacheny A, Arnould T, Renard P. miR-212/132 expression and functions: within and beyond the neuronal compartment. Nucleic Acids Res. 2012;40:4742–4753. doi: 10.1093/nar/gks151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt F, Molloy PL. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988;2:1136–1143. doi: 10.1101/gad.2.9.1136. [DOI] [PubMed] [Google Scholar]
- Wendtner CM. Cocktail of eternity: HDAC meets miR. Blood. 2012;119:1095–1096. doi: 10.1182/blood-2011-12-394494. [DOI] [PubMed] [Google Scholar]
- Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y. DNA methylation mediated by a microRNA pathway. Mol Cell. 2010;38:465–475. doi: 10.1016/j.molcel.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X. Methylation as a crucial step in plant microRNA biogenesis. Science. 2005;307:932–935. doi: 10.1126/science.1107130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Zhang JZ, Zhao CL, Zhang HY, Xu Q. Isolation and characterization of the CD133+ precursors from the ventricular zone of human fetal brain by magnetic affinity cell sorting. Biotechnol Lett. 2004;26:1131–1136. doi: 10.1023/B:BILE.0000035484.64499.ac. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Wang JY, Xu LY, Cai R, Chen Z, Luo BY. MicroRNA expression changes in the hippocampi of rats subjected to global ischemia. J Clin Neurosci. 2010;17:774–778. doi: 10.1016/j.jocn.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yuan P, Wang Y, Hunsberger JG, Elkahloun A, Wei Y, Damschroder-Williams P, Du J, Chen G, Manji HK. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology. 2009;34:1395–1405. doi: 10.1038/npp.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]