

Abstract
Combination therapy, which can optimize killing activity to cancers and minimize drug resistance, is a mainstream therapy against hormone-refractory prostate cancers (HRPCs). Rottlerin, a natural polyphenolic component, synergistically increased PC-3 (a HRPC cell line) apoptosis induced by camptothecin (a topoisomerase I inhibitor). Using siRNA technique to knockdown protein kinase C-δ (PKCδ), the data showed that rottlerin-mediated synergistic effect was PKCδ-independent, although rottlerin has been used as a PKCδ inhibitor. Rottlerin potentiated camptothecin-induced DNA fragmentation at S phase and ATM phosphorylation at Ser1981. The effect was correlated to apoptosis (r2 = 0.9). To detect upstream signals, the data showed that camptothecin acted on and stabilized topoisomerase I-DNA complex, leading to the formation of camptothecin-trapped cleavage complexes (TOP1cc). The effect was potentiated by rottlerin. To determine DNA repair capability, the time-related γH2A.X formation was examined after camptothecin removal. Consequently, rottlerin significantly inhibited camptothecin removal-mediated decline of γH2A.X formation at S phase, indicating the impairment of DNA repair activity in the presence of rottlerin. The combinatory treatment of camptothecin and rottlerin induced conformational change and activation of Bax and formation of truncated Bad, suggesting the contribution of mitochondria stress to apoptosis. In summary, the data suggest that rottlerin-mediated camptothecin sensitization is through the augmented stabilization of TOP1cc, leading to an increase of DNA damage stress and, possibly, an impairment of DNA repair capability. Subsequently, mitochondria-involved apoptosis is triggered through Bax activation and truncated Bad formation. The novel discovery may provide an anticancer approach of combinatory use between rottlerin and camptothecin for the treatment of HRPCs.
Keywords: Camptothecin, Rottlerin, Topoisomerase I-DNA cleavage complex, DNA damage, Hormone-refractory prostate cancer
1. Introduction
Prostate cancer is a major cause of cancer mortality in males. Hormone-refractory prostate cancer (HRPC), resistant to hormone therapy, is a major obstacle in clinical treatment. Accordingly, the approach for HRPC treatment is deeply demanded. However, the resistance developed in cancer cells is a major concern that largely diminishes the efficacy of cancer chemotherapeutic drugs. Combination therapy that combines two or more chemotherapeutic drugs is a well-recognized approach. With mechanism-based design of anticancer drugs, combination therapy can optimize the killing activity to cancer cells and minimize drug resistance [1,2].
Protein kinase C-δ (PKCδ) is a diacylglycerol-dependent but calcium-independent PKC isoform [3]. Several apoptotic stimuli have been identified to induce the translocation of PKCδ to plasma membrane, mitochondria and nucleus, leading to activation of apoptotic signaling cascades [4]. Rottlerin was originally discovered as a specific inhibitor of PKCδ. Recently, an increasing body of evidence suggests that rottlerin may trigger anticancer activities through PKCδ-independent pathways, such as inhibition of Cdc2 [5], uncoupling of mitochondria [6], induction of endoplasmic reticulum stress [7] and up-regulation of death receptors [8].
The nuclear proteins topoisomerases are enzymes responsible to determine DNA topology. Accordingly, topoisomerases are critical in the regulation of DNA replication, transcription and subsequent translation. Topoisomerase I creates a single-stranded nick in duplex DNA, forming topoisomerase I-DNA cleavage complexes (TOP1cc) and unwinding supercoiled DNA. As soon as DNA is relaxed, topoisomerase I religates the DNA nicks. Camptothecin and its derivative irinotecan are well-known topoisomerase I inhibitors, inducing apoptotic pathways through decelerating the dissociation of topoisomerase I-DNA to trap TOP1cc, which not only induces single strand breaks (SSBs) but also may cause double strand breaks (DSBs). As a result, these topoisomerase I inhibitors can cause severe DNA damage response and activate DNA damage-related molecules, leading to an ultimate cell death [9–12]. The clinical use of irinotecan is the treatment of metastatic colorectal cancers although the clinical trial designed for HRPC treatment had been studied [13]. The underlying mechanism that limits the use of topoisomerase I inhibitors in HRPC is highly anticipated to be identified and be overcome.
Rottlerin has been studied to combine with anticancer agents, including sorafenib, imatinib and tumor necrosis factor-related apoptosis-inducing ligands, in several cancer models [6,14–16]. Mitochondria-mediated pathways have been suggested to be responsible for the combined effect. However, the primary mechanism is yet to be elucidated. The present work was to study the combinatory effect and underlying mechanism of camptothecin and rottlerin in HRPC. The study also investigated why rottlerin compensated and overcame the less responsiveness of camptothecin in HRPC. The study may provide data supporting anticancer approach of topoisomerase I inhibitors in HRPC.
2. Materials and methods
2.1. Materials
RPMI 1640 medium and fetal bovine serum (FBS) were obtained from GIBCO/BRL Life Technologies (Grand Island, NY). Antibodies to PARP, PKCδ, topoisomerase I, Bax (6A7), Bad, cyclin D1, cyclin E, cyclin-dependent kinase 4 (Cdk4), Cdk2 and anti-mouse and anti-rabbit IgGs were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies to PKCδThr505, ATM, ATMSer1981, γH2A.X, Bid, p21Waf1/Cip1, cyclin A, cyclin B1 and Cdk1 were from Cell Signaling Technologies (Boston, MA). Caspase-3 was from Imgenex, Corp. (San Diego, CA). GAPDH and α-tubulin was form Epitomics, Inc. (Burlingame, CA). Rottlerin, camptothecin, etoposide, doxorubicin, docetaxel, vincristine, G06983, propidium iodide (PI) and all other chemical compounds were obtained from Sigma–Aldrich (St. Louis, MO). Ro-318220 was purchased from Calbiochem (La Jolla, CA).
2.2. Cell lines and cell culture
HRPC cell line PC-3 cells were from American Type Culture Collection (Rockville, MD). Cells were cultured in RPMI 1640 medium with 10% FBS (v/v), penicillin (100 units/ml) and streptomycin (100 μg/ml). Cultures were maintained in a 37 °C incubator with 5% CO2.
2.3. FACScan flow cytometric assay
After treatment, cells were harvested by trypsinization, fixed with 70% (v/v) alcohol at 4 °C for 30 min and washed with PBS. The cells were centrifuged and resuspended with 0.5 ml PI solution containing Triton X-100 (0.1%, v/v), RNase (100 μg/ml) and PI (80 μg/ml). DNA content was analyzed with the FACScan and CellQuest software (Becton Dickinson, Mountain View, CA).
2.4. Western blotting
After treatment, cells were harvested by trypsinization, centrifuged and lysed in 0.1 ml of lysis buffer (10 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 50 mM NaF and 100 μM sodium orthovanadate). For Western blot analysis, 40 μg proteins were separated by electrophoresis in a 10 or 15% polyacrylamide gel and transferred to a PVDF membrane. After a 1-h incubation at room temperature in PBS/5% non-fat milk, the membrane was washed with PBS/0.1% Tween 20 for another 1 h and overnight incubated with the indicated antibody at 4 °C. After three washings with PBS/0.1% Tween 20, the antimouse or antirabbit IgG (dilute 1:4000) was applied to the membranes for 1 h at room temperature. The membranes were washed with PBS/0.1% Tween 20 for 1 h and the detection of signal was performed with an enhanced chemiluminescence detection kit (Amersham, Buckinghamshire, UK).
2.5. Detection of Bax conformational change
After treatment, the cells were washed twice with ice-cold PBS, lysed in Chaps lysis buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 1% Chaps). Total proteins of 300 μg were incubated with 1 μg of Bax 6A7 antibody for 3 h at 4 °C. Then, 15 μl of A/G-agarose beads were added and incubated at 4 °C for 2 h. The beads were washed with Chaps lysis buffer for immunoblotting as describes.
2.6. TUNEL-PI double staining
TUNEL (terminal dUTP nick-end labeling) method identifies DNA fragmentation using TdT to transfer fluorescein-12-dUTP to the free 3′-OH of cleaved DNA. After treatment, the cells were washed, fixed and stained for the fragmented DNA detection according to the protocol provided by the suppliers (Promega, Madison, WI). Finally, the cells were suspended with 0.5 ml PI solution described above. The labeled DNA and cell cycle distribution were simultaneously quantified and analyzed by flow cytometric analysis.
2.7. γH2AX-PI double staining
After treatment, the cells were pelleted, fixed with 70% (v/v) alcohol at 4 °C for 30 min and incubated in PBS/0.2% Triton X-100 at room temperature for 5 min. The cells were washed with PBS and incubated with γH2A.X antibodies (dilute in PBS/1% BSA, 1:1000) at room temperature for 1 h. The cells were washed with PBS and incubated with FITC-conjugated secondary antibody (dilute in PBS/1% BSA, 1:1000) at room temperature for 1 h. Finally, the cells ware suspended with 0.5 ml PI solution. The expression of γH2A.X and cell cycle distribution were simultaneously quantified and analyzed by flow cytometric analysis.
2.8. In vivo complex of enzyme (ICE) assay
TOP1-DNA adducts were isolated using the ICE bioassay according to published study [17]. Cells (1 × 106) were lysed with 2 ml STE buffer (1% sarkosyl; 10 mM Tris–HCl pH 8.0, 1 mM EDTA) and passed through a 26-gauge needle. The cells were layered on the top of cesium chloride gradient. Step-gradients (2 ml each) containing CsCl solutions of the different densities (1.82, 1.72, 1.50 and 1.37 g/ml) were placed in a polyallomer tube (Beckman, Inc.). Tubes were centrifuged at 31,000 rpm in a Beckman SW40 rotor for 24 h at 20 °C. Then, 1 ml of each fraction was collected from the top of the tubes. Next, 100 μl of each fraction was diluted with an equal volume of SP buffer (25 mM sodium phosphate, pH 6.5), applied to a PVDF membrane with a dot-blot apparatus (BioRad, Inc.) and immunoblotted with anti-topoisomerase I antibody.
2.9. siRNA transfection
The siRNA against PKCδ is 5′-UGA UCU UGU CGA UGC AUU UCU UGU G-3′ which matches only to human PKCδ. The control sequence is 5′-UGA UUU GUU CAG CUA CGU CUU UGU G-3′. For transfection, PC-3 cells were seeded into 60-mm tissue culture dishes with 30% confluence and grown for 24 h to 50% confluence. Each dish was washed with serum-free Opti-MEM (Life Technologies, Ground Island, NY), and 2 ml of the same medium was added. Aliquots containing siRNA in serum-free Opti-MEM were transfected into cells using Lipofectamine 2000 following the manufacturer’s instructions. After incubation for 6 h at 37 °C, cells were washed with medium and incubated in 10% FBS-containing RPMI-1640 medium for 48 h. Then, the subsequent experiments were performed.
2.10. Measurement of mitochondrial membrane potential (ΔΨm)
JC-1, a mitochondrial dye staining mitochondria in living cells in a membrane potential-dependent fashion, was used to determine ΔΨm. Cells were treated with or without the agent for the indicated times. Thirty minutes before the termination of incubation, the cells were incubated with JC-1 (final concentration of 2 μM) at 37 °C for 30 min. The cells were finally harvested and the accumulation of JC-1 was determined using flow cytometric analysis.
2.11. Data analysis
Data are presented as the mean ± SEM for the indicated number of separate experiments. Statistical analysis of data was performed with one-way analysis of variance (ANOVA) followed by a t-test and P-values less than 0.05 were considered significant.
3. Results
3.1. Combination treatment of PC-3 cells with rottlerin and anticancer drugs
The effect of combination treatment with rottlerin and some other cancer chemotherapeutic drugs, including camptothecin (a topoisomerase I inhibitor), etoposide, doxorubicin (two topoisomerase II inhibitors), docetaxel (a microtubule stabilizing agent) and vincristine (a tubulin de-polymerizing agent), on apoptotic cell death was determined by FACScan flow cytometric analysis of PI staining. The data showed that the apoptosis induced by camptothecin, but not etoposide, doxorubicin and vincristine, was dramatically potentiated by rottlerin. On the contrary, rottlerin significantly decreased docetaxel-induced effect (Fig. 1A). The increased apoptosis caused by combination treatment was also detected in a concentration-dependent fashion (Fig. 1B). The microscopic examination showed that the combinatory treatment of rottlerin and camptothecin induced a morphological change to cell shrinkage, a phenomenon of apoptosis, and apoptotic bodies were apparent (Fig. 1C). To further confirm the effect by biochemical data, the caspase-3 and downstream substrate, PARP-1, were monitored. As a consequence, the combinatory treatment of rottlerin and camptothecin significantly induced the cleavage and activation of caspase-3 as well as PARP-1 cleavage (Fig. 1D).
Fig. 1.

Effect of rottlerin on apoptosis caused by several anticancer drugs PC-3 cells. Cells were treated with the indicated agents (2 μM rottlerin, 1 μM camptothecin, 10 μM etoposide, 3 μM doxorubicin, 0.03 μM docetaxel, 0.03 μM vincristine) (A), or treated with the graded concentrations of rottlerin and camptothecin (B) for 24 h. The cells were fixed and stained with propidium iodide to analyze apoptosis (subG1 population) by FACScan flow cytometric analysis. Or, the cell morphology was observed by microscopic examination (C). Scale bar, 20 μm. (D) The expression of caspase-3 and PARP-1 was detected by Western blot after a 9-h treatment. The data are representative of three independent experiments. ***p < 0.001 compared with the agent-free control.
3.2. Determination of PKCδ involvement in the combinatory treatment
To determine the functional role of PKCδ in the combinatory treatment, several approaches were used. First, the phosphorylation of PKCδ at Thr505 was detected. The data demonstrated that the combinatory treatment caused a profound PKCδ cleavage (Fig. 2A). Because PKCδ is a downstream substrate of caspase-3 [18], the PKCδ cleavage could be attributed to the caspase-3 activation in the combinatory treatment. Second, G06983 and Ro-318220 (two general PKC inhibitors) were used to examine whether pan-PKC inhibitors could mimic rottlerin-mediated effect. The data demonstrated that, instead of potentiating apoptosis, the pan-PKC inhibitors significantly decreased camptothecin-induced effect (Fig. 2B). Finally, the siRNA technique was used to knockdown PKCδ in this study. The data showed that the knockdown of PKCδ did not potentiate the effect mediated by camptothecin alone (lane 10 in Fig. 2C). Moreover, the knockdown of PKCδ did not modify the cleavage of procaspase-3 and PARP-1 induced by combinatory treatment of rottlerin and camptothecin (Fig. 2C). Altogether, the data suggest that rottlerin potentiated camptothecin-mediated effect through PKCδ-independent pathway.
Fig. 2.

Identification of the role of PKCδ in the combinatory treatment. (A) PC-3 cells were treated with the indicated agents for 9 (A) or 48 h (B). The cells were harvested and lysed for the detection of the indicated protein expression by Western blot or the cells were fixed and stained with propidium iodide to determine apoptosis by FACScan flow cytometric analysis. Data are expressed as mean ± SEM of three independent determinations. (C) PC-3 cells were transfected with control siRNA or PKCδ siRNA. The cells were harvested and lysed for the detection of the indicated protein expression by Western blot after a 9-h treatment. The data are representative of two independent experiments.
3.3. Effect of combinatory treatment on DNA fragmentation and ATM phosphorylation
Camptothecin serves as a topoisomerase I poison that binds to and stabilizes the topoisomerase I-DNA complex, preventing DNA religation and eventually causing DNA damage response. Using TUNEL and PI double staining to identify DNA fragmentation and to determine cell cycle progression, respectively, the data in Fig. 3A demonstrated that rottlerin potentiated camptothecin-induced DNA fragmentation at S phase (positive region in Fig. 3A). Based on the DNA damage response, the phosphorylation of ATM at Ser1981, a DNA damage sensor, was examined accordingly. As a consequence, camptothecin alone induced a time-dependent increase of ATM phosphorylation and activation. The effect was augmented by combinatory use of rottlerin (Fig. 3B). The synergistic effect on ATM phosphorylation was also detected in combinatory treatment of increasing rottlerin concentrations (Fig. 3C). Notably, the synergistic effect on ATM phosphorylation was highly correlated to cell apoptosis (r2 = 0.9, Fig. 3D), indicating that DNA damage response in the combinatory treatment played a crucial role in cell apoptosis.
Fig. 3.

Effect of camptothecin and rottlerin on DNA damage response. (A) PC-3 cells were treated with 1 μM camptothecin and/or 2 μM rottlerin for 9 h. The cells were harvested for the detection of DNA fragmentation and cell-cycle population using TUNEL apoptosis detection assay and PI staining of flow cytometric analysis, respectively. (B) and (C) The cells were treated with the agents for the indicated time. After the treatment, the cells were harvested and lysed for the detection of the indicated protein expression by Western blot. The expressions were quantified using the computerized image analysis system ImageQuant (Amersham Biosciences). (D) The correlation between p-ATM expression from (C) and apoptosis by PI staining of FACScan flow cytometric analysis was constructed.
3.4. Effect of rottlerin on camptothecin-induced formation of TOP1cc
Because camptothecin-mediated DNA damage was synergistically increased by rottlerin, the upstream event that resulted in DNA damage was determined. It has been well identified that camptothecin acted on and stabilized topoisomerase I-DNA complex, leading to the formation of camptothecin-trapped cleavage complexes (TOP1cc). The specific topoisomerase-genomic DNA cleavage complexes can be measured by ICE assay. As demonstrated in Fig. 4, topoisomerase I free form was partitioned to the fractions between 1 and 4. After the exposure to camptothecin (1 μM, 30 min), TOP1cc was trapped in fractions between 5 and 7 (Fig. 4). The data also showed that the level of TOP1cc was not modified by rottlerin alone but was increased by the combinatory treatment with 1 μM camptothecin and rottlerin. The increased level caused by the combinatory treatment was similar to that induced by 10 μM camptothecin alone (Fig. 4). The data suggest that rottlerin is able to potentiate the rapid formation of TOP1cc in camptothecin-treated cells.
Fig. 4.
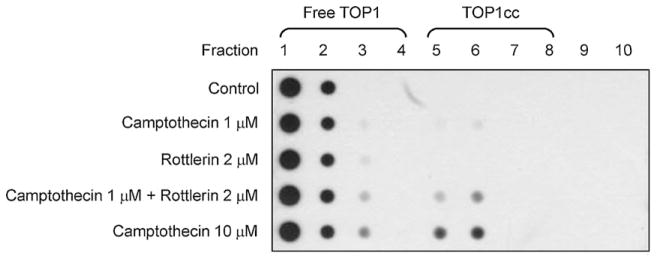
Effect of camptothecin and rottlerin on TOP1cc formation. PC-3 cells were treated with the indicated agent for 30 min. The ICE assay was used to detect topoisomerase I-DNA adducts formation as described in Section 2. Topoisomerase I free form was partitioned to the fractions between 1 and 4. TOP1cc was trapped in fractions between 5 and 7. The data are representative of three independent experiments.
3.5. Effect of rottlerin on camptothecin washout-mediated recovery from DNA damage
The level of DNA damage at S phase was determined by detecting the formation of γH2A.X because camptothecin-induced DNA damage is replication-mediated. H2A.X can be phosphorylated, termed γH2A.X, during DNA damage response. It has been well-recognized that γH2A.X may serve as a sensitive DNA damage marker. Using double staining of γH2A.X and PI, the data showed that camptothecin alone, but not rottlerin, induced a rapid formation of γH2A.X at S phase. The effect declined following the washout of camptothecin (Fig. 5 and Table 1). In contrast, the washout-mediated decline of γH2A.X formation at S phase was significantly prevented in the presence of rottlerin (Fig. 5 and Table 1). The data indicate that rottlerin may interrupt the endogenous DNA repair after camptothecin removal in PC-3 cell.
Fig. 5.

Effect of camptothecin and rottlerin on γH2A.X expression at S phase. After the treatment of PC-3 cells with 1 μM camptothecin (CPT) for 1 h, the cells were incubated in the absence or presence of 2 μM rottlerin for 8 or 24 h. γH2A.X-PI double staining was used to detect DNA damage in the S phase of the cell cycle as described in Section 2.
Table 1.
Effect of camptothecin (CPT) and rottlerin on γH2A.X expression at S phase.
| Treatment | Cell count of γH2A.X positive in S phase (%) |
|---|---|
| Control | 2.4 ± 0.6 |
| CPT 1 h | 85.7 ± 2.7 |
| Rottlerin 8 h | 13.5 ± 3.3 |
| Rottlerin 24 h | 19.4 ± 5.0 |
| CPT 1 h →washout 8 h | 47.8 ± 6.9 |
| CPT 1 h →rottlerin 8 h | 70.6 ± 1.0** |
| CPT 1 h →washout 24 h | 21.9 ± 14.5 |
| CPT 1 h →rottlerin 24 h | 59.2 ± 4.4* |
CPT, 1 μM; rottlerin, 2 μM.
p <0.05 compared with washout condition.
p <0.01 compared with washout condition.
3.6. Effect of combinatory treatment on the expression of several marker proteins
The progression of cell cycle is tightly regulated bycyclins and the binding partners, Cdks. Cyclin D and the associated Cdk 4 regulate G1-phase progression. Cyclin E/Cdk4 complex activity regulates late G1- and S-phase progression and cyclin A/Cdk2 complex activity dominates S and G2 phases. The progression from G2- to M-phase is driven by the activation of cyclin B1/Cdk1 complex. The combinatory treatment did not significantly modify the expression of these cell-cycle regulators caused by single use of camptothecin or rottlerin (Fig. 6A). P21 acts as an inhibitor of cell cycle progression. However, p21 can also directly inhibit DNA synthesis by binding to and inhibiting PCNA (proliferating cell nuclear antigen) [19]. Recently, p21 has been suggested to accumulate at DNA strand break site and is a detection marker of DNA damage sites [20]. The combinatory treatment of camptothecin and rottlerin dramatically up-regulated p21 expression (Fig. 6A), confirming the massive DNA damage. Furthermore, to functionally connect mitochondria-mediated signals between DNA damage and cell apoptosis, several ant-apoptotic and pro-apoptotic Bcl-2 family members were examined. The data showed that Bad was cleaved to generate a 15-kDa truncated Bad, a more potent apoptosis inducer [21] in response to combinatory treatment (Fig. 6A). Besides, although the level of Bax was not changed, the combinatory treatment increased the changes in Bax conformation, detected by immunoprecipitation with the conformation-specific antibody (Fig. 6A). The ΔΨm was determined using JC-1 staining. JC-1 aggregates (J-aggregates, red fluorescence) prefer higher ΔΨm in cells under normal condition. After the loss of ΔΨm, the JC-1 monomers are dominant with green fluorescence. The fluorescence ratio, therefore, is used to study ΔΨm. The data in Fig. 6B demonstrated that combinatory treatment of camptothecin and rottlerin significantly induced the loss of ΔΨm. Altogether, it has been suggested that mitochondria-mediated pathway may dominate apoptosis through the combinatory treatment-induced DNA damage response.
Fig. 6.

Effect of camptothecin and rottlerin on several protein expressions and ΔΨm. (A) PC-3 cells were treated without or with the indicated agent for 9 h. After the treatment, the cells were harvested and lysed for the detection of the indicated protein expression by Western blot. The data are representative of two independent experiments. (B) After the treatment, PC-3 cells were incubated with JC-1 for the detection of ΔΨm using FACScan flow cytometric analysis. The data are expressed as mean ± SEM of three independent experiments.
4. Discussion
Rottlerin, a natural polyphenol isolated from Mallotus philippinensis, is shown to inhibit PKC, in particular PKCδ [22]. Recent work has shown that rottlerin displays pharmacological activities, including anticancer activity, through PKCδ-independent pathways [5–8,23]. The data from the assays of PKCδ knockdown provided evidence supporting the PKCδ-independent pathway to rottlerin action in this study. However, the single use of rottlerin did not show promising efficacy against cancer cells. In contrast, rottlerin dramatically potentiated camptothecin-mediated apoptotic activity in HRPCs. The finding is particularly meaningful for clinical consideration since topoisomerase I inhibitors do not work well against HRPCs. In this study, the underlying mechanism of combinatory treatment of HRPCs with camptothecin and rottlerin has been elucidated.
Camptothecin is a highly selective topoisomerase I poison [24] although the water solubility is a major concern in use. Camptothecin reversibly binds TOP1cc while leads to irreversible DNA damage response when replication fork encounters a TOP1cc [10]. A lot of downstream signals can be, in turn, triggered to regulate and determine cell fate, including checkpoint arrest of the cell cycle, initiation of DNA repair and/or induction of apoptotic pathways [25]. The flow cytometric analysis with double staining of TUNEL and PI revealed that the combinatory treatment of camptothecin and rottlerin caused dramatic DNA fragmentation at DNA synthesis phase. ATM, a serine/threonine kinase, is responsible for cell cycle checkpoints, DNA repair and apoptosis [26]. ATM activation by phosphorylation on Ser1981 occurs in the presence of DSBs. The data showed that rottlerin extensively potentiated camptothecin-mediated ATM activation which was highly correlated to apoptosis. Altogether, the data highly suggest that rottlerin synergistically imposes DNA damage stress to camptothecin action. However, the synergistic effect did not happen to that combined with topoisomerase II inhibitors and anti-tubulin drugs (Fig. 1). It indicated that rottlerin selectively and very early acted on topoisomerase I-mediated targets. To further identify the underlying mechanism, the formation of TOP1cc was studied. The correlation between TOP1cc formation and DNA damage response has been reported in a wide variety of cancer cells when exposed to topoisomerase I inhibitors [27,28]. Moreover, it has been reported that in SN38 (an active metabolite of irinotecan) resistant cancer cell clones, the presence of topoisomerase I mutations did not change the expression level and activity of topoisomerase I, but decrease the TOP1cc associated with a reduction in DSBs when challenged with SN38 [29]. These studies suggest that the formation of TOP1cc is critical to DNA damage and apoptosis caused by topoisomerase I inhibitors. Accordingly, the synthesis of new topoisomerase I inhibitors that increase the stability of TOP1cc has been a potential approach [12]. Using ICE assay to determine TOP1cc, rottlerin was able to rapidly augment camptothecin-mediated TOP1cc formation. The effect may, at least partly, explain the synergistic effect on DNA damage and apoptosis caused by combinatory treatment.
One of the key regulators determining the sensitivity to topoisomerase I inhibitors is DNA repair capability, which also explains the selectivity between normal and cancer tissues that usually associated with intrinsically DNA repair defects. It has been suggested that mutations in genes required for homologous recombination DNA repair are hypersensitive to topoisomerase I inhibitors [30]. Targeting DNA repair may also increase the selectivity of topoisomerase I inhibitors for cancers [31,32]. Therefore, we elucidated the endogenous DNA repair capability after camptothecin exposure and washout. After transient exposure to camptothecin, γH2A.X level was decreased following the prolongation of camptothecin removal. It indicates that the DNA repair system works in HRPCs. In the presence of rottlerin, camptothecin removal-involved DNA repair was significantly decreased, leading to a sustained high level of γH2A.X expression. The data suggest that in the presence of rottlerin, DNA repair capability in HRPCs is impaired. However, it is complex that topoisomerase I functions at replication and transcription sites, and how the enzyme activity can influence chromatin and transcription [33–35]. Camptothecin specifically inhibits topoisomerase I and may thus affect several fundamental pathways in the cell. Therefore, rottlerin-mediated camptothecin sensitization may be partly attributed to other signaling pathways.
A large body of evidence suggests that mitochondria play a central role in the regulation of cell death processes. Moreover, Bcl-2 family of proteins is a key regulator for active control of mitochondrial membrane permeability that decides the apoptosis induction or not [36]. Bax is an important Bcl-2 family member and plays a critical role in apoptosis. In resting condition, Bax exists in cytoplasm although it loosely attaches to mitochondrial membrane. After apoptotic induction that causes Bax activation, Bax translocates and associates to mitochondrial membrane surface, leading to the exposure of an amino terminal epitope recognized by the monoclonal antibody 6A7 [37]. The combinatory treatment of camptothecin and rottlerin induced an increase of the excitatory conformational change of Bax, suggesting the involvement of Bax in apoptotic signaling pathway. Moreover, the combinatory treatment also induced the cleavage of Bad into a 15-kDa truncated fragment. The truncated Bad is more potent than the wild-type protein in apoptosis induction [21]. Because caspase-3 is well-recognized to cleave Bad into 15-kDa truncated form [38], the formation of truncated Bad may amplify the apoptosis induced by caspase-3.
In summary, the data suggest that rottlerin-mediated camptothecin sensitization is through the augmented stabilization of TOP1cc, leading to an increase of DNA damage stress and, possibly, an impairment of DNA repair system. Subsequently, mitochondria-involved signaling is triggered through the activation of Bax and, ultimately, cell apoptosis is executed by the activation of caspase-3 and amplified by the formation of truncated Bad. The novel discovery in this study may provide an anticancer approach of combinatory use between rottlerin and camptothecin for the treatment of HRPCs.
Supplementary Material
Acknowledgments
We acknowledge the support provided by the National Science Council of the Republic of China (NSC 99-2323-B-002-002 and NSC100-2320-B-002-006-MY3) and by the National Taiwan University (10R71809).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bcp.2012.03.023.
References
- 1.Stavridi F, Karapanagiotou EM, Syrigos KN. Targeted therapeutic approaches for hormone-refractory prostate cancer. Cancer Treat Rev. 2010;36:122–30. doi: 10.1016/j.ctrv.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Di Lorenzo G, Autorino R, Figg WD, De Placido S. Hormone-refractory prostate cancer: where are we going. Drugs. 2007;67:1109–24. doi: 10.2165/00003495-200767080-00002. [DOI] [PubMed] [Google Scholar]
- 3.Reyland ME. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front Biosci. 2009;14:2386–99. doi: 10.2741/3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase C delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- 5.Kim EH, Kim SU, Choi KS. Rottlerin sensitizes glioma cells to TRAIL-induced apoptosis by inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP. Oncogene. 2005;24:838–49. doi: 10.1038/sj.onc.1208241. [DOI] [PubMed] [Google Scholar]
- 6.Tillman DM, Izeradjene K, Szucs KS, Douglas L, Houghton JA. Rottlerin sensitizes colon carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis via uncoupling of the mitochondria independent of protein kinase C. Cancer Res. 2003;63:5118–25. [PubMed] [Google Scholar]
- 7.Lim JH, Park JW, Kim SH, Choi YH, Choi KS, Kwon TK. Rottlerin induces pro-apoptotic endoplasmic reticulum stress through the protein kinase C-delta-independent pathway in human colon cancer cells. Apoptosis. 2008;13:1378–85. doi: 10.1007/s10495-008-0264-z. [DOI] [PubMed] [Google Scholar]
- 8.Lim JH, Park JW, Choi KS, Park YB, Kwon TK. Rottlerin induces apoptosis via death receptor 5 (DR5) upregulation through CHOP-dependent and PKC delta-independent mechanism in human malignant tumor cells. Carcinogenesis. 2009;30:729–36. doi: 10.1093/carcin/bgn265. [DOI] [PubMed] [Google Scholar]
- 9.Teicher BA. Next generation topoisomerase I inhibitors: rationale and biomarker strategies. Biochem Pharmacol. 2008;75:1262–71. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Avemann K, Knippers R, Koller T, Sogo JM. Camptothecin, a specific inhibitor of type I DNA topoisomerase, induces DNA breakage at replication forks. Mol Cell Biol. 1988;8:3026–34. doi: 10.1128/mcb.8.8.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109:2894–902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, et al. Differential induction of topoisomerase I-DNA cleavage complexes by the indenoisoquinoline MJ-III-65 (NSC 706744) and camptothecin: base sequence analysis and activity against camptothecin-resistant topoisomerases I. Cancer Res. 2003;63:7428–35. [PubMed] [Google Scholar]
- 13.Reese DM, Tchekmedyian S, Chapman Y, Prager D, Rosen PJ. A phase II trial of irinotecan in hormone-refractory prostate cancer. Invest New Drugs. 1998;16:353–9. doi: 10.1023/a:1006120910380. [DOI] [PubMed] [Google Scholar]
- 14.Jane EP, Premkumar DR, Pollack IF. Coadministration of sorafenib with rottlerin potently inhibits cell proliferation and migration in human malignant glioma cells. J Pharmacol Exp Ther. 2006;319:1070–80. doi: 10.1124/jpet.106.108621. [DOI] [PubMed] [Google Scholar]
- 15.Kurosu T, Tsuji K, Kida A, Koyama T, Yamamoto M, Miura O. Rottlerin synergistically enhances imatinib-induced apoptosis of BCR/ABL-expressing cells through its mitochondrial uncoupling effect independent of protein kinase C-delta. Oncogene. 2007;26:2975–87. doi: 10.1038/sj.onc.1210117. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Liu N, Liu S, Liu Y, Zheng D. PKCdelta protects human breast tumor MCF-7 cells against tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis. J Cell Biochem. 2005;96:522–32. doi: 10.1002/jcb.20535. [DOI] [PubMed] [Google Scholar]
- 17.Fan JR, Peng AL, Chen HC, Lo SC, Huang TH, Li TK. Cellular processing pathways contribute to the activation of etoposide-induced DNA damage responses. DNA Repair (Amst) 2008;7:452–63. doi: 10.1016/j.dnarep.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Lee HZ. Protein kinase C involvement in aloe-emodin- and emodin-induced apoptosis in lung carcinoma cell. Br J Pharmacol. 2001;134:1093–103. doi: 10.1038/sj.bjp.0704342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flores-Rozas H, Kelman Z, Dean FB, Pan ZQ, Harper JW, Elledge SJ, et al. Cdk-interacting protein 1 directly binds with proliferating cell nuclear antigen and inhibits DNA replication catalyzed by the DNA polymerase delta holoenzyme. Proc Natl Acad Sci USA. 1994;91:8655–9. doi: 10.1073/pnas.91.18.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koike M, Yutoku Y, Koike A. Accumulation of p21 proteins at DNA damage sites independent of p53 and core NHEJ factors following irradiation. Biochem Biophys Res Commun. 2011;412:39–43. doi: 10.1016/j.bbrc.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 21.Condorelli F, Salomoni P, Cotteret S, Cesi V, Srinivasula SM, Alnemri ES, et al. Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol Cell Biol. 2001;21:3025–36. doi: 10.1128/MCB.21.9.3025-3036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gschwendt M, Müller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 23.Soltoff SP. Rottlerin: an inappropriate and ineffective inhibitor of PKCδ. Trends Pharmacol Sci. 2007;28:453–8. doi: 10.1016/j.tips.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Bjornsti MA, Benedetti P, Viglianti GA, Wang JC. Expression of human DNA topoisomerase I in yeast cells lacking yeast DNA topoisomerase I: restoration of sensitivity of the cells to the antitumor drug camptothecin. Cancer Res. 1989;49:6318–23. [PubMed] [Google Scholar]
- 25.Riches LC, Lynch AM, Gooderham NJ. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis. 2008;23:331–9. doi: 10.1093/mutage/gen039. [DOI] [PubMed] [Google Scholar]
- 26.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 27.Kiselev E, Deguire S, Morrell A, Agama K, Dexheimer TS, Pommier Y, et al. 7-azaindenoisoquinolines as topoisomerase I inhibitors and potential anticancer agents. J Med Chem. 2011;54:6106–16. doi: 10.1021/jm200719v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.You QD, Li ZY, Huang CH, Yang Q, Wang XJ, Guo QL, et al. Discovery of a novel series of quinolone and naphthyridine derivatives as potential topoisomerase I inhibitors by scaffold modification. J Med Chem. 2009;52:5649–61. doi: 10.1021/jm900469e. [DOI] [PubMed] [Google Scholar]
- 29.Gongora C, Vezzio-Vie N, Tuduri S, Denis V, Causse A, Auzanneau C, et al. New Topoisomerase I mutations are associated with resistance to camptothecin. Mol Cancer. 2011;10:64. doi: 10.1186/1476-4598-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malik M, Nitiss JL. DNA repair functions that control sensitivity to topoisomerase-targeting drugs. Eukaryot Cell. 2004;3:82–90. doi: 10.1128/EC.3.1.82-90.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pommier Y, Redon C, Rao VA, Seiler JA, Sordet O, Takemura H, et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res. 2003;532:173–203. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Chatterjee S, Cheng MF, Trivedi D, Petzold SJ, Berger NA. Camptothecin hypersensitivity in poly(adenosine diphosphate-ribose) polymerase-deficient cell lines. Cancer Commun. 1989;1:389–94. doi: 10.3727/095535489820875129. [DOI] [PubMed] [Google Scholar]
- 33.Capranico G, Marinello J, Baranello L. Dissecting the transcriptional functions of human DNA topoisomerase I by selective inhibitors: implications for physiological and therapeutic modulation of enzyme activity. Biochim Biophys Acta. 2010;1806:240–50. doi: 10.1016/j.bbcan.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Baranello L, Bertozzi D, Fogli MV, Pommier Y, Capranico G. DNA topoisomerase I inhibition by camptothecin induces escape of RNA polymerase II from promoter-proximal pause site, antisense transcription and histone acetylation at the human HIF-1alpha gene locus. Nucleic Acids Res. 2010;38:159–71. doi: 10.1093/nar/gkp817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solier S, Barb J, Zeeberg BR, Varma S, Ryan MC, Kohn KW, et al. Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Cancer Res. 2010;70:8055–65. doi: 10.1158/0008-5472.CAN-10-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baell JB, Huang DC. Prospects for targeting the Bcl-2 family of proteins to develop novel cytotoxic drugs. Biochem Pharmacol. 2002;64:851–63. doi: 10.1016/s0006-2952(02)01148-6. [DOI] [PubMed] [Google Scholar]
- 37.Peyerl FW, Dai S, Murphy GA, Crawford F, White J, Marrack P, et al. Elucidation of some Bax conformational changes through crystallization of an antibody–peptide complex. Cell Death Differ. 2007;14:447–52. doi: 10.1038/sj.cdd.4402025. [DOI] [PubMed] [Google Scholar]
- 38.Taghiyev AF, Guseva NV, Harada H, Knudson CM, Rokhlin OW, Cohen MB. Overexpression of BAD potentiates sensitivity to tumor necrosis factor-related apoptosis-inducing ligand treatment in the prostatic carcinoma cell line LNCaP. Mol Cancer Res. 2003;1:500–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.