

Abstract
Diminished trans-placental glucose transport plays an important role in prenatal calorie restriction induced reduction in fetal growth. Fetal growth restriction (FGR) has an impact in shaping the adult phenotype with trans-generational implications. To understand the mechanisms underlying prenatal calorie restriction induced trans-placental glucose transport, we examined epigenetic regulation of placental glucose transporter (Glut1 and Glut3) expression. We restricted calories by 50% in C57BL6 pregnant mice from gestational day 10 to 19 (CR; n=8) versus controls (CON; n=8) and observed a 50% diminution in placental Glut3 expression (p<0.05) with no effect on Glut1 expression by reverse transcription and quantitative real time PCR. CR enhanced DNA methylation of a CpG island situated ~1000 bp upstream from the transcriptional start site of the glut3 gene, with no such effect on the glut1 gene as assessed by methylation sensitive PCR and bisulfite sequencing. Chromatin immunoprecipitation (ChIP) assays demonstrated enhanced MeCP2 binding to the CpG island of the glut3 gene in response to CR versus CON (p<0.05). Sequential ChIP demonstrated that enhanced MeCP2 binding of the glut3-mCpG island enhanced histone deacetylase 2 (HDAC2) recruitment (p<0.05) but interfered with Sp1 binding (p<0.001) although not affecting Sp3 or Creb/pCreb interaction. We conclude that late gestation CR enhanced DNA methylation of placental glut3 gene. This epigenetic change augmented specific nuclear protein-DNA complex formation that was associated with prenatal CR induced reduction of placental glut3 expression and thereby trans-placental glucose transport. This molecular complex provides novel targets for developing therapeutic interventions aimed at reversing FGR.
Keywords: Fetal growth restriction, epigenetics, histone deacetylases, Sp1 transcription factor
Trans-placental facilitative glucose transport is essential for fetal growth and survival. This process is mediated by placental glucose transporter proteins. Of the 14 isoforms cloned, Glut1 and Glut3 are the predominant isoforms expressed in mammalian placentas. While in the human hemochorial monochorial placentas, both Glut1 and Glut3 are expressed by syncytiotrophoblasts and cytotrophoblasts respectively, in the mouse, these two isoforms are found in the syncytiotrophoblasts lining the labrynthine region of the hemochorial trichorial placentas. The murine labrynthine region mediates materno-fetal glucose transport. Mouse knock down experiments with antisense technology targeting Glut1 revealed significant fetal compromise consisting of growth restriction and developmental anomalies (1), while complete knock out caused embryonic demise (2). Similarly, null homozygous glut3 led to early embryonic loss, while null heterozygosity slowed fetal growth (3). Heterozygous null glut3 pregnant mice expressed reduced transplacental glucose transport supporting an important function of this isoform despite the presence of normal concentrations of placental Glut1.
Human condition of intra-uterine growth restriction revealed no change in placental Glut1 (4) with differing results related to placental Glut3 concentrations (5,6,7). In contrast fetal growth restriction in a mouse caused by prenatal calorie restriction reduced placental Glut3 protein concentrations with diminution of trans-placental glucose transport (8). The mechanism linking prenatal calorie restriction to reduced placental Glut3 protein concentrations remains unknown. Previous studies have demonstrated a role for epigenetic regulation of certain placental genes (9,10). More recently experiments involving genome-wide differential methylation of genes expressed by the murine placenta subjected to calorie restriction revealed a general hypomethylation except for some genes. One such gene was glut3 which was hypermethylated (11) in the 5'-flanking region. However, this observation has not been systematically validated. We therefore hypothesized that prenatal calorie restriction will epigenetically alter the transcriptional machinery responsible for placental Glut3 expression thereby adversely affecting trans-placental glucose transport. We tested this hypothesis by employing our well characterized prenatal calorie restriction during mid- and late gestation murine model and examined DNA methylation of placental glut3 and glut1 genes along with recruitment of key nuclear factors consisting of repressors and activators.
Materials and Methods
Animals
C57/BL6 mice were housed in 12:12 hour light-dark cycle with ad libitum access to standard rodent chow (Harlan Teklad 7013) and water. At eight weeks of age female mice were mated with a male mouse. Presence of a vaginal plug was designated gestational day 1. At gestation day 10, the pregnant mice were either continued on the ad libitum feeding schedule or restricted by 50% of their daily chow intake. On gestational d19, mice were euthanized with phenobarbital (100 mg/kg i.p.) and the placentas and fetuses individually collected, weighed and snap frozen immediately and stored at −80°C. The study protocol was approved by the Animal Research Committee of the University of California Los Angeles (UCLA) in accordance with guidelines of the National Institutes of Health.
DNA methylation and Bisulfite conversion
Genomic DNA was isolated from placental tissue using the DNeasy® Blood and Tissue Kit (Qiagen,Valencia, California) following the manufacturer's recommended protocol specific for DNA methylation experiments. CG Genome Universal Methylated and Unmethylated DNA (Millipore, Temecula, California) were also modified to serve as positive and negative standards (100% values). Extracted genomic DNA (1.5 μg) was modified by sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen, valencia, California).
Methyl sensitive quantitative PCR
Hundred nanograms of bisulfite converted DNA was amplified by MethySYBR quantitative methylation specific PCR (MSP) using primers that were designed by the MethPrimer software specific for either fully methylated (mglut3 or mglut1) or unmethylated (uglut3 or uglut1) CpG island of the glut3 (12) or glut1 5'-flanking region (primers for glut3, methylated forward [mF]: 5'TTTAGTGTTTTTAGGAAAGAAAAATGAC3', m reverse [R]: 5'AAAAAAAATCTTTACCAAATCGAA3'; unmethylated [um]F: 5'TTTAGTGTTTTTAGGAAAGAAAAATGAT3'. umR: 5'AAAAAAAATCTTTACCAAATCAAA3'; glut1, mF: 5'TTTATATTTTAGAATTAATGGCGGC3', mR: 5'CTAACTATACCGACTACGAAACGAA3'; umF: 5'TATATTTTAGAATTAATGGTGGTGG3', umR: 5'TCTAACTATACCAACTACAAAACAAA3'). Designed primers also carried either the converted (ActB) or not converted (ActG) sequences of β-actin special sequence containing no CpG sites as reported previously (13,14) which were used as negative control to correct the CT values. MethySYBR qMSPs were performed in triplicate for each sample and the CT values of mglut3 or mglut1 and uglut3 or uglut1 were normalized using corresponding standard curves and further corrected to ActB. Percent of fully methylated (PMR) and unmethylated (PUR) glut3 or glut1 DNA amounts were calculated assuming mglut+uglut = 100% as previously described (12).
Bisulfite converted DNA sequencing
Primers corresponding to sequences outside the CpG sites on either side but not covering any of the CpG sites were employed to amplify and detect both the modified and unmodified DNA which contain CpG sites with this same primer set. The amplified intervening methylated and unmethylated DNA was subsequently used for bisulfite genomic sequencing PCR (BSP). The PCR products were cloned into pCR 2.1 TOPO vector (Invitrogen, Carlsbad, California) following the manufacturer's instructions. Plasmid DNA was isolated from at least 6 clones (QIAprep spin miniprep) and confirmed by EcoR I digestion (New England Biolabs, Ipswich, Massachusetts), sequenced using M13 forward primer and the Big Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, California), and analyzed on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, California) (12).
SYBR Green real time PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, California) following the manufacturer's instructions. Glut3 and Glut1 mRNAs were quantified using SYBR GreenER-RT-qPCR Assay (Invitrogen, Carlsbad, California) according to the manufacturer's protocol. PCRs were conducted in independent triplicates for each sample. CT values of each copy were determined using default threshold settings. The gene expression level was normalized using endogenous control gene GAPDH, and the relative gene expression was determined using the 2(−delta deltaCT) method (15).
Chromatin immunoprecipitation (ChIP)
ChIP was performed according to ChIP-IT Express kit (Active Motif, Carlsbad, California) with some modifications. Frozen placental tissues were homogenized and then fixed in 1% formaldehyde at room temperature for 10 minutes, then 1 ml of 10% Glycine Stop Fix Solution was added followed by centrifugation at 720 RCF for 5 min. After washing twice with cold PBS, cells were re-suspended in cell lysis buffer supplemented with 7.5 μl protease inhibitor cocktail and PMSF and homogenized with a tissue homogenizer. Cells were then centrifuged and the nuclear pellet was separated and sonicated on ice in 0.5 ml of complete shearing buffer using a Fisher Scientific Sonic Dismembrator 100 with 20 pulses of 20 seconds each with 20 second intervals to obtain chromatin fragments of 300–500 bp size as determined by 2% agarose gel electrophoresis. The sample was then centrifuged at high speed (15,000 rpm for 15 min) to remove cell debris from the crude chromatin lysate. Ten μl of the chromatin lysate was used as the input control for PCR and 50 μl of sheared chromatin was added to a final volume of 100 μl in preparation for the ChIP assay.
The ChIP assay was initiated by adding protein G magnetic beads (25 μl), ChIP buffer (10 μl), sheared chromatin (50 μl), protease inhibitor cocktail (1 μl) and 2 g of antibody against MeCP2 (am 61285, Active Motif, Carlsbad, California), Dnmt1, Dnmt3A, Dnmt3B (am- 39204, am -3906, am-3907, Active Motif, Carlsbad, California), HDAC1 (ab7028-50, Abcam, Cambridge, Massachusetts), HDAC2 (sc 7899, Santa Cruz Biotechnology Inc, Santa Cruz, California), Creb (cs-203204, Millipore Corp., Temecula, CA, USA) or pCrebser133 (cs-204400, Millipore Corp., Temecula, CA, USA) and incubating overnight at 4°C. The magnetic beads were separated from antibody-antigen-DNA binding and the antibody-protein-DNA complex was eluted with an elution buffer from the protein A agarose beads. This was followed by reverse cross-linking of this complex by using 50 μl of reverse crosslink buffer to elute the chromatin. The eluted chromatin was then treated with 5M NaCl at 95°C for 15 minutes in a thermocycler followed by incubating with 2 μl Proteinase K at 37°C for 1 hour. The supernatant which contained the DNA was then centrifuged and used as the template for PCR.
Sequential chromatin immunoprecipitation (SeqChIP)
SeqChIP was performed using the Re- Chip-IT kit (Active Motif, Carlsbad, California). After the DNA-chromatin complex was first immunoprecipitated with MeCP2 antibody (2 μg) and dissociated from the protein G magnetic beads, the chromatin-MeCP2 complex was re-immunoprecipitated using either anti-Sp1 (sc-59), anti-Sp3 (sc-644, Santa Cruz Biotechnology), CREB (cs-203204, Millipore Corp., Temecula, CA), pCrebser133 (cs-204400, Millipore Corp., Temecula, CA, USA) or HDAC2 (sc 7899, Santa Cruz Biotechnology) antibodies (2 μg each) linked to magnetic G beads and the identical ChIP procedure followed as described above.
Real Time qPCR
Real-time PCR amplification was performed in triplicate using SYBR green on a Step One real Time qPCR thermocycler (Applied Biosystem, Foster City, California). ChIP-isolated genomic DNA was used as the template and lamin A was used as the internal control. The amplification cycles consisted of a hot start at 95°C for 10min, followed by 40 cycles at 95°C for 15 sec (denaturation) and 56°C for over 60 sec (annealing), using primers specific for mouse glut3 (forward: 5'ACTGGCCTTTGGGCTTTACT, reverse: 5'-AGCAAACCTGGTCCCTTTCT) that encompass the CpG island, and mouse Lamin A (forward: 5'-AGCCTCTGTCCTTCTGTCCA, reverse: 5'-TGAACTCCTCACGCACTTTG). The fold change in the CR group relative to control samples was determined by the comparative CT method described by Livak et al (15).
Nuclear Protein studies
Solubulized protein homogenates (30 μg) were subjected to electrophoresis on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Transblot; Bio-Rad, Hercules, CA). Membranes were blocked in 5% bovine milk for an hour then probed overnight at 4°C with antibodies against either MeCP2 (1:500 dilution), Sp1 (1:500), Sp3 (1:1000), CREB (1:500), pCREBSer133 (1:1000) or HDAC2 (1:500). The secondary antibody consisted of a horseradish peroxidase-conjugated antibody (1:2500) that allowed detection of the immunoreactive protein bands by enhanced chemiluminescence. Proteins were normalized to vinculin and quantification was performed using the image Quant 5.2 software (GE Healthcare Biosciences, Piscataway, NJ).
Data Analysis
Data are presented as Mean ± SEM. All statistical analyses were performed using Sigmastat 3.5 software (Systat, Point Richmond, CA). Two groups were compared by Student's t-test with normal distribution, while the Mann-Whitney rank sum test was employed in the absence of a normal distribution. Significance was assigned at a p < 0.05.
Results
Tissue-specific methylation of the glut3 gene
Our rationale was to initially establish tissue-specific differences in methylation of the CpG island in the 5'-flanking region of the glut3 gene. Hence we examined three tissues, one which does not express Glut3 (liver), one that highly expresses Glut3 (brain) and the third one being placenta which expresses Glut3 to a lesser extent than that of the brain and is the tissue of interest for the present study (16). This experimental design confirmed the specificity of our methylated and unmethylated primers as well. Figure 1A schematically demonstrates the 5'-flanking region of the murine glut3 gene. The region --1101 to --984 bp encompasses the CpG island which contains 9 CpG sites (UCSC Genome Browser at http://genome.ucsc.edu/; mm9 with TSS of Slc2a3 situated in the negative strand of chr6:122677827 – 122692763; RNA accession number: AK147258.1). The location of the primer sets employed in methylation-specific PCR and in ChIP associated PCR are shown. Figure 1B shows the amplified 5'-flanking region of the glut3 gene using methylated and unmethylated primers flanking this CpG region present upstream to the glut3 transcriptional start site (TSS). Genomic DNA was obtained from the liver (negative control), brain (positive control) and placenta. A ~130 bp band is seen only in the amplified product obtained using methylated primers in the liver with no such band noted with unmethylated primers in keeping with the liver not expressing Glut3. The brain on the other hand that highly expresses Glut3, demonstrates more unmethylated versus methylated glut3 bands. In contrast, the placenta expresses comparable amplification product amounts with both the methylated and unmethylated primers. Thus it appears that the extent of methylation of the glut3 CpG island in each tissue reflects the tissue-specific gene expression pattern previously reported (16).
Figure 1.

A. Schematic representation of the putative CpG island in relation to the transcriptional start site (TSS) within the 5' flanking region of the glut3 gene. Location of primers employed in the methylation specific PCR (MSP) and chromatin immunoprecipitation (ChIP) associated PCR are represented by arrows. B. Representative gel demonstrating the glut3 amplification product (~130 bp) obtained by employed either methylated or unmethylated set of forward and reverse primers on genomic DNA obtained from liver (does not express Glut3), brain (expresses high amounts of Glut3) and placenta (expressed lesser amounts of Glut3 compared to brain). CON = negative PCR control.
Effect of calorie restriction on methylation of the placental glut3 gene
We next explored the possibility of maternal calorie restriction enhancing methylation of the placental glut3 CpG island as a mechanism of reducing Glut3 expression. Figure 2 demonstrates glut3 methylation noted by methylation specific PCR (densitometry; Figure 2A) and bisulfite conversion and sequencing assay of genomic DNA (Figure 2B) along with methylation specific real time qPCR (Figure 2C), and reverse transcription and real time PCR quantification of Glut3 mRNA (Figure 2D) obtained from placentas of CON and CR groups. As can be seen, increased methylation of the glut3 gene was observed in placentas from CR versus CON groups by all three methods. Figure 2A demonstrates the ratio as a percent between methylated (M) and unmethylated (U) amplification products in CON (Left lower panel; M = 38.9±0.45 versus U = 61.09±0.45; p<0.0001; n=6 each) and CR (Middle lower panel; M = 48.75±1.43 versus U = 51.25±1.43; p=0.24; n=6 each) placentas. Comparison of methylated amplification products between CON and CR placentas (Right lower panel; CON = 38.91±0.45 versus CR = 48.75±1.45; p<0.0001; n=6 each) revealed increased methylation in the latter. Similarly bisulfite conversion with sequencing revealed methylation frequency (%) (Figure 2B; CON = 26.73±1.6 versus CR = 48.46±1.21; p<0.0001; n= 6 each) and ΔΔCT (Figure 2C; CON = 0.8±0.1 versus CR = 1.25±0.13; p = 0.043; n=6 each) in CON and CR placentas. An associated reduction in Glut3 mRNA was evident in CR versus CON groups (Figure 2D; CON = 3.0±0.4 versus CR = 1.2±0.3; p = 0.001; n=6 each). Hence, these results suggest that maternal calorie restriction increases methylation of the glut3 CpG island while reducing Glut3 mRNA concentrations.
Figure 2.
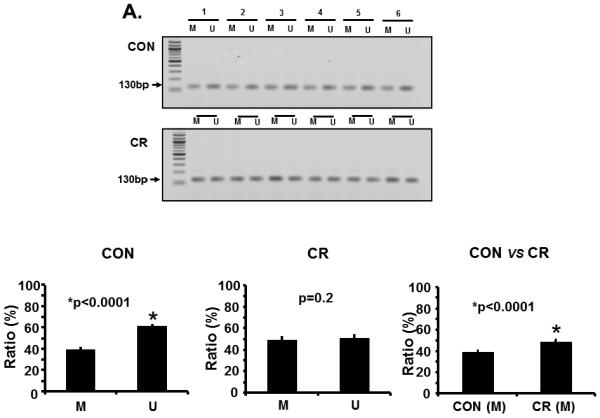
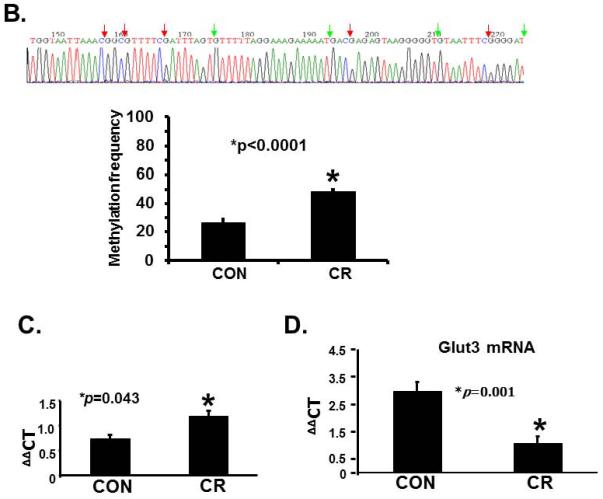
A. Representative gels demonstrating the amplification products of the glut3 5'-flanking region in control (CON; top panel) and calorie restricted (CR; bottom panel) groups by methylation specific PCR (MSP) are shown above in the inset. The numbers on top (1 to 6) represent DNA obtained from placentas of different pregnant mice. Amplified products recognized by methylated (M) and unmethylated (U) primers are seen. Relative frequency of methylation or unmethylation was calculated based on the optical density of the amplification products (arbitrary units) as a ratio of M+U value for a given sample (i.e. M=M/M=U or U=U/M+U). n=6/group, *p value as shown in each graph when compared to the corresponding CON group. The graphs shown below depict the relative frequency of methylation (M) and unmethylation (U) in CON (left) and CR (middle) groups with the methylation frequency compared between the CON and CR groups (right). B. Bisulfite sequencing of the glut3 5'-flanking region containing the CpG island. The top panel demonstrates the sequencing data with red arrows depicting the methylated and green arrows the unmethylated CpG sites within the CpG island of genomic DNA recovered from CR placentas. The bar graph in the bottom panel demonstrates the quantification of the methylation frequency in CON versus CR groups. *p value shown, n=6 clones in each experimental group. C. The bar graph demonstrates quantification of CpG island methylation by methylation specific qPCR of genomic DNA obtained from CON and CR placentas and shown as ΔΔCT values. *p value is shown, n = 6 per each experimental group. D. The bar graph demonstrates placental Glut3 mRNA concentrations assessed by reverse-transcription and real time quantitative PCR. *p value is shown. n=6 in each experimental group.
Effect of calorie restriction on methylation of the placental glut3 gene
In addition to Glut3, significant amounts of Glut1 are also expressed in the placenta (8), hence we next questioned whether maternal calorie restriction also affected methylation of CpG islands detected in the placental glut1 gene. Figure 3 demonstrates quantification of methylated (M) and unmethylated (U) glut1 amplification products obtained from CON (Left lower panel; M = 39.54±0.74 versus U = 60.46±0.74; p<0.0001; n=5 each) and CR (Middle lower panel; M = 42.16±0.41 versus U = 57.84±0.41; p<0.0001; n=5 each) placentas. Next methylated (M) glut1 amplification products were compared between CON and CR groups (Right lower panel; CON = 39.54±0.74 versus CR = 42.16±0.41; p=0.01; n=5 each). Thus, in contrast to glut3, minimal differential methylation was evident with the glut1 gene between the two groups by methylation specific PCR. Definitive confirmation by bisulfite conversion and sequencing, revealed all CpG sites within the proximal CpG island overlapping the transcriptional start site and 5'- to the ATG translational start site (UCSC Genome Browser at http://genome.ucsc.edu/; mouse gene Slc2a1 - chr4:119,108,745–119,137,329) converted to TpGs demonstrating no differential methylation between CON and CR groups.
Figure 3.

Schematic representation of the 5'-flanking region of the murine glut1 gene is shown above with locations of the proximal CpG island and the primers (p) employed in methylation specific PCR with the amplification product size in parenthesis. Representative gels demonstrating the amplification products of the glut1 5'-flanking region in control (CON; top panel) and calorie restricted (CR; bottom panel) groups by methylation specific PCR (MSP) are shown (n=5 placentas from different pregnant mice). Amplified products recognized by methylated (M) and unmethylated (U) primers are seen. Relative frequency of methylation and unmethylation in CON (bottom left panel) and CR (bottom middle panel) groups was calculated based on the optical density of the amplification products (arbitrary units) as a ratio of M+U value for a given sample (i.e. M=M/M=U or U=U/M+U) represented as a percent. n=5/group, *p value as shown in each graph when compared to the corresponding CON group. The bottom right panel shows a graph that depicts the relative frequency of methylation (M) in CON and CR groups with the methylation frequency compared between the CON and CR groups, n=5/group. This was followed by bisulfite conversion and sequencing which demonstrated no methylated CpG sites located 5'- to the ATG region in response to CR (negative data not shown).
Protein interaction with the glut3 gene
Figure 4A demonstrates qualitatively the amplification products which contain the CpG island of the glut3 gene eluted from ChIP of MeCP2, Dnmt1, Dnmt3A, Dnmt3B, HDAC1 and HDAC2. In contrast, Creb or pCrebser133 were unable to bind DNA generating no amplification product while Sp1 and Sp3 demonstrated barely detectable binding to the CpG island of the glut3 gene. No amplification product is noted in the presence of a non-specific IgG (negative control) but detected within the input chromatin (positive control). Quantification of the amplification product (~239bp) was undertaken employing ChIP assay with real time qPCR which demonstrated a significant increase in MeCP2 recruitment (CON = 1±0.09 versus CR = 1.66±0.24; p<0.05; n=7 each) to the CpG island of the glut3 gene in CR compared to CON placentas. There was also a trend towards an increase (~46%; CON = 1±0.14 versus CR = 1.47±0.23; p<0.1; n=13 each) in binding of Dnmt3B to the glut3-CpG in CR versus CON placentas. In contrast, no difference between the two groups was observed in the case of Dnmt1 (CON = 1±0.22 versus CR = 0.82±0.27; p<0.4; n=9 each), Dnmt3A (CON = 1±0.19 versus CR = 0.89±0.22; p<0.4; n=13 each), HDAC1 (CON = 1±0.11 versus CR = 0.85±0.12; p<0.4; n=13 each) or HDAC2 (CON = 1±0.13 versus CR = 0.96±0.12; p<0.4; n=9 each). While the undetectable CREB and pCREB could not be quantified, a trend towards a reduction in Sp1 (CON = 1±0.33 versus CR = 0.37±0.16; p<0.2; n=6 each) with a trend towards an increase in Sp3 (CON = 1±0.15 versus CR = 1.5±0.17; p<0.2; n=6 each) was observed that did not achieve statistical significance. These observations prompted our next set of experiments.
Figure 4.

A. Representative 2% agarose gels (top panel) demonstrate the glut3 5'-flanking region that contains the CpG island amplified by PCR (239 bp) from chromatin immunoprecipitation (ChIP) assays conducted in control (CON) and calorie restricted (CR) placentas in the presence of specific antibodies (anti-MeCP2, Dnmt1, Dnmt3a, Dnmt3b, HDAC1, HDAC2, Sp1, Sp3, CREB and pCREB; n=6–13/group), non-specific antibody (IgG) or no antibody (input). B. Sequential ChIP was conducted in control (CON) and calorie restricted (CR) placentas first with the anti-MeCP2 antibody followed by anti-Sp1, Sp3 or HDAC2, CREB and pCREB antibodies denoted by (s). ChIP (A) or sequential ChIP (B) followed by real time quantitative PCR denoted as relative quantification (RQ) and shown as a fraction of the corresponding CON group which assessed the recruitment of various nuclear proteins as depicted in bar graphs (bottom panels), n=3,9,12/group.*p<0.05, **p<0.001 and #p<0.1 (not significant) compared to the corresponding control group. C. Representative Western blots demonstrating the MeCP2, HDAC2, Sp1, Sp3, CREB and pCREB with vinculin (internal loading control) protein bands (top panel) and the quantification depicted in the bar graph (bottom panel) as a ratio to vinculin and presented as a percent of the corresponding CON group (n=6–8/group).
Protein-MeCP2 interaction with the glut3 gene
We questioned whether the enhanced recruitment of MeCP2 to the glut3-mCpG in response to CR may be responsible for mediating either increased recruitment of repressors or interfere with recruitment of activators to the glut3 gene. Hence, we designed sequential ChIP experiments. However, as seen in Figure 4B sequential ChIP with qPCR following MeCP2 recruitment to the glut3-CpG island demonstrated enhanced recruitment of HDAC2 (CON = 1±0.25 versus CR = 2.67±0.29; p<0.05; n=3 each) with diminished recruitment of Sp1 (CON = 1±0.11 versus CR = 0.36±0.1; p<0.001; n=12 each) but not Sp3 (CON = 1±0.34 versus CR = 1.25±0.23; p<0.4; n=9 each), CREB or pCREB (not detectable and unable to quantify) in CR versus CON placentas. These changes were in the face of no differences in nuclear protein amounts of total MeCP2 (CON = 100±21.8 versus CR = 110±15.7; p<0.4; n=8 each), Sp1 (CON = 100±15.8 versus CR = 107±22.4; p<0.4; n=6 each), Sp3 (CON = 100±10.27 versus CR = 111±9.2; p <0.4; n=8 each), pCREB (CON = 100±17.46 versus CR = 119±26.5; p<0.4; n=8 each) (low amounts of CREB could not be quantified) or HDAC2 (CON = 100±12.57 versus CR = 103±20.16; p<0.4; n=8 each) concentrations between CR and CON placentas (Figure 4C).
Molecular mechanisms responsible for reduction of placental Glut3 expression in response to maternal calorie restriction
Based on these results, we schematically summarized a proposed molecular model of placental glut3 gene transcription under control (Figure 5A) and calorie restricted (Figure 5B) conditions.
Figure 5.

Schematic representation of the glut3-CpG DNA-protein-protein interaction under control (CON) (A) and calorie restricted (CR) (B) conditions demonstrating the proposed mechanism responsible for placental glut3 gene transcription.
Discussion
We have for the first time validated an in-vivo role for DNA methylation of the CpG island present within the 5'-flanking region of the placental glut3 gene. Taking a candidate gene approach which is known to yield results that are sensitive and tissue-specific, we demonstrated that prenatal caloric restriction enhances DNA methylation of the murine placental glut3 gene while not affecting the predominantly expressed glut1 gene. This change is associated with a diminution in placental Glut3 mRNA while not changing Glut1 mRNA. Our prior genome-wide studies in the prenatal calorie restricted mouse placenta also revealed differential hypermethylation on the negative strand of the glut3 gene (11). Our present study is the first to validate this bioinformatic analysis of genome-wide DNA methylation studies. Previous investigations demonstrate that prenatal caloric restriction caused global DNA hypomethylation in the placenta (11) and embryos (17,18). Other investigations focused on whole-genome wide DNA methylation in response to prenatal high fat diet demonstrated a differential response uncovering groups of placental genes that were hypermethylated (19,20), the opposite finding of what was reported with prenatal caloric restriction.
Using two methods, namely methylation sensitive PCR and either semi-quantification of the gel isolated amplification products or quantification by real time qPCR and bisulfite conversion followed by sequencing of genomic DNA obtained from placentas, both revealed increased methylation of the CpG island in the 5'-flanking region of the glut3 gene in CR versus CON placentas. This differential methylation of the glut3 gene yielded reduced placental Glut3 expression, while the absence of such differential methylation of the glut1 gene was associated with no change in placental Glut1 expression between CR and CON placentas (8). This specific reduction in Glut3 mRNA in response to prenatal CR translates into a diminution in placental Glut3 protein concentrations which in turn reduces trans-placental glucose transport (8). Thus, the molecular mechanism responsible for reduction of placental Glut3 expression and function (glucose transport) in prenatal calorie restriction is DNA methylation of the CpG island within the 5'-flanking region of the glut3 gene.
There is an accumulation of information regarding the role of imprinted genes expressed in the placenta playing a significant role in trans-placental nutrient transport. Placenta specific Igf-2 gene null mice expressed a reduction in trans-placental system A amino acid transport with a compensatory increase in placental glut3 gene expression (21). In contrast, when placental glut3 gene was reduced in a null heterozygous glut3 mouse, trans-placental glucose transport decreased despite normal placental Glut1 concentrations, with a compensatory increase in System A amino acid transport (3). Placental Glut3 expressed in syncytiotrophoblasts that line the labrynthine region which forms the materno-fetal tissue barrier, has a significant role in mediating trans-placental glucose transport in the murine hemochorial and trichorial placenta.
In the human hemochorial and monochorial placenta, recently a gestation dependent increased methylation of CpGs noted in the glut3 gene along with a corresponding reduction in Glut3 expression with no similar effect on the glut1 gene or Glut1 expression was reported (22). However, studies involving the human intra-uterine growth restricted (IUGR) condition demonstrated no change in placental GLUT1 protein concentrations (4) but a reduction in GLUT3 protein (6). The prenatal calorie restricted murine model provides the ability to isolate the nutrient restrictive stimulus that causes fetal growth restriction and examine its impact on placental glucose transporter gene expression and function. We have previously observed that prenatal caloric restriction in the mouse reduced placental Glut3 protein concentrations which translated into a diminution in trans-placental glucose transport (8). This reduction in trans-placental glucose transport closely mimics that reported previously in the null heterozygous glut3 pregnant mouse (3). Thus our present finding of hypermethylation of the placental glut3 gene in response to calorie restriction has functional significance in-vivo whereby it alters trans-placental glucose transport and reduces fetal growth (8).
Calorie restricted reduction in glut3 transcription and the resultant mRNA due to epigenetic mechanisms involves relative gene silencing by enhanced DNA methylation and recruitment of certain histone modifying enzymes, in particular histone deacetylases which contribute to the ultimate partial silencing of a gene (23, 24). Alternatively transcription factors which may include reduced recruitment of activators or increased recruitment of repressors could also inhibit glut3 gene transcription (25,26). Hence in the present study, we examined both these molecular mechanisms as a consequence of enhanced DNA methylation of the glut3 promoter.
In the presence of a trend towards a ~46% increased involvement of Dnmt3B, a 1.8-fold enhanced recruitment of MeCP2 to the methylated CpG island of the glut3 gene occurred. This key recruitment of a nuclear protein attracted HDAC2 by 2.8-fold which mediates histone deacetylation. Histone deacetylation depending on the amino acid residues involved can further contribute towards the partial silencing of the downstream gene expression (27). This DNA-protein-protein complex consisting of glut3-mCpG-MeCP2-HDAC2 appears to have an in-vivo repressor effect on glut3 gene expression, reflected by the associated mRNA concentrations. Previously employing human placental explants or trophoblasts in-vitro a role for histone deacetylation on partially silencing gene expression was reported (28, 29). Our present study suggests an in-vivo role for histone deacetylation in regulating placental glut3 gene expression.
In addition, the enhanced formation of the glut3-mCpG-MeCP2-HDAC2 complex negatively affected the recruitment of Sp1 similar to the previous observation of hypermethylation of the Keap1 promoter in lung cancer cells (30). Sp1 has been described as a trans-activator of various downstream genes (31) including glut3 in muscle (32) and glut1 in myocardium (33) and trophoblasts (34). Previously, employing drosophila Schneider cells that lack endogenous murine genes including Sp1 and Sp3 revealed that exogenous Sp1 bound the Sp1-binding site (between −203 to −177 bp) on the murine glut3 gene and reduced glut3-luciferase transcriptional activity. In contrast, Sp3 did not bind the Sp1 site directly nor Sp1, but perhaps by interacting with pCREB led to enhanced murine glut3-luciferase activity. These investigations were pursued in-vitro with non-mammalian cultured cells reflecting the role of exogenous transcription factors. However, the function of transcription factors is tissue-specific varying based on interaction with other nuclear proteins and the cell-specific environment (25). Our present investigation is the first to examine the role of Sp1/Sp3/pCREB in placental tissue under in-vivo conditions of prenatal calorie restriction. Under such conditions, reduction in the recruitment of Sp1 due to the augmented formation of the glut3-mCpG-MeCP2-HDAC2 complex with no change in either Sp3 or CREB/pCREB along with a diminution in Glut3 mRNA is suggestive of an in-vivo trans-activating role for Sp1 in the case of placental glut3 transcription under control conditions.
In conclusion, our present observations are novel on three fronts: 1) mid- to late gestation CR enhances DNA methylation of the placental glut3 gene, 2) MeCP2 binds the methylated CpG island of the placental glut3 gene, and 3) differential recruitment of MeCP2 that enhances HDAC2 but interferes with Sp1 recruitment to the glut3 gene is associated with reduced Glut3 mRNA concentrations. These molecular findings are reflected in prior observations of a diminution in placental Glut3 protein concentrations and trans-placental glucose transport associated with fetal growth restriction (8). This biological activity of the molecular machinery related to hypermethylation of the placental glut3 gene mimics that observed with 50% reduction in placental glut3 gene expression encountered in the pregnant null heterozygous glut3 mouse (3). All these findings collectively set the stage for epigenetic regulation of placental glut3 gene in response to prenatal caloric restriction. Our studies pave the way for future studies to unravel whether glut3 is an imprinted gene with a differentially methylated region that is parentally silenced under certain circumstances and secondly whether the CR induced differential glut3 DNA methylation can be reversed with dietary manipulations that overcome prenatal caloric restriction. Additionally whether this differential DNA methylation of the placental glut3 gene acquired during mid- to late- gestation has any trans-generational consequences warrants future study.
Acknowledgments
This work was supported by grants from NIH HD 46979 and HD 33997.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heilig CW, Saunders T, Brosius FC, 3rd, Moley K, Heilig K, Baggs R, Guo L, Conner D. Glucose transporter-1 deficient mice exhibit impaired development and deformities that are similar to diabetic embryopathy. Proc Natl Acad Sci USA. 2003;100:15613–15618. doi: 10.1073/pnas.2536196100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J, Noebels JL, De Vivo DC. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet. 2006;15(7):1169–79. doi: 10.1093/hmg/ddl032. [DOI] [PubMed] [Google Scholar]
- 3.Ganguly A, McKnight RA, Raychaudhuri S, Shin BC, Ma Z, Moley K, Devaskar SU. Glucose transporter isoform-3 mutations cause early pregnancy loss and fetal growth restriction. Am J Physiol Endocrinol Metab. 2007;292:E1241–1255. doi: 10.1152/ajpendo.00344.2006. [DOI] [PubMed] [Google Scholar]
- 4.Jansson T, Ylvén K, Wennergren M, Powell TL. Glucose transport and system A activity in syncytiotrophoblast microvillous and basal plasma membranes in intrauterine growth restriction. Placenta. 2002;23(5):392–9. doi: 10.1053/plac.2002.0826. [DOI] [PubMed] [Google Scholar]
- 5.Jansson T, Wennergren M, Illsley NP. Glucose transporter expression and distribution in the human placenta throughout gestation and in intrauterine growth retardation. J Clin Endocr Metab. 1993;77:1554–1562. doi: 10.1210/jcem.77.6.8263141. 1993. [DOI] [PubMed] [Google Scholar]
- 6.Kainulainen H, Jarvinen T, Heinonen PK. Placental glucose transporters in fetal intrauterine growth retardation and macrosomia. Gynecol Obstet Invest. 1997;44(2):89–92. doi: 10.1159/000291493. [DOI] [PubMed] [Google Scholar]
- 7.Janzen C, Lei MY, Cho J, Sullivan P, Shin BC, Devaskar SU. Placental glucose transporter 3 (GLUT3) is up-regulated in human pregnancies complicated by late-onset intrauterine growth restriction. Placenta 2013 Aug 28. doi: 10.1016/j.placenta.2013.08.010. Pii: S0143-4004(13)00695-4. Doi: 10.1016/j.placenta.2013.08.012010. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganguly A, Collis L, Devaskar SU. Placental glucose and amino acid transport in calorie-restricted wild-type and Glut3 null heterozygous mice. Endocrinology. 2012;153(8):3995–4007. doi: 10.1210/en.2011-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelissen EC, van Montfoort AP, Dumoulin JC, Evers JL. Epigenetics and the placenta. Hum Reprod Update. 2011;17(3):397–417. doi: 10.1093/humupd/dmq052. [DOI] [PubMed] [Google Scholar]
- 10.Yuen RK, Penaherrera MS, von Dadelszen P, McFadden DE, Robinson DP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet. 2010;18(9):1006–1012. doi: 10.1038/ejhg.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen PY, Ganguly A, Rubbi L, Orozco LD, Morselli M, Ashraf D, Jaroszewicz A, Feng S, Jacobsen SE, Nakano A, Devaskar SU, Pellegrini M. Intra-Uterine Calorie Restriction Affects Placental DNA Methylation and Gene Expression. Physiol Genomics. 2013 May;21 doi: 10.1152/physiolgenomics.00034.2013. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Shin BC, Thamotharan S, Devaskar SU. Creb1-Mecp2-(m)CpG complex transactivates postnatal murine neuronal glucose transporter isoform 3 expression. Endocrinology. 2013;154(4):1598–611. doi: 10.1210/en.2012-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo PK, Watanabe H, Cheng PC, Teo WW, Liang X, Argani P, Lee JS, Sukumar S. MethySYBR, a novel quantitative PCR assay for the dual analysis of DNA methylation and CpG methylation density. J Mol Diagn. 2009;11:400–414. doi: 10.2353/jmoldx.2009.080126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hattermann K, Mehdorn HM, Mentlein R, Schultka S, Held-Feindt J. A methylation-specific and SYBR-green-based quantitative polymerase chain reaction technique for O6-methylguanine DNA methyltransferase promoter methylation analysis. Anal Biochem. 2008;377:62–71. doi: 10.1016/j.ab.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. “Analysis of relative gene expression data using real-timequantitative PCR and the method.”. Methods. 25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Thamotharan S, Stout D, Shin BC, Devaskar SU. Temporal and spatial distribution of murine placental and brain GLUT3-luciferase transgene as a readout of in vivo transcription. Am J Physiol Endocrinol Metab. 2013;304(3):E254–66. doi: 10.1152/ajpendo.00214.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdg GC. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br J Nutr. 2008;100:278–282. doi: 10.1017/S0007114507894438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nut. 2007;97:1064–1073. doi: 10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabory A, Ferry L, Fajardy I, Jouneau L, Gothié JD, Vigé A, Fleur C, Mayeur S, Gallou-Kabani C, Gross MS, Attig L, Vambergue A, Lesage J, Reusens B, Vieau D, Remacle C, Jais JP, Junien C. Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PLoS One. 2012;7(11):e47986. doi: 10.1371/journal.pone.0047986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallou-Kabani C, Gabory A, Tost J, Karimi M, Mayeur S, et al. Sex- and Diet-Specific Changes of Imprinted Gene Expression and DNA Methylation in Mouse Placenta under a High-Fat Diet. PLoS One. 2010;5(12):e14398. doi: 10.1371/journal.pone.0014398. doi: 10.1371/journal.pone.0014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Constância M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A, Sibly C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417:945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- 22.Novakovic B, Gordon L, Robinson WP, Desoye G, Saffery R. Glucose as a fetal nutrient: dynamic regulation of several glucose transporter genes by DNA methylation in the human placenta across gestation. J Nutr Biochem. 2013;24(1):282–288. doi: 10.1016/j.jnutbio.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Goldberg AD, David Allis' C, Bernstein E. Epigenetics: A Landscape Takes Shape. Cell. 2007;128(4):635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Devaskar SU, Raychaudhuri S. Epigenetics--a science of heritable biological adaptation. Pediatr Res. 2007;61(5 Pt 2):1R–4R. doi: 10.1203/pdr.0b013e31805cdbd8. [DOI] [PubMed] [Google Scholar]
- 25.Rajakumar A, Thamotharan S, Raychaudhuri N, Menon RK, Devaskar SU. Trans-activators regulating neuronal glucose transporter isoform-3 gene expression in mammalian neurons. J Biol Chem. 2004;279(25):26768–79. doi: 10.1074/jbc.M402735200. [DOI] [PubMed] [Google Scholar]
- 26.Rajakumar RA, Thamotharan S, Menon RK, Devaskar SU. Sp1 and Sp3 regulate transcriptional activity of the facilitative glucose transporter isoform-3 gene in mammalian neuroblasts and trophoblasts. J Biol Chem. 1998;273(42):27474–83. doi: 10.1074/jbc.273.42.27474. [DOI] [PubMed] [Google Scholar]
- 27.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4(1):4–5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi JC, Holtz R, Murphy SP. Histone deacetylases inhibit IFN-gamma-inducible gene expression in mouse trophoblast cells. J Immunol. 2009;182(10):6307–6315. doi: 10.4049/jimmunol.0802454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munro SK, Mitchell MD, Ponnampalam AP. Histone deacetylase inhibition by trichostatin A mitigates LPS induced TNFα and IL-10 production in human placental explants. Placenta. 2013 Apr 25; doi: 10.1016/j.placenta.2013.04.004. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 30.Guo D, Wu B, Yan J, Li X, Sun H, Zhou D. A possible gene silencing mechanism: hypermethylation of the Keap1 promoter abrogates binding of the transcription factor Sp1 in lung cancer cells. Biochem Biophys Res Commun. 2012;428(1):80–5. doi: 10.1016/j.bbrc.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Li L, He S, Sun JM, Davie JR. Gene regulation by Sp1 and Sp3. Biochem Cell Biol. 2004;82(4):460–71. doi: 10.1139/o04-045. [DOI] [PubMed] [Google Scholar]
- 32.Copland JA, Pardini AW, Wood TG, Yin D, Green A, Bodenburg YH, Urban RJ, Stuart CA. IGF-1 controls GLUT3 expression in muscle via the transcriptional factor Sp1. Biochim Biophys Acta. 2007;1769(11–12):631–40. doi: 10.1016/j.bbaexp.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Santalucia T, Boheler KR, Brand NJ, Sahye U, Fandos C, Vinals F, Ferre J, Testar X, Palacin M, Zorzano A. Factors involved in GLUT-1 glucose transporter gene transcription in cardiac muscle. J Biol Chem. 1999;274(25):17626–34. doi: 10.1074/jbc.274.25.17626. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto Y, Sakata M, Yamamoto T, Nishio Y, Adachi K, Ogura K, Yamaguchi M, Takeda T, Tasaka K, Murata Y. Involvement of nuclear transcription factor Sp1 in regulating glucose transporter-1 gene expression during rat trophoblast differentiation. Biochem Biophys Res Commun. 2001;288(4):940–8. doi: 10.1006/bbrc.2001.5860. [DOI] [PubMed] [Google Scholar]