

Abstract
Mesenchymal stem cells (MSCs) possess unique paracrine and immunosuppressive properties, which make them useful candidates for cellular therapy. Here, we address how cellular senescence influences the therapeutic potential of human MSCs (hMSCs). Senescence was induced in bone marrow-derived hMSC cultures with gamma irradiation. Control and senescent cells were tested for their immunoregulatory activity in vitro and in vivo, and an extensive molecular characterization of the phenotypic changes induced by senescence was performed. We also compared the gene expression profiles of senescent hMSCs with a collection of hMSCs used in an ongoing clinical study of Graft Versus Host disease (GVHD). Our results show that senescence induces extensive phenotypic changes in hMSCs and abrogates their protective activity in a murine model of LPS-induced lethal endotoxemia. Although senescent hMSCs retain an ability to regulate the inflammatory response on macrophages in vitro, and, in part retain their capacity to significantly inhibit lymphocyte proliferation, they have a severely impaired migratory capacity in response to proinflammatory signals, which is associated with an inhibition of the AP-1 pathway. Additionally, expression analysis identified PLEC, C8orf48, TRPC4, and ZNF14, as differentially regulated genes in senescent hMSCs that were similarly regulated in those hMSCs which failed to produce a therapeutic effect in a GVHD trial. All the observed phenotypic alterations were confirmed in replicative-senescent hMSCs. In conclusion, this study highlights important changes in the immunomodulatory phenotype of senescent hMSCs and provides candidate gene signatures which may be useful to evaluate the therapeutic potential of hMSCs used in future clinical studies.
Keywords: Mesenchymal stem cells, Senescence, Cellular therapy, Immunotherapy
Introduction
Mesenchymal stem cells (MSCs) are multipotent precursors of nonhematopoietic mesodermal lineages that are capable of clonal differentiation into both mesenchymal and nonmesenchymal lineages. Although isolated predominantly from bone marrow and subcutaneous fat, MSCs can be found in virtually all mammalian connective tissues (reviewed in Bernardo et al. [1]). Human MSCs (hMSCs) can be isolated easily from small tissue biopsies, and expanded ex vivo for many cell passages without significant loss of differentiation potential. Together, these properties mark hMSCs as ideal candidates for cell and genetic therapy. Indeed, in the last decade, hMSCs have been used to treat a variety of traumatic and degenerative disorders, including bone fractures, cartilage lesions, and myocardial infarction.
In addition to their robust differentiation potential, MSCs have a remarkable capacity for inhibiting the immune response; an activity known generally as immunomodulation or immunoregulation (reviewed in Gebler et al. [2]). This activity bestows MSCs with a hypoimmunogenic phenotype, allowing them to evade, at least temporally, the alloreactive immune response, even in xenogeneic conditions. This immunoregulatory capacity of hMSCs includes the inhibition of host T-cell proliferation and cytokine production, B-cell proliferation, NK cell activation, and dendritic cell maturation. By exploiting these characteristics, hMSCs have been used successfully in different experimental models to prevent allogeneic transplant rejection, and to treat various experimental autoimmune/inflammatory disorders such as graft versus host disease (GVHD), experimental autoimmune encephalitis, collagen-induced arthritis, and septic shock. This immunoregulatory capacity is also being tested in clinical studies of GVHD, Crohn’s disease, and other human inflammatory disorders.
While the immunoregulatory capacity of MSCs is recognized, the specific molecular mechanisms that govern this ability are poorly understood. Several cell-to-cell and soluble signals have been identified as potential drivers of immune suppression by MSCs, including prostaglandin E, indoleamine 2,3-dioxygenase, NO, and IL-10, but none of these can explain fully the diverse immune modulatory activities of MSCs observed in vivo. Moreover, the immunosuppressive ability of MSCs is not constitutive, but requires proinflammatory signals such as IFNc, which induce important phenotypic changes in MSCs [3,4].
Increasing evidence suggests that the regenerative capabilities of transplanted MSCs in damaged tissues, such as infarcted myocardium, are linked more to their paracrine activity (including anti-inflammatory actions), than to their potential for differentiation into specific cell lineages (reviewed in Liang et al. [5]). Accordingly, understanding the physiological and pathological factors that affect the immune modulatory activity of hMSCs is not only relevant for the treatment of autoimmune/inflammatory disorders, but should also be considered a key step in the development of effective hMSC strategies for the treatment of degenerative pathologies.
Previously, we validated the use of xenogeneic murine models of autoimmune and inflammatory diseases to study the immunoregulatory activity of hMSCs in vivo. We found that systemic infusion of hMSCs in mice significantly reduced the incidence and severity of experimental arthritis [6], colitis [7, 8], and sepsis [8], to a degree similar to that of syngeneic murine MSCs. These therapeutic effects were mediated through downregulation of both Th1-driven autoimmune and inflammatory responses; as hMSCs migrate to secondary lymphoid organs, they reduce the production of inflammatory cytokines and chemokines, and induce de novo generation of CD41CD251FoxP31 Treg with capacity to suppress self-reactive T effector responses [9].
Although hMSCs are available from different tissues, their quantity in the body is relatively low. As most cell therapy protocols use a minimum of 20–100 million hMSCs per treatment (autologous transplantation), hMSCs need to be expanded in vitro for at least 4–8 weeks prior to transplantation. The length of this period, and the overall quality of the hMSC product, depends both on the isolation and culture methods used, and also the clinical history, age, and the genetic makeup of the donor. Still longer expansion regimens would be required for more clinically useful off-the-shelf allogenic hMSC therapies.
Recently, it has been demonstrated that human cells (fibroblasts and normal and tumorigenic epithelial cells), induced to senesce through replicative exhaustion, DNA damage, or other stresses, mount a severe proinflammatory response through the activation of a senescence-associated secretory phenotype (SASP, reviewed in Tchkonia et al. [10]). This SASP is characterized by a 10–800-fold overexpression and secretion of various proinflammatory chemokines including IL-8, IL-6, Granulocyte macrophage (GM)-CSF, GROa, GROb, GROc, ICAM-1, and MCP1–4. Additionally, it is becoming more accepted that all ex vivo cell expansion procedures favor the accumulation of aneuploid cells, which is intimately associated with the progression of senescence [11–13]. Other studies describe a direct correlation between telomerase activity (which blocks cell senescence) and stem cell function (reviewed in Tumpel and Rudolph [14]). Therefore, it is evident that a pre-expansion test, which can anticipate the behavior of the hMSCs, would be a valuable tool to help improve expansion yields and also to guarantee biosafety and functional value of the clinical product.
Considering that immunosuppressive activity is induced upon exposure of MSCs to proinflammatory signals, we hypothesized that this response will be dependent on the functional state of the MSCs. We show here that cell senescence strongly impairs the immunoregulatory capacity of hMSC in vivo, and suggest that examination of cell senescence markers could be a useful approach to evaluate the clinical potential of hMSCs.
Materials and Methods
Biological Samples
The study was carried out in accordance with guidelines of the Instituto de Salud Carlos III (Madrid, Spain). For the induction of senescence and related experiments, four independent hMSC lines (listed as #19, #33, #44, and #45), isolated from bone marrow tissue of male or female donors aged 18–29 years, were acquired from Inbiobank Stem Cell Bank (San Sebasti an, Spain). For some experiments, we also used eight bone marrow-derived hMSC samples obtained from an ongoing clinical study of graft-versus-host disease (GVHD). A brief description of the clinical study is included in Supporting Information.
Cell Culture
hMSCs (4 × 103 cells per square centimeter) were cultured in low glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, penicillin 100 U/ml, and streptomycin 1,000 U/ml (all culture reagents were from Sigma-Aldrich, St. Louis, MO). Cells were cultured in a humidified 37°C incubator at 5% CO2, and were passaged once per week. Media were changed twice weekly.
Induction of Cell Senescence
Cell senescence was induced by exposing cultured hMSCs to ionizing radiation (10 Gy) produced by a high-voltage X-ray-generator tube (Mark I-68, J.L. Shepherd & Associates, San Fernando, CA). Senescence was assessed 10 days after irradiation by cytochemical staining for β-galactosidase, as described in Supporting Information. The obtained radiation-induced senescent cells were denoted SEN+. Alternatively, in some experiments, cells were grown until reaching replicative exhaustion (replicative-senescent cells).
TERT Lentiviral Transduction
Primary hMSCs at passage 5 were transduced as described [15] with a lentiviral vector encoding the human telomerase reverse transcriptase catalytic subunit (pRRL.hTERT) [16]. Cells were evaluated for long-term cell proliferation, telomerase expression and activity, and telomere length after >10 passages (described in Supporting Information). Immortalized cells were denoted SEN−.
Induction of Endotoxemia and Sepsis
Endotoxemia was induced in 7–10-week-old BALB/c male mice (Harlan Laboratories, Gannat, France) by i.p. injection of 400 μg/mouse lipopolysaccharide (LPS, from Escherichia coli serotype 055:B5; Sigma-Aldrich). At 30 minutes after LPS injection, mice received an i.p. injection of phosphate buffered saline (PBS) or 1 million hMSCs in PBS. Survival after LPS injection was monitored every 12 hours for 6 days.
Serum, liver, lungs, and small intestines were collected 6 hours after LPS administration and used for protein extraction and cytokine measurement. All experiments were performed in accordance with institutional guidelines for the Care and Use of Laboratory Animals in Research, and approved by CNIC.
Lymphocyte Proliferation Assay
Buffy coat preparations were obtained from whole blood of healthy volunteers following the guidelines of the Centro de Transfusion de la Comunidad de Madrid, Spain. Human peripheral blood mononuclear cells (PBMCs) were isolated from the buffy coats by density sedimentation on Ficoll-Hypaque (Sigma-Aldrich) gradients (20 minutes, 2,000 rpm, at room temperature). Cells recovered from the gradient interface were washed twice in Roswell Park Memorial Institute (RPMI) complete medium (consisting of RPMI 1640 medium supplemented with 10% FBS, 2 mM glutamine, penicillin 100 U/ml, and streptomycin 1,000 U/ml) and immediately used for culture.
PBMCs (105) were cultured in duplicate with RPMI complete medium in the presence of phytohemagglutinin (PHA, 10 μg/ml, Sigma-Aldrich) with or without various quantities of hMSCs (2 × 103 to 5 × 104) in flat-bottomed 96-well plates. After 72 hours culture, proliferation was evaluated using a colorimetric method of BrdU incorporation (Roche Applied Science, Mannheim, Germany).
Macrophage Culture
Peritoneal exudate mouse cells were elicited by i.p. injection with 2 ml of 3% sterile sodium thioglycolate (Sigma-Aldrich) in 8-week-old BALB/c male mice (Harlan Laboratories). Peritoneal cells were obtained 3 days later by peritoneal lavage with cold PBS, washed in cold RPMI medium, and cultured in RPMI complete medium at a concentration of 106 cells per milliliter. After 2 hours at 37°C, nonadherent cells were removed by extensive washing. At least 95% of the adherent cells were macrophages as judged by morphological and phagocytic criteria, and by flow cytometry analysis (CD11b and F4/80). Macrophage monolayers (1 × 106 cells per well) were incubated with RPMI complete medium in the absence or presence of LPS (1 μg/ml), and hMSCs (1:5 hMSC/macrophage cell ratio). To determine the cell-contact dependence of the coculture response, LPS-stimulated macrophages (1 × 106) were placed in the upper insert of a transwell system (0.8 μm pore, Corning, Inc., Corning, NY), and hMSCs (2 × 105) were placed in the lower well. Cell-free supernatants were collected after 24 hours incubation, and cytokine levels were determined. For the generation of macrophage-CM, macrophages were cultured for 24 hours at 80% confluence in RPMI complete medium containing LPS (1 μg/ml).
Cell Migration Assay
To determine the in vitro migratory potential of hMSCs in response to signaling by stimulated macrophages, hMSCs (1.5 × 104) were cultured in RPMI complete medium in a 24-well tissue culture insert with an 8 μm pore size membrane (Corning). Inserts were placed on top of wells containing macrophage-conditioned media (macrophage-CM). For the generation of macrophage-CM, macrophages were cultured for 24 hours at 80% confluence in RPMI complete medium containing LPS (1 μg/ml). After 3 hours, the filter membranes were washed with PBS, and nonmigrated cells were removed from the upper side using a cotton swab. Finally, membranes were fixed with 4% paraformaldehyde and mounted in mounting medium containing DAPI. Migration of hMSCs was determined by counting the number of DAPI-stained nuclei on the underside of the membrane under ×200 magnification, using Cell-Profiler image analysis software (http://www.cellprofiler.org).
Cytokine Determination
For cytokine determination in mouse tissues, protein extracts were isolated by homogenization of tissue fragments (50 mg tissue per milliliter) in 50 mM Tris-HCl, pH 7.4, with 0.5 mM dithiothreitol, and 10 μg/ml of a cocktail of proteinase inhibitors containing phenylmethylsulfonyl fluoride, pepstatin, and leupeptin (Sigma-Aldrich). Samples were centrifuged at 30,000g for 20 minutes and stored at −80°C. Cytokine levels in the serum, tissue protein extracts, and culture supernatants were determined by specific sandwich ELISAs using BD OptEIA ELISA Sets (BD Biosciences, Mississauga, Canada).
Secretome Analysis
Subconfluent cultures (10,000 cells per square centimeter) were washed and incubated in serum-free DMEM for 24 hours to generate conditioned medium (CM), which was collected and cells counted. CM was filtered (0.2 μm pore), frozen at −80°C, and later analyzed using a custom human 51-plex Luminex assay (Affymetrix, Santa Clara, CA), as described in Supporting Information.
Microarray Analysis
Total RNA was isolated from cultured cells with the miRNeasy Mini Kit (Qiagen, Valencia, CA). RNA was quantified with a NanoDrop-1000 spectrophotometer and quality was monitored with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Agilent Whole Human Genome 4×44K V2 Microarray Kit (G4845A, Agilent Technologies) and Agilent Human miRNA Microarray V3 (G4470C, Agilent Technologies) were used to measure gene and miRNA expression, respectively. A full description of the samples, experimental procedures, data processing, and statistical analysis used for both types of microarrays is included in Supporting Information. All microar-ray results have been submitted to the Gene Expression Omnibus database at http://www.ncbi.nlm.nih.gov/geo; accession number GSE48662.
Gene and Protein Expression Analysis
Total RNA was isolated and quantified as described for the microarray analysis. Human transcripts were quantified by real-time reverse transcriptase polymerase chain reaction (RT-PCR) using the corresponding TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA). GAPDH was used as endogenous normalization control. Western blot and immunofluorescence analyses were performed as described in Supporting Information.
Statistical and Functional Analysis
Statistical analysis of experimental data was performed with Prism 5.0 (Graphpad Software, Inc., San Diego, CA). All values are expressed as mean ± SE of mice/experiment. Unless otherwise stated, differences between groups were analyzed by double-tailed t test. Survival curves were analyzed by the Mantel-Cox log-rank test. Results were considered statistically significant at p < .05. Gene (or gene product) functional analysis was generated as described in Supporting Information.
Results
Cell Senescence Inhibits the Lymphocyte-Inhibitory Activity of hMSCs
Cell senescence was induced in human bone marrow-derived hMSCs by gamma-irradiation (10 Gy). Ten days after irradiation, 90% of cells displayed a senescent phenotype as measured by β-gal expression (Supporting Information Fig. S1A). These cells were denoted “SEN+.”
To inhibit (prevent) senescence, hMSCs were immortalized by lentiviral transduction of hTERT. Both expression of hTERT and telomerase activity were positive in the transduced cells after >10 passages in culture (Supporting Information Fig. S1B, S1C). Compared with wild-type, nontransduced hMSCs, cumulative population doublings of the transduced cells showed an absence of proliferative arrest more than 12 cell passages (Supporting Information Fig. S1E), together with significant telomere elongation (Supporting Information Fig. S1D). These telomerase-immortalized cells were denoted “SEN−.”
To determine the extent to which cell senescence could impair the inhibitory effect of hMSCs on lymphocyte proliferation, we first performed an in vitro PBMC proliferation assay. Human allogeneic PBMCs were cultured with PHA for 3 days in the presence or absence of wild-type or senescent (gamma-irradiated) hMSCs (SEN+). We found that, although the basal inhibitory activity was variable in the four samples tested, the inhibition of lymphocyte proliferation by hMSCs was significantly reduced in all samples cultured with SEN+ cells, compared to nonirradiated cells (Fig. 1). However, SEN+ hMSCs still retained a significant lymphocyte inhibitory capacity in vitro (between 25% and 53% at a PBMC/MSC ratio of 5:1).
Figure 1.
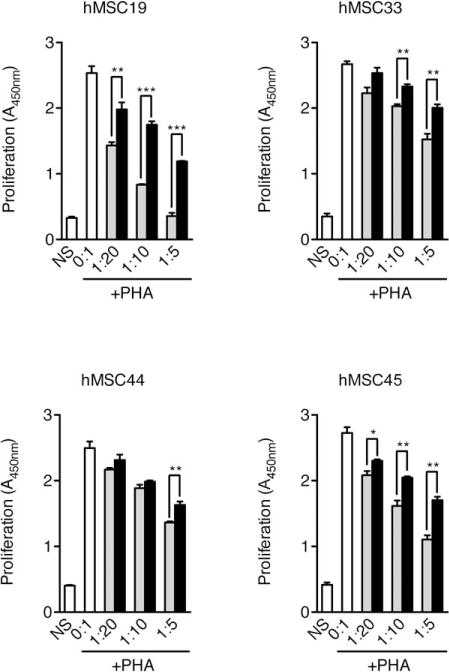
Senescent hMSCs have a reduced capacity to inhibit lymphocyte proliferation. Human PBMCs (105) were cultured in triplicate with complete RPMI medium in the presence of PHA (10 μg/ml), and with or without various numbers of bone marrow-derived hMSCs (0:1 to 1:5 MSC/PBMC ratios) from different donors (hMSC19, 33, 44, and 45) in flat-bottomed 96-well plates. After 72 hours of culture, proliferation was evaluated by colorimetric measurement of BrdU incorporation. White bars, PBMC without hMSC; gray bars, PBMC cocultured with wild-type hMSC; black bars, PBMC cocultured with senescent hMSC; NS, non-stimulated PBMC. All data correspond to experiments performed using PBMCs from the same donor. *, p < .05; **, p < .01; ***, p < .005, versus cocultures with nonsenescent hMSCs. Error bars represent SEM. Abbreviations: hMSC, human mesenchymal stem cell; PHA, phytohemagglutinin.
Senescence Abolishes the Therapeutic Immunoregulatory Activity of hMSCs In Vivo
We and others have previously described that infusion of hMSCs has significant therapeutic efficacy in experimental-induced sepsis, and protects against mortality caused by endotoxin [8, 17]. We thus investigated whether cell senescence influences the protective effect of hMSCs in a murine model of lethal endotoxemia induced by high-dose endotoxin. Using three independent bone marrow hMSC isolates, we tested the protective effect of the administration with: wild-type short passage (<6 culture passages) nonsenescent (WT) cells, short passage gamma-irradiated SEN+ cells, and long-term passage (>20 culture passages) telomerase-expressing hMSCs (SEN−). Interestingly, compared to WT cells, SEN+ hMSCs lost the capacity to rescue mice from LPS-induced endotoxemic death, while SEN− cells showed a similar protective effect to WT cells (Fig. 2A). As demonstrated previously, the protective effect of hMSCs to endotoxemic death is mediated by downregulating the exacerbated inflammatory response, which is characteristic of this condition [8, 17]. As anticipated, administration of WT hMSCs resulted in a significant reduction of the inflammatory mediators TNFa, and IL-6, in the serum and lungs of septic animals (Fig. 2B). Similar findings were observed with SEN− cells (Fig. 2B). Furthermore, IL-10 was significantly decreased in the serum, but not lung, of animals administered with WT or SEN− cells. In contrast, animals administered SEN+ hMSCs had similar levels of inflammatory mediators to those detected in untreated septic mice (Fig. 2B). No significant changes in cytokine levels were detected in the liver or intestine of the treated animals compared to untreated septic mice (data not shown).
Figure 2.
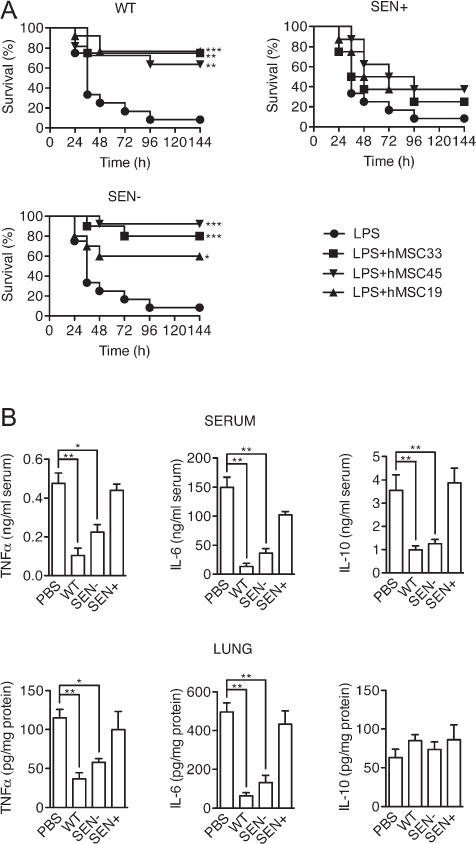
Senescent hMSCs fail to protect against lethal sepsis. Mice (10 mice/group) were injected i.p. with LPS (400 μg/mouse) and treated i.p. with PBS or 106 hMSCs 30 minutes later. Three different hMSC isolates (hMSC19, 33, and 44) were used. WT, wild-type primary hMSCs; SEN+, gamma-irradiated hMSCs; SEN−, telomerase-immortalized hMSCs. (A): Survival was monitored every 12 hours. (B): Cytokine levels were determined by ELISA in protein extracts from blood serum and lung collected 6 hours after LPS injection (n = 5). *, p < .05; **, p < .01; ***, p < .005, versus controls with LPS alone. Error bars represent SEM. Abbreviations: hMSC, human mesenchymal stem cell; LPS, lipopolysaccharide PBS, phosphate buffered saline.
Radiation-Induced Senescent hMSCs Maintain Their Immunomodulation Activity on Stimulated Macrophages, but Have a Reduced Migratory Capacity
We next asked whether SEN+ hMSCs were regulating the inflammatory response through acting directly on inflammatory cells, as has been reported previously for wild-type hMSCs [8]. Thus, we cocultured hMSCs with mouse macrophages activated with 1 μg/ml LPS, and measured inflammatory cytokine production. Consistent with our findings in vivo, coculture of macrophages with WT or SEN-hMSCs inhibited the production of TNFa and IL-6 by macrophages under LPS stimulation, and this was accompanied by a significant increase in secretion of IL-10 (Fig. 3A). Notably, these results were mirrored with SEN+ cocultures (Fig. 3A). However, when hMSCs were cocultured with macrophages on transwells (0.8 μm pore size), macrophage TNFa and IL-6 productions were inhibited similarly to that observed with direct contact, but IL-10 levels were unchanged, underscoring the requirement for cell contact [17]. Given that a central component of the anti-inflammatory activity of hMSCs is their capacity for migration to sites of inflammation (reviewed in Spaeth et al. [18]), we then used transwell assays (8 μm pore size) to assess the migration of hMSCs in the presence of LPS-stimulated mouse macrophages. Interestingly, compared with wild-type cells, SEN+ hMSCs had a significantly reduced migratory capacity (approximately 85% reduction) in response to stimulated macrophages (Fig. 3B). Collectively, these results suggest that although SEN+ hMSCs appear to maintain most of their intrinsic modulatory activity on macrophages, their capacity for performing this in vivo is probably limited due to their reduced migratory potential. Similar results (more than 75% reduction in migratory capacity) were obtained using replicative-senescent (rSEN) hMSCs (Fig. 3C; refer Supporting Information Fig. S7 for characterization of replicative-senescent hMSCs).
Figure 3.
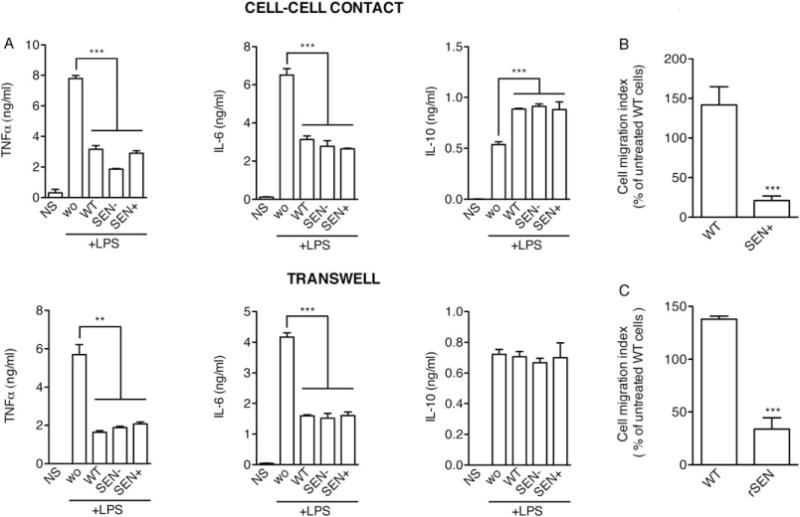
Senescent human mesenchymal stem cells (hMSCs) maintain their intrinsic immunomodulatory activity on stimulated macrophages, but fail to migrate in response of proinflammatory signals. (A): Macrophages (1 × 106 cells/well) were cultured in the presence of LPS (1 μg/ml), alone (wo), or with hMSCs (2 × 105 cells/well). Macrophages cultured in the absence of LPS and hMSCs (NS) were used as negative controls. The experiment was performed with macrophages and hMSCs in the same well (upper panels) or with the macrophages and hMSCs separated by a 0.8 μm-pore transwell (lower panels). Cytokine levels in the medium were determined after 24 hours, by ELISA. WT, wild-type presenescent hMSCs; SEN+, gamma-irradiated hMSCs; SEN−, telomerase-immortalized hMSCs. (B): 1.5 × 104 presenescent (WT) or gamma-irradiated (SEN+) hMSCs were used in transwell migration assays in response to conditioned medium from LPS-stimulated macrophages. (C): 1.5 × 104 presenescent (WT) or replicative-senescent (rSEN) hMSCs were used in trans-well migration assays in response to conditioned medium from LPS-stimulated macrophages. **, p < .01; ***, p < .005, versus controls. Error bars represent SEM (n = 3). Abbreviations: LPS, lipopolysaccharide; WT, wild type.
A Senescence-Associated Secretory Phenotype in hMSCs
Previous work has described that irradiated fibroblasts undergo permanent senescence growth arrest, and develop a senescence-associated secretory phenotype (SASP) 4–7 days after irradiation [19]. To determine the secretory phenotype of SEN+ hMSCs, we induced senescence in four independent isolates of human bone marrow-derived hMSCs by exposure to a similar dose of ionizing radiation (10 Gy). Ten days following irradiation, wild-type (WT, nonirradiated) and senescent (SEN+, gamma-irradiated) cells were cultured in serum-free medium for 24 hours and this conditioned medium (CM) was collected. The CM was analyzed using Luminex antibody arrays designed to detect 51 secreted human proteins involved in intercellular signaling during inflammation (Supporting Information Table S1). Signals were calculated and quantified as described in Supporting Information Methods, and presented as the median fluorescence intensity log 2-fold changes between SEN+ and WT samples for each analyte. Consistent with studies in other cell types [19], we found that, although the profiles were in part donor dependent, senescent hMSCs (SEN+) secreted higher levels of numerous proteins compared to WT cells (Fig. 4A). Of the 51 proteins interrogated by the arrays, 27 were significantly altered (t test, p < .05) in the CM of SEN+ cells and were oversecreted in comparison to CM from WT cells. These 27 identified SASP components ranged from (normalized to 105 cells per milliliter) a low concentration of 0.92 pg/ml for IL-17F, to the highest concentration of 716.87 pg/ml for IL-6, in the CM of SEN+ cells (Fig. 4B). Furthermore, nine of the proteins (LEPTIN, TGFA, IL8, EOTAXIN, IFNG, VCAM1, IFNB, IL4, and MCP1) were secreted greater than 10-fold more from SEN+ cells compared to WT cells (Fig. 4C). The immune system processes (Gene Ontology, GO terms) associated to all the identified SASP components are shown in Supporting Information Table S2.
Figure 4.
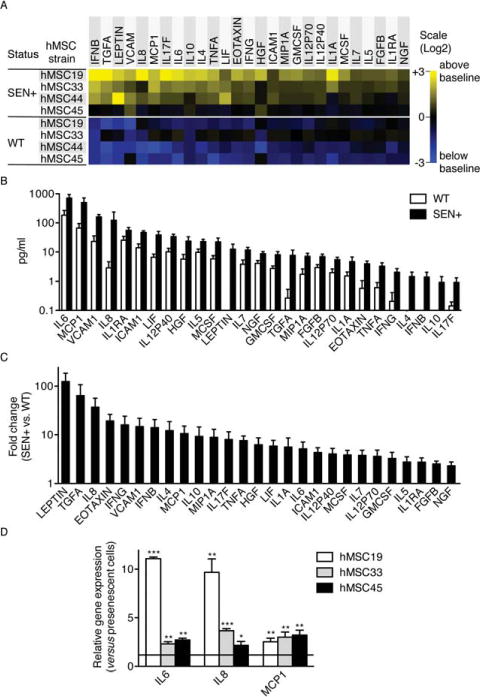
Senescent hMSCs secrete higher levels of inflammatory mediators compared to WT cells. (A): Soluble factors secreted by the indicated cells were detected by antibody arrays and analyzed as described in the text, Materials and Methods, and Supporting Information. WT, wild-type primary hMSCs; SEN+, gamma-irradiated hMSCs. For each protein, all signals were averaged and used as the baseline. Signals higher than baseline are displayed in yellow; signals below baseline are displayed in blue. The numbers in the heat map key (right) indicate log 2-fold changes from the baseline. Only proteins significantly oversecreted in SEN+ cells (p < .05; compared with WT cells) are represented. (B): Levels of proteins (normalized to 105 cells per milliliter) significantly oversecreted in SEN+ cells compared with WT cells. (C): Fold changes in the levels of proteins significantly oversecreted in SEN+ cells compared with WT cells. When protein levels where under the detection limit, a baseline of 0.1 pg/ml was considered. (D): Relative expression levels of the major SASP components IL6, IL8, and MCP1 were quantified by real-time RT-PCR in three different isolates of presenescent and replicative-senescent hMSCs. α-Tubulin was used as endogenous control. *, p < .05; **, p < .01; ***, p < .005, versus controls. Error bars represent SEM (n = 3). Abbreviations: hMSC, human mesenchymal stem cell; WT, wild type.
In order to verify whether the alterations found in radiation-induced senescent hMSCs were representative for replication-induced senescence, we analyzed the expression of three major components of the SASP (IL6, IL8, and MCP1) by real-time RT-PCR. Results demonstrated that all three genes were also overexpressed in replicative-senescent hMSCs in comparison with presenescent cells (Fig. 4D).
Genome-Wide Gene Expression Profile Analysis of Radiation-Induced Senescent hMSCs
To complement the secretome profile of hMSCs, we also measured global gene expression of hMSCs in a parallel study. Microarray analysis (Agilent Whole Human Genome Microarray Kit) indicated that a total of 5,975 protein-coding genes (of which 4,102 corresponded to annotated genes) were significantly regulated (adjusted p < .05) in hMSCs 10 days after irradiation, when compared with nonirradiated cells. Gene set enrichment analysis using the PANTHER program revealed significant enrichment (Bonferroni multiple test-corrected p-value < .05) for a wide variety of biological processes, including cell communication and immune system process (Supporting Information Fig. S2). A detailed analysis using the IPA software showed a predominant enrichment (Benjamini-Hochberg multiple test-corrected p-value < .05) for biological functions involved in gene expression, cell cycle, and cancer (Fig. 5A). Other significantly enriched functions included cell death, cellular growth and proliferation, and cellular movement (Supporting Information Fig. S3). The IPA analysis also showed a significant enrichment of canonical pathways involved in cancer, cell cycle, and DNA repair (Fig. 5B). The regulated genes were mapped to a total of 25 networks (Supporting Information Table S5). The top functions related to the altered networks were: cell assembly and morphology, cell cycle, and DNA replication, recombination, and repair.
Figure 5.
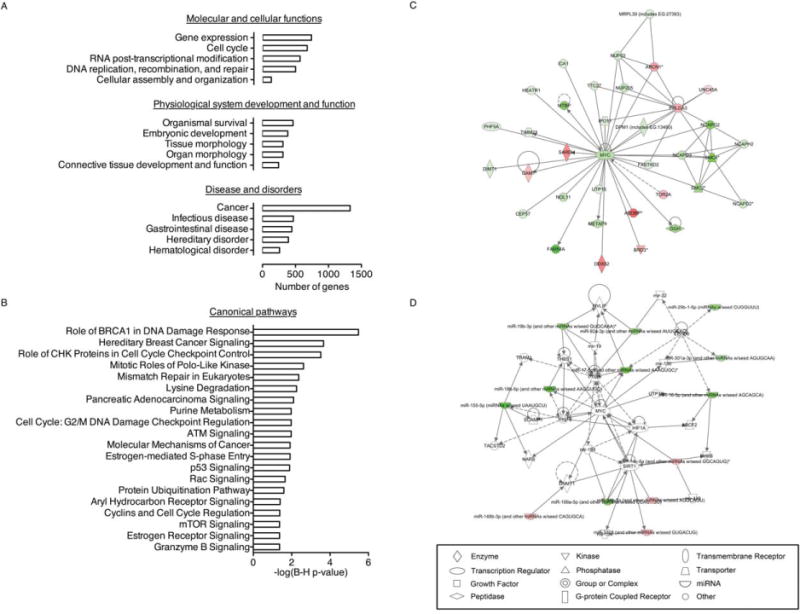
Gene function alterations found in SEN+ human mesenchymal stem cells (hMSCs). (A): Genes significantly regulated in SEN+ hMSCs (multiple-test adjusted p-value < .05) in comparison with nonirradiated (WT) cells were analyzed using IPA (Ingenuity Systems, www.ingenuity.com). Graphs show the top five functional categories altered in each of the three IPA major BioFunction groups. The full list of biological functions significantly regulated is shown in Supporting Information Figure S3. (B): Significantly regulated IPA canonical pathways (Benjamini-Hochberg multiple test-corrected p-value < .05). (C): Top network generated using IPA from the list of differentially expressed miRNAs (SEN+ vs. WT hMSCs). Major functions associated with this network are: cancer, reproductive system disease, and connective tissue disorders. Regulated genes appear shaded in green (downregulated) or red (upregulated). (D): Second most significant network found in the list of differentially expressed genes (SEN+ vs. WT hMSCs). Major functions associated with this network are: cellular assembly and organization, cell cycle, and DNA replication, recombination, and repair (Supporting Information Table S5).
To identify miRNAs potentially regulated in senescent hMSCs, we obtained differential miRNA expression profiles of SEN+ hMSCs versus nonirradiated cells (Agilent Human microRNA Microarray v2.0). A total of 31 miRNAs were significantly altered in the SEN+ cells (adjusted p < .05, Supporting Information Table S6). The most prominently regulated family of miRNAs was the hsa-miR-17 cluster; strikingly, all members of this family were downregulated in SEN+ cells. Those miR-NAs prominently upregulated included miR-629-3p, miR-572, and miR-135a-3p (Supporting Information Table S6). Analysis of the miRNA microarray data with the IPA software showed a predominant enrichment (Benjamini-Hochberg multiple test-corrected p-value < .05) for cellular functions involved in cellular development, cell growth and proliferation, cell cycle, cell death, and DNA replication, recombination, and repair (Supporting Information Fig. S4A, refer Supporting Information Fig. S5 for a full list of enriched biological functions). We also used the RNA22 tool to generate a list of putative targets for all the regulated miRNAs, and obtained a nonredundant list of 5,148 genes (4,713 annotated). Of these, 3,543 were assigned to 17 known GO biological processes by the PANTHER program (Supporting Information Fig. S4B). Many of these processes could play a relevant role during the development of hMSC senescence. Interestingly, the second-highest score network identified through the use of IPA in the gene expression microarray data share MYC as their nodal molecule (Fig. 5C), in a network including 13 of the regulated miRNAs (Fig. 5D). Taken together, our results show an extensive alteration in pathways related with cell proliferation and DNA damage response in SEN+ hMSCs, which is consistent with injury through irradiation.
A Strong Inhibition of the AP-1 Pathway Is Associated with the Migratory Defect in Senescent hMSCs
Based on our unpublished results, we hypothesized that the clear migration defect observed in senescent hMSCs could be related to the inactivation of AP-1. AP-1 mediates the response to a variety of extracellular stimuli and plays a key role in the regulation of diverse processes such as differentiation, proliferation, and migration (reviewed in Angel et al. [20]). When we measured the expression of the AP-1 components FOS and JUN (and their phosphorylated forms) after stimulation of presenescent and both radiation- and replicative-senescent hMSCs with macrophage-conditioned medium (previously used as a migratory stimuli), we found that senescent hMSCs showed a marked decrease in the expression of these proteins (Fig. 6A).
Figure 6.
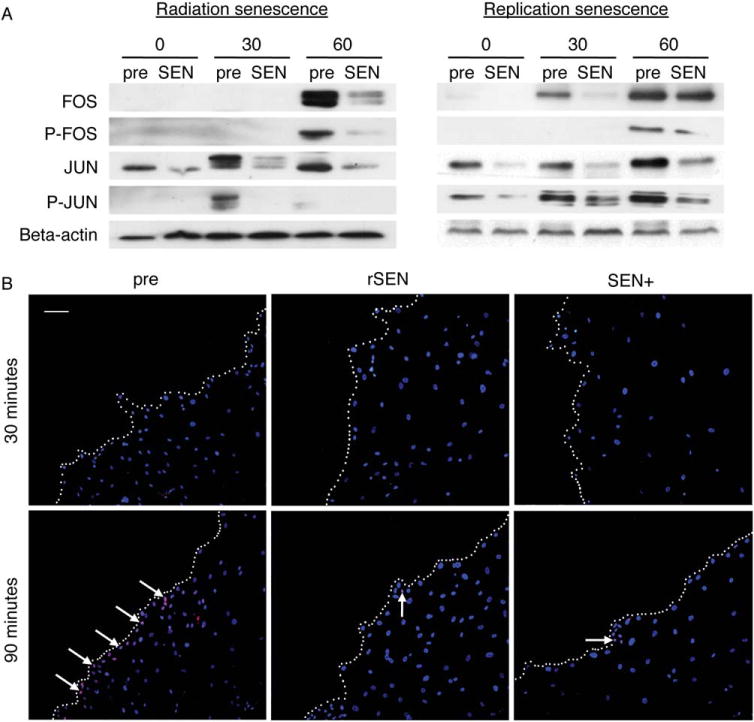
Senescent human mesenchymal stem cell (hMSC) shows a strong inhibition of the AP-1 pathway in response to migratory stimuli. (A): Western blot analysis of AP-1 components FOS, JUN, and their phosphorylated forms after treatment of presenescent (pre) or senescent (SEN) hMSCs with conditioned medium from LPS-stimulated macrophages at indicated times (minutes). Representative results from at least three experiments are shown. (B): Analysis of FOS activation at the wound edge. A scratch in a monolayer of presenescent (pre), radiation-induced senescent (SEN+), and replication-induced senescent (rSEN) hMSCs was made as described in Supporting Information. At the indicated times after scratching (30 or 90 minutes), the cells were fixed, permeabilized, and stained with an anti-FOS antibody and the corresponding Cy5-labeled secondary antibody (red). Cell nuclei were counterstained using DAPI (blue). Representative fluorescence microscopy images with merged DAPI and Cy5 signals are shown. White dotted lines indicate the cell migration front (wound edge). White arrows point to some cell nuclei expressing FOS. Scale bar = 100 μm.
To ascertain whether this defect in AP-1 activation could be related to the migratory impairment observed in senescent hMSCs, we evaluated FOS expression in an in vitro wound-healing assay (see Supporting Information). The majority of presenescent wound-edge cells showed a high FOS expression 90 minutes after the scraping of the cell monolayer, whereas both irradiation- and replication-induced senescent wound-edge hMSCs exhibited comparatively weak FOS levels after the same time (Fig. 6B). These results demonstrate a close association of AP-1 inhibition with the impaired response of senescent hMSCs to migratory stimuli.
Clinical Efficacy of hMSCs Correlates with the Expression Levels of Senescence-Regulated Genes
Reminiscent of the results for our secretome profiling, it is recognized in the field that there is significant donor-dependent variability in the biological properties of hMSCs, which may limit their clinical value in allogeneic applications. Given our findings so far, we thought it instructive to determine whether the differentially regulated gene functions in experimentally induced cell senescence could correlate with the clinical efficacy of hMSCs. We therefore performed microarray analyses of gene and miRNA expression in eight different hMSCs isolates (C1 through C8) which are being used in an ongoing clinical study (EudraCT number: 2009-011164-11). In this study, allogeneic MSCs obtained from bone marrow samples of healthy donors are being used to treat patients diagnosed of refractory severe acute or extensive GVHD.
For statistical analysis, the eight donor hMSC samples were classified in two groups depending on their clinical efficacy (see Supporting Information for a summary of the clinical study and the major variables analyzed). Group 1 samples (Gr1: C1, C2, C4, C5, and C7) were those that induced a therapeutic response, either complete (C5), or partial (C1, C2, C4, and C7) at the lowest dose used in the study. Group 2 samples (Gr2: C3, C6, and C8) were those that did not elicit an effect that could be classified as a complete or partial response (Supporting Information Table S7). As might be expected from cells isolated from healthy donors and expanded under highly reproducible Good Manufacturing Practices cell culture conditions, no significantly regulated (adjusted p < .05) gene functions (neither in protein-coding genes nor miRNAs) were found when comparing Gr1 and Gr2. However, a hierarchical cluster analysis of gene expression data revealed that two of the Gr2 samples (C4 and C8) were more similar to the SEN+ samples than to the control (WT) samples (Supporting Information Fig. S6). We then searched for genes differentially upregulated or downregulated in both SEN+ and Gr2 samples compared with WT samples, but not differentially expressed in Gr1 compared with WT or SEN+ samples. As shown (Fig. 7A), PLEC (upregulated; logFC = 1.303) and C8orf48, TRPC4, and ZNF14 (downregulated; log FC = −1.202, −3.005, and −1.220, respectively) were selectively regulated in both Gr2 and SEN+ samples compared with WT hMSCs. None of these genes were similarly regulated in Gr1 cells compared to SEN+ or WT cells, which precludes the possibility of these differences being dependent on the slightly different culture conditions used for the clinical (Gr1 and Gr2) and nonclinical samples (WT and SEN+).
Figure 7.
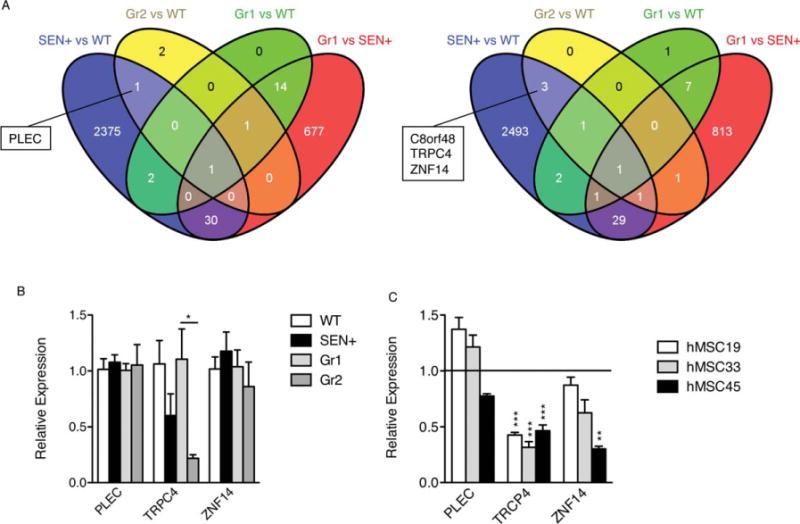
Four genes in the microarray analyses appear selectively regulated in both senescent and therapeutically ineffective hMSCs. We searched for genes differentially upregulated or downregulated in both SEN+ and Gr2 samples (therapeutically ineffective) compared with WT samples, but not differentially expressed in Gr1 (therapeutically effective) compared with WT or SEN+ samples. (A): Venn diagrams show the number of RNA probes differentially expressed (adjusted p < .05) in each cell population, analyzed with the Agilent Whole Human Genome Microarray Kit. For SEN+ and WT, n = 4; for Gr1, n = 5; for Gr2, n = 3. (B): Relative expression levels of PLEC, TRPC4, and ZNF14 (endogenous control GAPDH) measured by real-time RT-PCR. The samples used were the same as those used for the microarray analysis. WT (average) was used as a calibrator for SEN samples, and Gr1 (average) was used as a calibrator for Gr2 samples. (C): Relative expression levels of PLEC, TRPC4, and ZNF14 (endogenous control GAPDH) measured by real-time RT-PCR in three different isolates of replicative-senescent hMSCs. *, p < .05; **, p < .01; ***, p < .005, versus presenescent samples. Error bars represent SEM (n = 3). Abbreviations: hMSC, human mesenchymal stem cell; WT, wild type.
When real-time RT-PCR was performed with the same RNA samples used for the microarray experiments, only TRPC4 gave a statistically significant differential expression (Fig. 7B). Similar results were obtained when the relative expression of PLEC, TRPC4, and ZNF14 was evaluated in replication-induced senescent hMSCs (Fig. 7C). These results indicate that TRCP4 deserves further study as a potential quantitative marker for pre-evaluating the clinical efficacy of individual hMSCs samples used in future allogeneic clinical applications.
Discussion
Human MSCs (hMSCs) display a potent in vivo immunoregulatory activity, and are used with increasing frequency for cell therapy in the setting of autoimmune/inflammatory responses. Therefore, understanding the physiological and pathological factors that affect the immunoregulatory activity of hMSCs is of critical importance to develop safe and effective strategies for the treatment of degenerative disorders with an inflammatory component. Previous studies have clearly established that organismal aging correlates with a certain level of functional decline in hMSCs (reviewed in Sethe et al. [21]), including the reduction of proliferation, differentiation [22], and angiogenic [23] potential. This process has been related to phenotype alterations observed in culture, such as: morphological changes [24], reduction of telomere length [24, 25], expression of cell senescence markers, for example, p53 and beta-galactosidase [26–28], reduction in a subset of signaling molecules, for example, TGF-β and BMP2/4 [29], increased levels of proinflammatory molecules such as IL-6 [30], differential expression of several miRNAs [31], increased genetic damage [32], and decreased protection mechanisms such as repair [33] and antioxidant enzyme activities [34], and heat shock proteins [31]. However, a clear understanding of this process and the specific mechanisms involved in age/senescence-related loss of therapeutic potential in hMSCs is still lacking.
Here, we studied the potential relationship between the cell senescence process and the immunoregulatory activity of hMSCs, and attempted to assess the relevance of cell senescence markers as quantitative indicators for the clinical efficacy of hMSCs. We focused our investigations on cell senescence because: (a) due to the prolonged expansion regimens that are needed in the clinic to obtain sufficient amounts of hMSCs for therapy, it is quite likely that a substantial proportion of cells may undergo senescence, and (b) it is recognized that senescent human cells display important changes in their transcriptional and secretory states that confer them with strong proinflammatory characteristics [19].
Cellular senescence is a normal biological process which functions to prevent the proliferation of damaged cells, thus avoiding the transmission of damage to daughter cells. It consists of an irreversible cell growth arrest initiated in response to a range of intrinsic and extrinsic factors such as aging and exposure to stress. Ionizing radiation is a genotoxic agent that is able to induce cell senescence in a reproducible and persistent manner [35], without triggering cell death mechanisms such as apoptosis [36]. Our data demonstrate that radiation-induced senescence abrogates the protective immunoregulatory effect of hMSCs in a murine model of sepsis. Previous studies have shown that the mechanisms involved in the strong in vivo anti-inflammatory effects of hMSCs include the reduction of inflammatory cytokines and chemokines, the impairment of Th1 cell expansion, the induction of immunomodulatory macrophages, and the induction of regulatory T cells [6–10, 17, 37]. In agreement with these findings, systemic administration of hMSCs elicited a significant reduction in the serum and lung concentrations of TNFa and IL-6 in our murine model. In contrast to data from other experimental models, we also observed a significant decrease in serum levels of IL-10 [6–8, 17]. The absence of a protective phenotype in senescent hMSCs correlated with the failure of these cells to cause significant reductions in cytokine levels in animals compared to those administered with hMSCs. However, senescent hMSCs retained some capacity for lymphocyte inhibition in vitro, and were comparable in their capacity to modulate the activation of macrophages, both by paracrine action and by cell-to-cell contact. Nevertheless, the migratory response of senescent hMSCs to signals from activated macrophages was severely reduced. Collectively, these results strongly suggested that the loss of in vivo immunoregulatory activity in senescent hMSCs is probably due mainly to their reduced migratory potential, rather than a dysfunction in their signaling mechanisms. Thus, administration of a senescent population of hMSCs in an inflammatory context could be biologically comparable to the administration of a very low dose of WT hMSCs. Infused senescent cells may modulate to a degree, the function of lymphocytes and macrophages, but too few cells would be able to reach the sites of inflammation to achieve clinical benefit. This reduction in the ability of senescent MSCs and other cell types to migrate has been reported previously [38–41], and may be related to changes in the cytoskeleton [42]. Our results suggest that this senescence-related migration defect could have important implications for the clinical use of hMSCs, and could be mediated by the inactivation of the AP-1 signaling pathway.
Human cells induced to senesce through extended proliferation or by genotoxic stress secrete many factors associated with inflammation (reviewed in Tchkonia et al. [10]). This senescence-associated secretory phenotype (SASP) develops over several days, and is triggered by persistent DNA-damage signaling [43]. The SASP described here for hMSCs was complex, and showed significant variability between donors, making interpretation of its potential biological effects challenging. Nevertheless, three proteins (IL-8, VCAM-1, and MCP1) were secreted more than 10-fold in senescent cells compared to nonirradiated cells, reaching levels greater than 100 pg/ml. IL-8 induces chemotaxis in neutrophils and other granulocytes [44], and is a potent promoter of angiogenesis [45]. VCAM-1 mediates the adhesion of leukocytes to vascular endothelium, and the level of its soluble form has been frequently related to systemic inflammation [46, 47]. Finally, MCP1 (CCL2) is a chemoattractant for monocytes and basophils and has a prominent role in several inflammatory diseases, such as multiple sclerosis [48] and inflammatory bowel disease [49]. The increased levels of these three molecules alone, notwithstanding other secreted proinflammatory mediators, strongly suggest that in vivo administration of senescent hMSCs could exacerbate the inflammatory response at a systemic level and/or counteract the anti-inflammatory effect of these cells as measured in vitro.
Cellular senescence changes profoundly the transcriptional profile of hMSCs [50, 51]. Our microarray analysis detailed more than 5,000 genes, including 31 miRNAs, differentially expressed (adjusted p < .05) in senescent hMSCs compared to control cells. Cellular functions prominently altered in senescent cells included cell growth and proliferation, cell cycle, cell death, and cellular movement. Furthermore, profiling of miRNA expression revealed an upregulation of the miR-34 family, which has been previously reported to be regulated by p53 and to induce cell senescence [52, 53]. In contrast, the miR-17 cluster was downregulated in senescent cells, mirroring its expression in human aging [54]. In our study, the pattern of senescence-regulated miRNAs was different from that previously reported in similar studies [31, 51, 55]. Whether these differences are related to the different species used, or are dependent on culture conditions or methods used to induce senescence (replicative versus radiation), and so forth, remains to be determined.
Notably, a significantly downregulated gene in senescent hMSCs was the proto-oncogene MYC, which also appeared as the nodal molecule in two of the highest scoring genetic networks identified from the gene expression microarray. MYC is a transcription factor whose activation by diverse mitogenic signals such as WNT, SHH, and EGF results in cell proliferation. Moreover, reduced MYC signaling triggers telomere-independent senescence by regulating BMI1 and CDKN2A [56]. Indeed, the relevance of MYC downregulation in hMSC senescence has been highlighted in previous data [57]. Thus, our results support the idea that radiation-induced MYC downregulation in hMSCs could be an important mechanism for limiting the proliferation of damaged cells.
The complex transcriptional response triggered by senescence in hMSCs prevented any meaningful analysis to identify specific markers that could be relevant for the therapeutic efficacy of these cells. Also, our microarray data from hMSC cells used in an allogeneic clinical study failed to identify a single regulated gene when comparing therapeutically effective hMSC (Gr1) cells to clinically ineffective cells (Gr2). However, we did find that PLEC, C8orf48, TRPC4, and ZNF14 were similarly regulated in the latter group and in senescent (SEN+) hMSCs, when compared with WT control cells. PLEC (plectin, upregulated in both Gr2 and SEN+ compared to WT) is a cytoskeletal cross-linking protein whose inhibition has been reported to bypass cellular senescence in rat embryo fibroblasts [58]. It is also described to be a major early substrate for caspase-8, which is required for actin reorganization during apoptosis [59]. The downregulation of TRPC4 (a nonselective calcium-permeable cation channel) is also biologically relevant in the context of senescence, since it is known that replicative senescence leads to the suppression of calcium-dependent membrane currents in human fibroblasts [60]. Reassuringly, real-time RT-PCR validated the differential expression of TRPC4 in both radiation- and replication-induced senescent hMSCs. Therefore, we tentatively suggest that these four genes should be considered in further studies as potential markers for identifying hMSC samples with a low therapeutic potential in allogeneic clinical applications.
Conclusions
This study provides evidence that cell senescence abrogates the protective activity of hMSCs in an experimental model of lethal sepsis. Interestingly, this loss of efficacy was not associated with a defect of hMSC-related inflammatory responses in vitro, but was possibly the result of a reduction in their migratory capacity associated with an inhibition of the AP-1 pathway. Senescent hMSCs also adopted a senescence-associated-secretory phenotype, directed to promote systemic inflammation. Candidate gene pathways dysregulated in senescent hMSCs included MYC, miR-34 family, and miR-17 cluster. In particular, four genes (PLEC, C8orf48, TRPC4, and ZNF14) were similarly regulated in senescent hMSCs, and in hMSC cells that were therapeutically ineffective in an allogeneic GVHD clinical study. The potential of these gene expression variations to act as markers for hMSC pre-evaluation will be addressed in future studies to gauge the therapeutic potential of hMSCs intended for use in clinical allogeneic applications.
Supplementary Material
Acknowledgments
We thank Dr. E. Samper for his valuable help in data analysis and interpretation; S. Calleja and R. Alvarez (CNIC) for microarray hybridization; A. García and J.C. Ramírez (CNIC) for production of lentiviral stocks; F. Cabo (CNIC) for bioinformatics and statistical support, and K. McCreath for helpful discussions. This work was supported by a grant to MAG from the Spanish Ministry of Science and Innovation (PLE2009-0112), and by grants to A.B. from the Ministry of Economy and Competitiveness (SAF 2008-02099; PLE2009-0147, and PSE-010000-2009-3), Comunidad Autonoma de Madrid (S2010/BMD-2420), Red de Terapia Celular del Instituto de Salud Carlos III (TerCel), and the European Commission (FP7-HEALTH-2009/CARE-MI). M.A.G. is also supported by the “Miguel Servet” Program (CP07/00306) of the Instituto de Salud Carlos III (Ministry of Economy and Competitiveness, Spain). J.C.S. was supported by PLE2009-0112. M.T. is currently a predoctoral fellow funded by the Spanish Programa de Formación del Profesorado Universitario (Ministry of Education, Culture, and Sports, Spain). The CNIC is supported by the Spanish Ministry of Economy and Competitiveness and the Pro-CNIC Foundation.
Footnotes
Author Contributions
J.C.S.: collection and/or assembly of data and manuscript writing; M.T.: collection and/or assembly of data; M.E.F.: provision of study material or patients; M.D. and J.C.: data analysis and interpretation; A.B.: financial support, editing, and final approval of manuscript; M.A.G.: conception and design, financial support, data analysis and interpretation, and manuscript writing. A.B. and M.A.G. share senior authorship.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
See www.StemCells.com for supporting information available online.
References
- 1.Bernardo ME, Locatelli F, Fibbe WE. Mesenchymal stromal cells. Ann N Y Acad Sci. 2009;1176:101–117. doi: 10.1111/j.1749-6632.2009.04607.x. [DOI] [PubMed] [Google Scholar]
- 2.Gebler A, Zabel O, Seliger B. The immunomodulatory capacity of mesenchymal stem cells. Trends Mol Med. 2012;18:128–134. doi: 10.1016/j.molmed.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Polchert D, Sobinsky J, Douglas G, et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol. 2008;38:1745–1755. doi: 10.1002/eji.200738129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stagg J, Pommey S, Eliopoulos N, et al. Interferon-gamma-stimulated marrow stromal cells: A new type of nonhematopoietic antigen-presenting cell. Blood. 2006;107:2570–2577. doi: 10.1182/blood-2005-07-2793. [DOI] [PubMed] [Google Scholar]
- 5.Liang X, Ding Y, Zhang Y, et al. Paracrine mechanisms of mesenchymal stem cell-based therapy: Current status and perspectives. Cell Transplant. 2013 doi: 10.3727/096368913X667709. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez MA, Gonzalez-Rey E, Rico L, et al. Treatment of experimental arthritis by inducing immune tolerance with human adipose-derived mesenchymal stem cells. Arthritis Rheum. 2009;60:1006–1019. doi: 10.1002/art.24405. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez MA, Gonzalez-Rey E, Rico L, et al. Adipose-derived mesenchymal stem cells alleviate experimental colitis by inhibiting inflammatory and autoimmune responses. Gastroenterology. 2009;136:978–989. doi: 10.1053/j.gastro.2008.11.041. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Rey E, Anderson P, Gonzalez MA, et al. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut. 2009;58:929–939. doi: 10.1136/gut.2008.168534. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Rey E, Gonzalez MA, Varela N, et al. Human adipose-derived mesenchymal stem cells reduce inflammatory and T cell responses and induce regulatory T cells in vitro in rheumatoid arthritis. Ann Rheum Dis. 2010;69:241–248. doi: 10.1136/ard.2008.101881. [DOI] [PubMed] [Google Scholar]
- 10.Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izadpanah R, Kaushal D, Kriedt C, et al. Long-term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Res. 2008;68:4229–4238. doi: 10.1158/0008-5472.CAN-07-5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarte K, Gaillard J, Lataillade JJ, et al. Clinical-grade production of human mesenchymal stromal cells: Occurrence of aneuploidy without transformation. Blood. 2010;115:1549–1553. doi: 10.1182/blood-2009-05-219907. [DOI] [PubMed] [Google Scholar]
- 13.Ueyama H, Horibe T, Hinotsu S, et al. Chromosomal variability of human mesenchymal stem cells cultured under hypoxic conditions. J Cell Mol Med. 2012;16:72–82. doi: 10.1111/j.1582-4934.2011.01303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tumpel S, Rudolph KL. The role of telomere shortening in somatic stem cells and tissue aging: Lessons from telomerase model systems. Ann N Y Acad Sci. 2012;1266:28–39. doi: 10.1111/j.1749-6632.2012.06547.x. [DOI] [PubMed] [Google Scholar]
- 15.Reiser J. Production and concentration of pseudotyped HIV-1-based gene transfer vectors. Gene Ther. 2000;7:910–913. doi: 10.1038/sj.gt.3301188. [DOI] [PubMed] [Google Scholar]
- 16.Beausejour CM, Krtolica A, Galimi F, et al. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemeth K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spaeth E, Klopp A, Dembinski J, et al. Inflammation and tumor microenvironments: Defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–738. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 19.Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001;20:2413–2423. doi: 10.1038/sj.onc.1204380. [DOI] [PubMed] [Google Scholar]
- 21.Sethe S, Scutt A, Stolzing A. Aging of mesenchymal stem cells. Ageing Res Rev. 2006;5:91–116. doi: 10.1016/j.arr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Stolzing A, Jones E, McGonagle D, et al. Age-related changes in human bone marrow-derived mesenchymal stem cells: Consequences for cell therapies. Mech Ageing Dev. 2008;129:163–173. doi: 10.1016/j.mad.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Efimenko A, Starostina E, Kalinina N, et al. Angiogenic properties of aged adipose derived mesenchymal stem cells after hypoxic conditioning. J Transl Med. 2011;9:10. doi: 10.1186/1479-5876-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baxter MA, Wynn RF, Jowitt SN, et al. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells. 2004;22:675–682. doi: 10.1634/stemcells.22-5-675. [DOI] [PubMed] [Google Scholar]
- 25.Lee JJ, Nam CE, Kook H, et al. Constitution and telomere dynamics of bone marrow stromal cells in patients undergoing allogeneic bone marrow transplantation. Bone Marrow Transplant. 2003;32:947–952. doi: 10.1038/sj.bmt.1704253. [DOI] [PubMed] [Google Scholar]
- 26.Stenderup K, Justesen J, Clausen C, et al. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. 2003;33:919–926. doi: 10.1016/j.bone.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Park JS, Kim HY, Kim HW, et al. Increased caveolin-1, a cause for the declined adipogenic potential of senescent human mesenchymal stem cells. Mech Ageing Dev. 2005;126:551–559. doi: 10.1016/j.mad.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 28.Vacanti V, Kong E, Suzuki G, et al. Phenotypic changes of adult porcine mesenchymal stem cells induced by prolonged passaging in culture. J Cell Physiol. 2005;205:194–201. doi: 10.1002/jcp.20376. [DOI] [PubMed] [Google Scholar]
- 29.Moerman EJ, Teng K, Lipschitz DA, et al. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: The role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. 2004;3:379–389. doi: 10.1111/j.1474-9728.2004.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheleuitte D, Mizuno S, Glowacki J. In vitro secretion of cytokines by human bone marrow: Effects of age and estrogen status. J Clin Endocrinol Metab. 1998;83:2043–2051. doi: 10.1210/jcem.83.6.4848. [DOI] [PubMed] [Google Scholar]
- 31.Yu JM, Wu X, Gimble JM, et al. Age-related changes in mesenchymal stem cells derived from rhesus macaque bone marrow. Aging Cell. 2011;10:66–79. doi: 10.1111/j.1474-9726.2010.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, DiGirolamo CM, Navarro PA, et al. Telomerase deficiency impairs differentiation of mesenchymal stem cells. Exp Cell Res. 2004;294:1–8. doi: 10.1016/j.yexcr.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 33.Lombard DB, Chua KF, Mostoslavsky R, et al. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 34.Edwards MG, Sarkar D, Klopp R, et al. Age-related impairment of the transcriptional responses to oxidative stress in the mouse heart. Physiol Genomics. 2003;13:119–127. doi: 10.1152/physiolgenomics.00172.2002. [DOI] [PubMed] [Google Scholar]
- 35.Le ON, Rodier F, Fontaine F, et al. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell. 2010;9:398–409. doi: 10.1111/j.1474-9726.2010.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muthna D, Soukup T, Vavrova J, et al. Irradiation of adult human dental pulp stem cells provokes activation of p53, cell cycle arrest, and senescence but not apoptosis. Stem Cells Dev. 2010;19:1855–1862. doi: 10.1089/scd.2009.0449. [DOI] [PubMed] [Google Scholar]
- 37.Anderson P, Souza-Moreira L, Morell M, et al. Adipose-derived mesenchymal stromal cells induce immunomodulatory macrophages which protect from experimental colitis and sepsis. Gut. 2013;62:1131–1141. doi: 10.1136/gutjnl-2012-302152. [DOI] [PubMed] [Google Scholar]
- 38.Kasper G, Mao L, Geissler S, et al. Insights into mesenchymal stem cell aging: Involvement of antioxidant defense and actin cytoskeleton. Stem Cells. 2009;27:1288–1297. doi: 10.1002/stem.49. [DOI] [PubMed] [Google Scholar]
- 39.Reed MJ, Ferara NS, Vernon RB. Impaired migration, integrin function, and actin cytoskeletal organization in dermal fibroblasts from a subset of aged human donors. Mech Ageing Dev. 2001;122:1203–1220. doi: 10.1016/s0047-6374(01)00260-3. [DOI] [PubMed] [Google Scholar]
- 40.Sandeman SR, Allen MC, Liu C, et al. Human keratocyte migration into collagen gels declines with in vitro ageing. Mech Ageing Dev. 2000;119:149–157. doi: 10.1016/s0047-6374(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 41.Schneider EL, Mitsui Y. The relationship between in vitro cellular aging and in vivo human age. Proc Natl Acad Sci USA. 1976;73:3584–3588. doi: 10.1073/pnas.73.10.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishio K, Inoue A. Senescence-associated alterations of cytoskeleton: Extraordinary production of vimentin that anchors cytoplasmic p53 in senescent human fibroblasts. Histochem Cell Biol. 2005;123:263–273. doi: 10.1007/s00418-005-0766-5. [DOI] [PubMed] [Google Scholar]
- 43.Rodier F, Coppe JP, Patil CK, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992;307:97–101. doi: 10.1016/0014-5793(92)80909-z. [DOI] [PubMed] [Google Scholar]
- 45.Li A, Dubey S, Varney ML, et al. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 46.Spronk PE, Bootsma H, Huitema MG, et al. Levels of soluble VCAM-1, soluble ICAM-1, and soluble E-selectin during disease exacerbations in patients with systemic lupus erythematosus (SLE); a long term prospective study. Clin Exp Immunol. 1994;97:439–444. doi: 10.1111/j.1365-2249.1994.tb06107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blankenberg S, Rupprecht HJ, Bickel C, et al. Circulating cell adhesion molecules and death in patients with coronary artery disease. Circulation. 2001;104:1336–1342. doi: 10.1161/hc3701.095949. [DOI] [PubMed] [Google Scholar]
- 48.Tanuma N, Sakuma H, Sasaki A, et al. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006;112:195–204. doi: 10.1007/s00401-006-0083-7. [DOI] [PubMed] [Google Scholar]
- 49.Spoettl T, Hausmann M, Herlyn M, et al. Monocyte chemoattractant protein-1 (MCP-1) inhibits the intestinal-like differentiation of monocytes. Clin Exp Immunol. 2006;145:190–199. doi: 10.1111/j.1365-2249.2006.03113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geissler S, Textor M, Kuhnisch J, et al. Functional comparison of chronological and in vitro aging: Differential role of the cytoskeleton and mitochondria in mesenchymal stromal cells. PLoS One. 2012;7:e52700. doi: 10.1371/journal.pone.0052700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner W, Horn P, Castoldi M, et al. Replicative senescence of mesenchymal stem cells: A continuous and organized process. PLoS One. 2008;3:e2213. doi: 10.1371/journal.pone.0002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tazawa H, Kagawa S, Fujiwara T. Micro-RNAs as potential target gene in cancer gene therapy of gastrointestinal tumors. Expert Opin Biol Ther. 2011;11:145–155. doi: 10.1517/14712598.2011.542749. [DOI] [PubMed] [Google Scholar]
- 54.Hackl M, Brunner S, Fortschegger K, et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell. 2010;9:291–296. doi: 10.1111/j.1474-9726.2010.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zou Z, Zhang Y, Hao L, et al. More insight into mesenchymal stem cells and their effects inside the body. Expert Opin Biol Ther. 2010;10:215–230. doi: 10.1517/14712590903456011. [DOI] [PubMed] [Google Scholar]
- 56.Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a) Proc Natl Acad Sci USA. 2006;103:3645–3650. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jung JW, Lee S, Seo MS, et al. Histone deacetylase controls adult stem cell aging by balancing the expression of poly-comb genes and jumonji domain containing 3. Cell Mol Life Sci. 2010;67:1165–1176. doi: 10.1007/s00018-009-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tarunina M, Alger L, Chu G, et al. Functional genetic screen for genes involved in senescence: Role of Tid1, a homologue of the Drosophila tumor suppressor l(2)tid, in senescence and cell survival. Mol Cell Biol. 2004;24:10792–10801. doi: 10.1128/MCB.24.24.10792-10801.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stegh AH, Herrmann H, Lampel S, et al. Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol Cell Biol. 2000;20:5665–5679. doi: 10.1128/mcb.20.15.5665-5679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu S, Thweatt R, Lumpkin CK, Jr, et al. Suppression of calcium-dependent membrane currents in human fibroblasts by replicative senescence and forced expression of a gene sequence encoding a putative calcium-binding protein. Proc Natl Acad Sci USA. 1994;91:2186–2190. doi: 10.1073/pnas.91.6.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.