

Abstract
Many organisms survive fluctuating and extreme environmental conditions by manifesting multiple distinct phenotypes during adulthood by means of developmental processes that enable phenotypic plasticity. We report on the discovery of putative plasticity-enabling genes that are involved in transforming the gill of the euryhaline teleost fish, Fundulus heteroclitus, from its freshwater to its seawater gill-type, a process that alters both morphology and function. Gene expression that normally enables osmotic plasticity is inhibited by arsenic. Gene sets defined by antagonistic interactions between arsenic and salinity show reduced transcriptional variation among individual fish, suggesting unusually accurate and precise regulatory control of these genes, consistent with the hypothesis that they participate in a canalized developmental response. We observe that natural selection acts to preserve canalized gene expression in populations of killifish that are most tolerant to abrupt salinity change and that these populations show the least variability in their transcription of genes enabling plasticity of the gill. We found that genes participating in this highly canalized and conserved plasticity-enabling response had significantly fewer and less complex associations with transcriptional regulators than genes that respond only to arsenic or salinity. Collectively these findings, which are drawn from the relationships between environmental challenge, plasticity, and canalization among populations, suggest that the selective processes that facilitate phenotypic plasticity do so by targeting the regulatory networks that gives rise to the response. These findings also provide a generalized, conceptual framework of how genes might interact with the environment and evolve toward the development of plastic traits.
Keywords: phenotypic plasticity, canalization, evolution, gene regulatory networks, natural populations
Introduction
Phenotypic plasticity is a condition-dependent form of development that allows an organism to transform its physical traits in response to changes in environmental conditions (Debat and David 2001; Parsons et al. 2011). There are many well-studied examples of phenotypic change allowing organisms to thrive in fluctuating environments. These include invertebrates that develop armored defensive morphologies in the presence of predators (Dodson 1989), butterflies that acquire seasonal wing patterns (Watt 1969), and fish that alter gill structure in response to changes in oxygen levels, temperature, and salinity (Copeland 1950; Sollid and Nilsson 2006). Despite these and other classic examples of phenotypic plasticity and its importance for the survival of many organisms in variable environments, limited understanding of its genetic underpinnings has prevented study of its regulatory mechanisms and evolution (Debat and David 2001).
The adaptive evolution of populations to changing environments relies on the expression of heritable genetic variation among individuals, with natural selection promoting those individuals that are most fit and that reproduce consistent phenotypes. Under these conditions, selection for the stabilization of developmental gene regulatory programs that control the expression of morphological traits, as is seen in early development, results in reduced interindividual phenotypic variation, classically defined as canalization (Waddington 1942; Gibson and Wagner 2000). For example, regulation of Gap gene expression was recently shown to limit variation of segment determination in the Drosophila blastoderm during morphogenesis (Manu et al. 2009). Because phenotypic plasticity is also a developmental process that depends on the environment to constrain individual traits (Debat and David 2001), and because variation in the regulation of gene expression is a known target of natural selection (Whitehead and Crawford 2006), transcriptional regulation of these gene regulatory programs may also be subject to selection favoring canalization, as long as plasticity is under the control of discrete sets of genes (Debat and David 2001; Valladares et al. 2002; Oleksiak and Crawford 2012).
The killifish, Fundulus heteroclitus, provides a unique opportunity for investigating the evolution of phenotypic plasticity. It is a euryhaline teleost adapted to life in estuaries along the east coast of the United States (Whitehead 2010). Its tidal niche produces extreme osmotic gradients, requiring some populations of killifish to tolerate daily changes in salinity that can range from freshwater to marine (Shaw et al. 2008; Whitehead et al. 2011). As a result, some populations have evolved to rapidly and reversibly transform their adult gills between seawater and freshwater gill types to compensate for changes in salinity (Copeland 1950; Philpott and Copeland 1963; Wilson and Laurent 2002), while other populations facing less severe osmotic variation have not (Whitehead et al. 2011). Because killifish populations that vary in their ecology are also easily sampled, they provide a popular model system to study naturally occurring variation of gene expression and evolved phenotypes in outbred populations (Oleksiak et al. 2002, 2005; Whitehead and Crawford 2006; Whitehead et al. 2011).
Here, we used killifish as a model of phenotypic plasticity to characterize genes that enable the development of plastic traits and to investigate how evolution preserves their regulatory controls. The plastic response of the gills to salinity change can be inhibited by arsenic. Our previous work demonstrates that exposure to nontoxic levels of arsenic produces no physiological effects in fish maintained in stable freshwaters or seawater environments, but inhibits the animal’s capacity to acclimate to increasing salinity (Stanton et al. 2006; Shaw, Gabor, et al. 2007; Shaw et al. 2010). To identify putative phenotypic plasticity genes, we exposed fish to arsenic and performed genome expression studies to identify arsenic-dependent gene expression in the gills of killifish following a rapid and dramatic change in salinity. We then tested if the expression of these putative plasticity-enabling genes is characterized by reduced interindividual variation compared with other differentially expressed (DE) genes. We also quantified the variation of these genes in natural populations that differ in their ability to acclimate to different salinities to discover how the regulation of transcriptional variation contributes to changes in osmotic tolerance. Finally, we measured the complexity of networks constructed with these plasticity-enabling genes, and their transcriptional regulators to reveal possible mechanisms controlling transcriptional variation.
Results
Putative Plasticity-Enabling Genes Identified
Genome expression studies identified gene sets thought to be critical for establishing osmotic plasticity based on their regulatory dependence on arsenic during salinity change (i.e., arsenic–salinity interaction). Genome expression profiles from gills of Northern killifish, which exhibit extreme osmotic plasticity, were compared between fish acclimated to freshwater and directly challenged with seawater for 1 and 24 h in the presence and absence of arsenic (100 µg/l)—a concentration observed in natural environments (USEPA 1980). Linear models were used to determine genes differentially regulated by arsenic, salinity and their combination. The models specified “arsenic,” “salinity 1 h,” and “salinity 24 h,” as main effects and included interaction terms to identify genes in which arsenic interacts with salinity 1 h or salinity 24 h in a nonadditive manner (results for all genes in supplementary fig. S1, Supplementary Material online).
Analysis of the gene expression data using linear models revealed 496 DE genes with significant (P < 0.05, fold change >+ 2 or <−2) salinity-arsenic interactions. Of these, 367 were uniquely associated with the interaction, that is, they were not significant in any main effect. Linear models detected 203 and 221 DE genes for salinity 1 h and salinity 24 h, respectively (fig. 1A and B). Arsenic alone had a minor effect on gene expression (129 DE genes). A majority (73%) of the 496 DE interaction genes showed reduced expression (supplementary fig. S1, Supplementary Material online) and all of these interactions were antagonistic (i.e., less than additive; fig. 1C and D) producing expression values that more closely resembled those of killifish living in stable freshwater environments.
Fig. 1.

Arsenic–seawater interactions reveal phenotypic plasticity-enabling genes. Differences in gene expression (GE) from gill tissue of four male killifish, Fundulus heteroclitus, for each of six treatments were analyzed by measuring quantile normalized log2 expression values using a two-factor linear model that includes two categorical variables (presence of arsenic and time spent in seawater) as well as their interactions. DE genes were defined by P values < 0.05 and fold changes >2 for at least one treatment. The y axis shows differences in GE from freshwater (FW) in units of SD for (A) genes significantly DE at 1 h, (B) genes significantly DE at 24 h, (C) genes with significant interactions between arsenic and salinity at 1 h, and (D) genes with significant interactions between arsenic and salinity at 24 h. Genes upregulated compared with FW appear in red; down in blue. DE genes at 1 h (A) and 24 h (B) diverge from FW, yet the divergence at 1 h (A) is not seen at 24 h (A). Arsenic’s inhibition of gene expression associated with salinity acclimation is seen by observing the interaction gene sets. DE genes of the two treatments, arsenic–seawater 1 h (C) and arsenic–seawater 24 h (D), deviate from expectations of additivity. For example, the arsenic–seawater 1 h DE genes (C) respond to both arsenic and 1 h seawater treatments when compared with FW. An additive model predicts that the combined effect of both arsenic (As) and seawater acclimation at 1 h (SW 1 h) should result in a greater (red) or lesser (blue) GE response (As + 1 h SW). However, this combined effect produces an antagonistic interaction GE pattern that is similar to that observed in a stable environment (FW). Genes showing interaction effects at 24 h (D) recapitulate this pattern.
Putative biological functions for the interaction gene sets were characterized by defining orthologous zebrafish genes with OrthoDB (Waterhouse et al. 2011) and evaluating known functions and enrichments within biological pathways using Ingenuity Pathways Analysis (IPA) software (Ingenuity Systems, Inc., Redwood City, CA). Pathway analyses of the interaction gene sets (1 h) revealed significant associations with tissue development, endocrine system development, nucleic acid metabolism, cellular development, and hematological development. At 24 h, pathway analyses of the interaction gene sets revealed significant associations with cell morphology, cellular assembly, renal development, cell death, and cellular growth and proliferation.
Plasticity-Enabling Genes Are Characterized by Reduced Interindividual Variation
To assess differences in interindividual gene expression variation we compared the coefficient of variation (COV) of the interaction genes to the COV of all other DE genes (i.e., main effects). Variation in the expression of these putative plasticity-enabling genes among individual fish was reduced compared with those from the main effects gene set (Wilcoxon rank sum test, P < 2 e-16; fig. 2A). The COV for the DE interaction gene set was only 83% as large as the COV for the DE main effects gene set. In addition, interindividual variation in gene expression for the 367 uniquely interaction DE genes (1 and 24 h) differed between the three experimental time points (Wilcoxon rank sum tests, P < 5.8 e-06). Maximum variation was observed in freshwater, a reduction in variation occurred during the early onset of plasticity (1 h), and variation increased again after 24 h as the fish initially acclimate to seawater (fig. 2B).
Fig. 2.

Plasticity in gene expression and physiology are canalized. Box plots illustrate the data range (whiskers are 1.5 times interquartile range), interquartile range (box), and median (line). Unique letters indicate significance, as defined below for each plot. (A) Genes revealed through the combined sets of arsenic and seawater interactions exhibit reduced interindividual variation as evidenced by their significantly lower coefficient of variation (COV; Wilcoxon rank sum test; n = 496 interaction genes, 365 main effects genes; P < 2 e-16) compared with the combined main effects of arsenic, seawater 1 h, and seawater 24 h. (B) The reduced variation in putative phenotypic plasticity gene expression observed in the unique interaction gene set persists across the time-course of acclimation as revealed by the distribution of their SD. Wilcoxon rank sum tests identified significant differences (P < 5.8 e-6) between time points in interindividual variation of interaction gene expression. (C) Significant differences in interaction gene expression variation (SD) were observed between freshwater and mesohaline populations (P < 0.03) and freshwater and coastal populations (P < 0.001) using Wilcoxon rank sum tests. The expression of these genes is more accurately and precisely regulated in the freshwater population and variation increases with salinity of native environments (freshwater<mesohaline<coastal). (D) The functional significance of the canalized gene expression is explored by comparing interindividual variation in plasma chloride levels between the three populations during the early stages (0–24 h) that reflect the onset of acclimation. Variation in the distribution of plasma chloride levels is significantly lower in the freshwater population (n = 17 fish per population, modified robust Levene’s test, P < 0.04).
Population Plasticity Range Corresponds to Reduced Interindividual Variation
Natural populations known to differ in their evolved plasticity to osmotic challenge were examined to determine whether natural selection imposed by a steep salinity cline preserves the tight regulation of the putative plasticity-enabling genes. For these studies genome expression data reported in Whitehead et al. (2011) were reanalyzed. These authors investigated adaptive differences in tolerance to hypo-osmotic shock in three populations residing in freshwater (<0.1‰ salinity), mesohaline (10–12‰ salinity), and coastal (>30‰ salinity) environments of the Chesapeake Bay, which cross a critical physiological boundary (<1‰ salinity). They identified DE genes among the three populations at four time points during osmotic acclimation (6, 24, 72, and 168 h). We characterized interindividual variation in expression of the interaction genes identified in the present study from a Northern population of killifish that is extremely plastic to salinity change across the populations studied by Whitehead et al. (2011) that vary in their osmotic plasticity.
The interaction genes identified as important to hyper-osmotic plasticity in the present study were also differentially regulated during the onset of hypo-osmotic tolerance in the studies by Whitehead et al. (2011) (fig. 3). Bidirectional mapping (≥94% identical, ≥70 bp) of probe sequences between the microarray platform used in the present study and the one used by Whitehead et al. (2011) identified 1,255 shared genes that were used in these comparisons. Gene-set enrichment analysis (GSEA) (Subramanian et al. 2005) of ranked expression values for the genes shared between the two studies revealed that only fish from the freshwater population (<0.1‰ salinity) described by Whitehead et al. (2011) shared similar expression patterns with the interaction gene sets described in the present study, and this enrichment was only observed during early acclimation (6 and 24 h; fig. 3B). In contrast, no population-specific or distinct temporal pattern of shared expression was observed for the main effect gene set (fig. 3A).
Fig. 3.
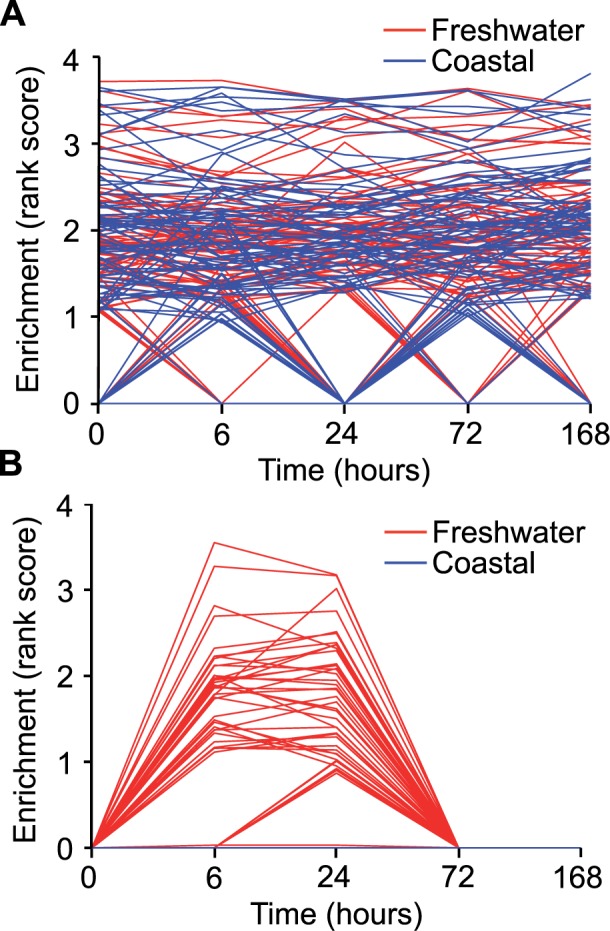
GSEA reveals general plastic response of interaction genes. GSEA (Subramanian et al. 2005) was used to investigate local enrichments of the main effects gene set and interaction gene sets across time (0, 6, 24, 72, and 168 h) in the freshwater and coastal populations reported in Whitehead et al. (Whitehead et al. 2011). For these analyses, the data from Whitehead et al. were ranked by mean expression value for each time point within a population and time point, and enrichment by the interaction or main effects gene sets identified in the current manuscript interrogated. For gene sets exhibiting significant enrichment, the Enrichment Rank Scores for genes from the comparison group (i.e., interaction, main effects) that were overrepresented in the top or bottom of these sets (i.e., the leading-edge subsets) that account for a gene set’s enrichment signal are provided as individual lines for seawater 1 h (A) and arsenic–seawater 1 h interactions (B) treatments. These are plotted across time points (0 h, seawater control, through 168 h in freshwater) for populations living in coastal (blue) and freshwater (red) environments. These plots reveal that only fish from the freshwater population (red) shared similar expression patterns with the interaction gene sets (B) during early acclimation (6 and 24 h).
Regulatory accuracy and precision of the putative plasticity-enabling genes increased as a function of a population’s plasticity range, which in Whitehead et al. (2011) was greatest for the freshwater population and decreased from mesohaline to coastal populations. Significantly less interindividual variation was observed in the expression of the interaction genes when fish were confronted with an osmotic challenge in freshwater populations (fig. 2C) compared with both the mesohaline (P < 0.03) and coastal populations (P < 0.001). By contrast, no differences in the transcriptional variation of the interaction genes were detected across the three populations when maintained in common seawater conditions (P < 0.49). In addition, the freshwater population also maintained tighter control of ion homeostasis during the early phase of the plastic response (≤24 h) as measured by plasma chloride levels compared with mesohaline and coastal populations (modified robust Levene’s test, P < 0.04; fig. 2D).
Reduced Network Complexity May Explain Reduced Interindividual Variation
Gene regulatory networks were constructed using IPA from the interaction versus main effects gene sets and their known transcriptional regulators (fig. 4A and B and supplementary fig. S2, Supplementary Material online). Network analysis comparing interaction and main effects gene sets revealed a significant reduction in the complexity of gene regulatory networks formed from interaction genes and their known upstream regulators (fig. 4A) compared with main effects genes and their known upstream regulators (fig. 4B and C; P < 0.05). To validate these findings, we also constructed negative-control reference networks between these two sets of genes, and downstream molecules they are reported to regulate (supplementary fig. S3B and C, Supplementary Material online). No differences in network complexity were observed between the two gene sets and their regulatory targets.
Fig. 4.

Phenotypic plasticity-enabling genes display reduced transregulatory complexity. Gene regulatory networks constructed using IPA software (Ingenuity Systems) between the uniquely interaction gene sets (A) and the uniquely main effect gene sets (B) are highlighted in orange, and their known, direct and indirect upstream transregulating molecules are highlighted in blue. Visual inspection of the networks suggests that the interaction gene sets form less complex networks (A) than noninteraction gene sets (B). Density distribution of these relationships (C) is significantly reduced in the unique interaction gene set. These networks are scale free and the probability that a vertex in the network interacts with k other vertices decays as a power law: P(k) ∼ k−g. Analysis of covariance comparing log values of P(k) to log values of k determines that the slope (g) for the interaction gene sets is significantly different (n = 13 levels of k for interaction genes, 26 levels of k for main effects genes; P < 0.05) indicating less connectivity among the genes.
Discussion
This study provides empirical evidence supporting the predicted existence of plasticity genes and suggests that canalization of their expression contributes to the evolution of plastic traits (Debat and David 2001; Oleksiak and Crawford 2012). Our analyses of several data sets drawn from four separate natural populations of killifish converge to 1) identify putative plasticity-enabling genes, 2) demonstrate that they are tightly regulated during salinity acclimation, and 3) reveal that this canalization is likely the result of diminished complexity of gene regulatory networks. These findings suggest that natural selection preserves canalization by directly targeting the gene regulatory networks that underlie the plastic response.
Putative Plasticity-Enabling Genes Identified
A classic chemical inhibition approach (Wooley 1944) was applied to identify candidate killifish genes that appear to control condition-dependent gill-type morphology and function. Such chemical knockdown methods have been widely applied to identify candidate genes and gene sets associated with discrete physiological functions (Pichler et al. 2003). Arsenic was used in our studies to inhibit plasticity because it was shown to specifically inhibit salinity acclimation in killifish (Stanton et al. 2006; Shaw, Gabor, et al. 2007; Shaw et al. 2010). Previous studies demonstrated that when fish are stably maintained in either freshwater or seawater, concentrations of arsenic up to 12,000 μg/L, which is 120 times the subtoxic concentration used in the present study have no measurable physiological effects and do not illicit cellular defense mechanisms in the gills, for example, induction of heat shock proteins and the sodium potassium pump that are known indicators of stress (Shaw, Gabor, et al. 2007; Bers and Despa 2009). In contrast during salinity acclimation the concentration of arsenic used in the present study has been shown to interfere with ion transport and trafficking of chloride channels in the gill epithelium (Stanton et al. 2006; Shaw et al. 2010), and at the high concentrations (12,000 μg/L) that have no effect on killifish living in freshwater or seawater completely block their ability to acclimate to changes in salinity (Stanton et al. 2006; Shaw, Gabor, et al. 2007; Shaw et al. 2010). In this study, the number of genes that exhibited transcriptional dependence on the interfering effects of arsenic during salinity acclimation (i.e., arsenic–salinity interactions) was three times greater than when fish were exposed to arsenic alone (supplementary fig. S1, Supplementary Material online) and these gene expression patterns were universally antagonistic—more closely resembling the gene expression patterns of killifish living in stable freshwater environments (fig. 1). These findings suggest that arsenic targets and inhibits plasticity and from them we predict that the interaction genes whose regulation during salinity change is dependent on arsenic (i.e., chemically knocked down) are plasticity-enabling genes important for establishing osmotic tolerance.
The reversible transformation of the fish gill from a freshwater-type to a seawater-type phenotype or vice versa requires dynamic changes in salt transport and cellular ultrastructure of mitochondrion rich cells (MRC) (Wilson and Laurent 2002; Evans et al. 2005; Wood and Grosell 2008) that occur in two distinct phases, an acute early phase that begins <1 h and a second phase that commences after 24 h (Katoh and Kaneko 2003; Shaw, Gabor, et al. 2007; Shaw et al. 2008; Whitehead et al. 2011). For example, an increase in sodium and chloride secretion by MRC occurs rapidly (≤1 h) in the killifish gill following transfer from freshwater to seawater, as these cells reorganize and recruit vesicles containing cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels from an intracellular pool to the apical surface of the gill membrane (Daborn et al. 2001; Shaw et al. 2008). Katoh and Kaneko (2003) have demonstrated that changes in sodium and chloride transport and epithelium composition of the gill are accompanied by morphological changes of MRC that include changes in their surrounding accessory cells that occur within hours. Longer-term chronic changes (≥24 h) in gill structure are characterized by de novo production of ion-transporting proteins (Scott et al. 2004; Shaw, Gabor, et al. 2007; Whitehead et al. 2011), and ultimately through the formation of new MRC (Katoh and Kaneko 2003).
The patterns of differential expression of the interaction gene sets reflect this well-accepted view that salinity acclimation involves two distinct phases (Katoh and Kaneko 2003; Shaw, Gabor, et al. 2007; Shaw et al. 2008; Whitehead et al. 2011), and their inferred biological functions are consistent with the dynamic changes that occur as the gill alters its morphology and function (Evans 2008). The two sampled time points of the present study produced unique sets of interaction genes, as only a few were significant at both 1 and 24 h salinity time points (supplementary fig. S1, Supplementary Material online). Others have similarly noted dynamic patterns of gene expression in killifish gills during salinity acclimation and suggested that in changing environments natural selection is more likely to target these genes (Whitehead et al. 2011). Of the genes exhibiting dynamic expression during salinity acclimation, these authors identified 13 core genes they suggest are important for the evolution of salinity tolerance observed in killifish. Based on sequence homologies only four could be directly compared with the gene models used in the present study and all were observed in the interaction gene set and shared similar salinity dependent patterns of expression between the two studies. The inferred biological functions associated with the comprehensive interaction gene sets from our study also reflect the dynamic changes occurring in gill function and morphology, including enrichment of pathways associated with tissue development, endocrine systems development, nucleic acid metabolism, and cellular development at 1 h, and cell morphology, assembly, cell death, and cellular growth and proliferation at 24 h. We conclude from these associations that genes revealed through the interactions between arsenic and osmotic stress at both 1 and 24 h are critical for the plastic response and represent putative plasticity-enabling genes.
Expression of Putative Plasticity-Enabling Genes is Canalized
Transcriptional regulation is a variable trait that is subject to natural selection (Whitehead and Crawford 2006; Oleksiak and Crawford 2012) and of predicted importance for the evolution of phenotypic plasticity (Debat and David 2001). There are reports of evolved differences in expression variation across populations residing along environmental gradients (Whitehead and Crawford 2006; Levine et al. 2011). For example, Levine et al. (2011) studied whole-genome expression among Drosophila populations from temperate and tropical regions and found adaptive contraints on the expression levels of development genes across environmental gradients. Selection for the stabilization of such developmental programs reduces interindividual morphological variation during early development, classically defined as canalization (Waddington 1942; Gibson and Wagner 2000), and increased regulatory controls on transcription are known to provide such reproducibility of morphological outcomes (Hornstein and Shomron 2006; Manu et al. 2009). For example, regulation of Gap gene expression in Drosophila has been shown to underlie the limited variation observed in blastoderm segment determination during gastrulaton (Manu et al. 2009). Therefore transcriptional regulation of phenotypic plasticity—a condition-dependent developmental program, may also be subject to selection favoring canalization (Debat and David 2001; Oleksiak and Crawford 2012).
Given that canalization of gene experssion has been suggested to play a role in the stabilization of phenotypes, if the interaction genes identified in the present study are important for enabling the onset of plasticity it is predicted that their transcriptional regulation would be less variable than other DE genes during the time-course of plasticity. Indeed, interindividual variation in expression was significantly lower in the uniquely interaction gene sets compared with the main effects genes (fig. 2A), and the interaction genes were more acureately and precisely regulated during the onset of plasticity across the time-course of these expreiments (1 h, fig. 2B). Similarly, if gene expression canalization is required for the plastic changes occurring in the killifish gills, then selection should preserve the precise expression of interaction genes in the natural populations originally investigated by Whitehead et al. (2011) as a function of their salinity range and osmotic tolerance (i.e., freshwater > mesohaline >coastal). Canalization of these genes should also be predicted to functionally contribute to salinity acclimation by constraining phenotypic outcomes. Indeed, our results are consistent with these predictions. Interindividual variation in interaction gene expression (fig. 2C) and tighter control of ion homeostasis as measured by plasma chloride levels (fig. 2D) was more robust in the freshwater population compared with the mesohaline and coastal populations. Although the reduced transcriptional variation observed in the freshwater population following an osmotic challenge could arguably be a function of the lower heterozygosity reported for it (Whitehead et al. 2011), no differences were observed in the regulatory control of the interaction genes among the three populations when held in common conditions (seawater, 0 h). Therefore, a hallmark feature of these genes associated with transforming gill morphology and function is the canalization of their expression patterns during the early phases of the plastic response that selection has preserved in natural populations as a function of their plastic range.
Reduced Network Complexity May Explain Reduced Interindividual Variation
What mechanisms might account for the tight regulation of genes responsible for the expression of plastic traits? Canalized gene expression could plausibly result from a more streamlined association with upstream controlling genes as suggested by two studies. von Dassow et al. (2000) constructed networks of known interactions among segment polarity genes that regulate development of neuroblasts, denticle patterns, and appendage primordia to investigate their ability to accurately regulate their expression. Although they hypothesized that robustness would only be achieved by adding complexity to what they predicted would be a fragile core topology, their simulated modules clearly demonstrated that the most streamlined working network model was the most robust. Landry et al. (2007) used yeast accruing mutations under neutral conditions to demonstrate that genes associated with longer, and more connected gene regulatory networks are more sensitive to regulatory perturbation and increased transcriptional variation. These simulated and empirical models of robustness are found to equally apply to our present study. As predicted, the regulatory networks constructed from the interaction genes would have fewer transcriptional regulators than regulatory networks composed from main effects genes. This is in fact the case (fig. 4A and B and supplementary fig. S3, Supplementary Material online) as interaction gene networks and those drawn from main effects genes can be described as following different power laws, consistent with mathematical models that describe scale-free networks (Barabasi and Oltvai 2004; King et al. 2012). Thus, our observations drawn from natural killifish populations under selection lead to the same conclusions as the in silico study of von Dassow et al. (2000) and the laboratory study of Landry et al. (2007). If less complex gene regulatory networks result in reduced transcriptional variation in plasticity-enabling genes it follows that selection might target the regulatory network for the evolution of plastic traits.
Conclusion
This study using a model species for a mechanistic understanding of phenotypic plasticity provides multiple lines of evidence for the existence of plasticity-enabling genes. Observations in the killifish model suggest that natural selection operates along a steep salinity gradient to preserve canalized gene expression in these phenotypic plasticity genes, likely by reducing the complexity of their regulatory gene networks. A conceptual model that draws on these collective findings (fig. 5) now provides a framework for exploring the functional relationships between environmental challenge, plasticity, and canalization that are critical in shaping ecologically responsive traits, and may ultimately help explain why plasticity is a hallmark feature of some species and not others. In particular, our findings suggest that selection targets the regulatory network for the evolution of phenotypic plasticity. This finding may presage the next major research thrust in molecular evolution, namely, to apply a systems genetics perspective to the question of how genes interact with the environment and evolve toward the development of ecoresponsive phenotypes. Understanding the evolutionary mechanisms that allow some populations to compensate for abrupt and extreme environmental changes will also help to understand and prepare for emerging environmental challenges such as climate change (Dawson et al. 2011).
Fig. 5.

A conceptual model of the functional relationships between environmental challenge, plasticity, and canalization. We propose (center graph) that increasing environmental challenge requires greater plasticity; a trait that natural selection enables by reducing complexity in gene regulatory networks and results in decreased gene expression variation. This generalizable model is highlighted in the surrounding panels with the results observed in the current study. Salinity gradients—Killifish prefer seawater environments, yet some populations can inhabit a wide range of salinities. Northern populations in Maine, United States, experience daily tidal influxes of freshwater. Similarly, a population living in the Potomac River thrives in freshwater. Both of these populations face the greatest environmental challenge due to the increased energetic costs of maintaining osmotic balance compared with mesohaline and coastal populations living in brackish water and seawater, respectively. Salinity induced phenotype—phenotypic plasticity enables populations to osmoregulate by modulating gill morphology to either a seawater or freshwater type. The Potomac River and Maine populations are capable of extreme osmotic plasticity. Gene regulatory complexity—this morphological transformation is regulated by a discrete set of genes that show canalized expression patterns coinciding with plasma chloride levels, which are also canalized in these fish. Canalization of these phenotypic plasticity-enabling genes is underpinned by a streamlined interaction network of upstream regulatory genes.
Materials and Methods
This manuscript reports on the analyses of data obtained from two animal studies. Materials and methods from those experiments utilizing natural populations of Northern killifish F. heteroclitus and previously unreported methods for data reanalyzed from Whitehead et al. (2011) that used killifish populations the Chesapeake Bay are organized by subject headers that match those of the results.
Putative Plasticity-Enabling Genes Identified
Animals and Care
These animal studies were performed in compliance with Institutional Animal Care and Use guidelines approved by Mount Desert Island Biological Laboratory (MDIBL No. 07-03). Natural populations of Northern F. heteroclitus were collected from Northeast Creek (Mount Desert Island, ME) by minnow trap and held in aquaria containing running seawater (pH 8.1 ± 0.4; salinity 33 ± 0.5‰, 15 °C) for at least 2 weeks to ensure acclimation to seawater. Fish were maintained outdoors under natural light cycles (photoperiod 15:9-h light:dark) and fed commercial flake food (48% protein, 9% fat; Tetracichlid, Tetra, Blacksburg, VA) once a day that contained no detectable inorganic arsenic, or monomethyl- or dimethyl arsenic: Only arsenobetaine (Shaw et al. 2010).
Chemicals
All chemicals employed were reagent grade or above.
Experimental Exposures
Killifish were acclimated to freshwater, as previously described. To investigate gene regulatory changes that occur in the gill during seawater acclimation, adult male fish, four fish per group, were directly transferred to 100% seawater as described (Shaw, Gabor, et al. 2007; Shaw Jackson, et al. 2007; Shaw et al. 2008, 2010) and gill tissues sampled at 0, 1, and 24 h. A second, identical time-course was performed using fish that had been pre-exposed to arsenic (as sodium arsenite) at a concentration of 100 μg/L for 48 h and then maintained at that arsenic concentration throughout the time-course. We chose this arsenic dose because it is encountered in marine environments and it is not toxic to killifish held in stable freshwater or seawater conditions, but does limit their capacity to acclimate to changing salinity (Shaw, Gabor, et al. 2007; Shaw, Jackson, et al. 2007). Control fish (0 h in seawater) were maintained in freshwater, with or without the addition of arsenic, for the duration of the experiment. Background freshwater arsenic levels were below detection limit (<1 μg/L) and background seawater arsenic values were between 1.5 and 3 μg/L, which is normal for all coastal waters in the United States. Water to which arsenic had been added was tested by inductively coupled plasma-mass spectrometry (ICP-MS) as described in Shaw, Jackson, et al. (2007) and Shaw et al. (2010) to ensure that the arsenic concentrations were as expected (Shaw, Gabor, et al. 2007; Shaw et al. 2010). Test water was aerated and replaced daily, and no appreciable variation in general water quality parameters such as ammonia, pH, salinity, and temperature was detected. Following exposures, fish were anesthetized in tricane (0.2 g/l) and euthanized by double pithing (Shaw et al. 2010). Tissues were immediately isolated, rinsed, and placed in RNAlater according to manufacturer’s recommendations.
RNA Isolation
Total RNA was isolated using RNAeasy kits (Qiagen, Valencia, CA). Tissue samples were homogenized on ice in lysis buffer containing guanidine isothyocyanate using a Tissue-Tearor (Biospec Products, Barlesville, OK), and RNA was precipitated using an equal volume of ethanol. The RNA was loaded on glass fiber columns, washed, and eluted with nuclease free water. Samples were then DNAase treated (DNAfree, Ambion), quantified using spectrophotometric optical density (OD260/280) measurements (Nanodrop ND-1000, ThermoFisher Scientific, Waltham, MA) and their integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Wilmington, DE). All samples used in genome expression studies achieved a RNA integrity number (RIN) score >7.
Transcriptome Sequencing, Assembly, and Annotation
We sequenced, assembled and annotated a reference transcriptome for subsequent use in developing microarrays genome expression studies. Killifish transcriptomes, representing 57 different conditions (supplementary table S1, Supplementary Material online) were sequenced from two 454 (GS-Flx) sequencing runs, using the procedure describe in (Meyer et al. 2009). The two runs produced a total of 1,340,048 expressed sequence tags (EST) containing 273,469,835 bp. Of these ESTs 1,302,633 exceeded our minimal quality standards (supplementary table S2, Supplementary Material online), which represents 229,144,736 bp that were used for the assembly. Eighty-seven percent (1,132,718 EST) of the high quality ESTs assembled into 38,673 contigs (18,682,616 bp) with an average length of 483 bp and N50 of 646 bp (supplementary table S2 and fig. S3A, Supplementary Material online). The average depth of coverage of the contigs was 9.8× per nucleotide positions (supplementary fig. S3B, Supplementary Material online). Of these contigs, about one-third (11,873) were considered large (>500 bp) and within this group the average contig size was 950 bp and the N50 was 1,028 bp. The majority of contigs (35,261) contained predicted open-reading frames (ORFs) that averaged 196 bp (supplementary fig. S3C, Supplementary Material online) and there were more than 12,000 ORFs greater than 200 bp in length. The remaining 14% (169,915 ESTs) of sequence reads were not homologous with other sequences and were retained as singletons (supplementary table S3, Supplementary Material online). Assembled contigs and singletons were aligned against the database of orthologous genes (OrthoDB [Waterhouse et al. 2011]; cegg.unige.ch, last accessed August 25, 2014) recovering 22,505 OrthoDB genes. The OrthoDB genes contained 17,849 Danio rerio gene models, including 876 of the 934 conserved single copy genes observed in closely related fish species.
Genome Expression Experiments
The transcriptome information was used to construct a custom array (NimbleGen HX12 x 135 K format, design ID 090504_FH_JC_EXP_HX12), which included three unique temperature-balanced probes for each of 38,673 contigs and control and random probes (Roche NimbleGen) designed to reflect the transcriptome nucleotide frequencies and sequence by Markov modeling. The RNA for use in microarray experiments was amplified using MessageAmp II kits (Ambion) and aRNA labeled with Cy dyes (Cy3 and Cy5) using NimbleGen Dual Color Labeling Kit (Roche NimbleGen) as describe in Jeyasingh et al. (2011). The four biological replicates labeled for each of the six treatments (24 samples) were randomly hybridized using the Hybridization Kit (Roche NimbleGen) to a fractional, factorial designed, microarray experiment using standard two-color comparisons with dye swaps. Following hybridization, raw data were extracted using NimbleScan v2.4 software (Roche NimbleGen) and quantile-normalized across arrays. Normalized log2 expression values were analyzed using linear models implemented in R (Team 2008) that included two variables, main effects of arsenic and time spent in seawater (salinity 1 h and salinity 24 h), and interactions between arsenic and salinity 1 h, or salinity 24 h. Differential expression of probes responding to a treatment was determined according to recommendations of the Microarray Quality Control Consortium (Guo et al. 2006; Shi et al. 2006) using P values < 0.05 and an estimated fold change ≥2.
Validation of Genome Expression Experiments
The results from these genome expression studies were validated in different fish exposed to the same treatment groups, using quantitative polymerase chain reaction (qPCR) assays to measure transcript levels of seven genes. These experiments were highly correlated (r = 0.83, P < 1.089 e-09, supplementary table S3 and fig. S4, Supplementary Material online), demonstrating significant agreement between these two studies. These microarray findings were further supported through concordance with 21 genes previously identified as important for salinity acclimation (see figs. 3 and 4, Whitehead et al. 2011). Of these genes, five shared common targets based on probe homology with the microarray employed in the current experiment and all of these overlapping genes shared similar expression patterns in freshwater and seawater with those reported in Whitehead et al. (2011) that were derived from different killifish populations.
Pathway Enrichment Analysis
Putative functions of interaction gene-sets were inferred through the application of IPA (Ingenuity Systems). For these analyses, Danio rerio orthologs of the interaction genes were identified through OrthoDB (Waterhouse et al. 2011) and used as input files for IPA. Global functional analysis was used to measure the likelihood that the association between interactions genes, and a curated biological process was due to chance.
Plasticity-Enabling Genes Are Characterized by Reduced Interindividual Variation
Differences in interindividual variation in gene expression between the set of main effects genes and the total combined set of interaction genes were assessed (fig. 2A). The COV was determined for each DE gene across all individuals within a comparison group (main effects, interaction) by dividing its standard deviation (SD) by the group mean. A Wilcoxon rank sum test as implemented in R was used to establish differences in COV between interaction genes (n = 496) and main effects genes (n = 365).
Interindividual variation using SD determined for each gene in the combined interaction gene sets was assessed across time through pairwise comparisons using Wilcoxon rank sum tests implemented in R (Team 2008).
Population Plasticity Range Corresponds to Reduced Interindividual Variation
We assessed differences in the variation among fish for the expression of the interaction genes across three distinct populations using expression data collected by Whitehead et al. (2011). Briefly, these authors sampled three populations residing along a steep salinity cline that included freshwater, mesohaline and coastal. Fish from each population were field collected and acclimated in the laboratory to seawater for 6 months. Following acclimation to this common condition, fish were immediately transferred to freshwater and for each population, gills were sampled and RNA isolated from five fish at 0, 6, 24, 72, and 168 h. These samples were amplified using MessageAmp II amplification kits (Ambion), aRNA labeled with Alexa fluor dyes (Alexa Fluor 555 and 647; Molecular Probes), and samples competitively hybridized to custom arrays (Agilent 8X15K element design; design ID 021434), which included probes for 6,800 elements, each printed in duplicate, plus control elements. The five biological samples were hybridized in a loop design with paired samples balanced across treatments. A mixed model specifying “dye” as a fixed effect, “array” as a random effect, and time and population as main effects including an interaction term were used to identify genes DE (P < 0.01).
Bidirectional Mapping of Probes
Probe sequences were bidirectionally mapped between the two microarray platforms. Homology was determined for regions of overlap ≥70 bp in which the two probes shared ≥94 identity. This mapping revealed 1,255 probes shared between the two platforms.
Gene-Set enrichment analysis
GSEA (Subramanian et al. 2005) was used to investigate local enrichments of the main effect gene set and interaction gene set across time (0, 6, 24, 72, 168 and h) in the freshwater and coastal populations reported in Whitehead et al. (2011). For these analyses, the data from Whitehead et al (2011) were ranked by mean expression value for each time point within a population and analyzed for enrichment by the main effect gene set (seawater, 1 h) and the interaction gene set (1 h). For gene sets that showed a significant enrichment, the Enrichment Rank Scores for genes from the comparison groups (i.e., interaction, main effects) overrepresented in the top or bottom of these lists (i.e., the leading-edge subsets) that account for a gene set’s enrichment signal were generated for each time point and population (Subramanian et al. 2005). These Enrichment Rank Scores for genes contributing significantly to the core enrichment within enriched gene sets are plotted for each population for the main effects gene set and the interaction gene set (fig. 3).
Variation in Candidate Plasticity Gene Expression among Populations
We compared variation in the expression of the uniquely interaction genes that based on probe homology were also present in (Whitehead et al. 2011) among populations (Freshwater, Mesohaline, and Coastal) using SD calculated for each gene (fig. 3C). Differences in the SD of interaction gene expression among fish was determined through pairwise comparisons of populations (i.e., freshwater and mesohaline, and freshwater-coastal) using Wilcoxon rank sum tests implemented in R (Team 2008).
Variation in Plasma Chloride Levels Across Populations
Plasma chloride measurements were determined following direct transfer from seawater to freshwater as previously discussed for each of 17 replicate fish across the first 24 h of acclimation for the coastal, mesohaline, and freshwater populations (Whitehead et al. 2011). Visualization of the data suggested that the coastal and freshwater population followed a normal distribution, whereas the mesohaline population followed a Cauchy or heavy-tailed distribution. A modified robust Levene’s type test based on the absolute deviation from the trimmed mean was used to assess differences in variation between the populations (fig. 3D). A classical Levene’s test was also conducted excluding the mesohaline population based on the absolute deviations from the mean. Both tests returned similar results.
Reduced Network Complexity May Explain Reduced Interindividual Variation
We used IPA to establish how many genes in the IPA knowledge base play a regulatory role with genes we identified as DE. We first mapped differentially express killifish genes to zebrafish genes using ORTHODB (Waterhouse et al. 2011), as previously described. Next, we used the Ingenuity knowledge base to identify upstream or downstream regulatory relationships (fig. 4A and B; supplementary fig. S3, Supplementary Material online). We defined regulatory relationships as those Ingenuity considers to have a relation type in the set (“expression,” “transcription,” or “protein-DBA”) for molecules other than those ingenuity considers to be in the set (“drug” or “microRNA” or “mature microRNA”). Barabasi and Oltvai (2004) have observed that biological networks are scale free and the probability that a vertex in the network interacts with k other vertices decays as a power law: P(k) ∼ k. For example, networks with more hubs on which more edges terminate have smaller values of g. We used an analysis of covariance to compare log values of P(k) to log values of k to see whether the slope (g) for uniquely interaction gene sets is significantly different from the slope for uniquely main effects gene sets.
Supplementary Material
Supplementary tables S1–S3 and figures S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank C. Taub, M. Adamo, and R. Lehman for support with animal experiments; K. Mockaitis, Z. Smith, and the Center for Genomics and Bioinformatics (CGB) at Indiana University (IU) for assistance with transcriptome sequencing; J.-H. Choi, A. Buechlein, C. Jackson and H. Tang for contributions to the transcriptome assembly and bioinformatics support; B. Scavone and Kestril studios for artwork; J. Lopez for performing the microarray hybridization experiments; S. Tugendreich and Ingenuity Systems for enabling access to data associated with the network analysis; S. Zhang and J. Roach for helping with multipopulation experiments and data collection; and B. Oliver, A. Scoville, M. Pfrender and the ecological and evolutionary genomics class at University of Notre Dame for editorial comments. The microarray data have been deposited at Gene Expression Omnibus under BioProject PRJNA201069 and accession GSE47035. The 454 sequence data have been deposited at the NCBI Sequence Read Archive under BioProject PRJNA201069 and experiment accession SRX278446. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GAJO00000000. The version described in this article is the first version, GAJO01000000. Support for these studies was provided by the National Institute of Environmental Health Sciences (NIEHS) Superfund Basic Research Program (P42 ES007373 to B.A.S. and C.Y.C); NIEHS (R01 ES019324 to J.R.S.); the Department of Defense-Strategic Environmental Research and Development Program (ER1503 to C.Y.C. and J.R.S.); National Institute of Health (NIH) grant (RO1 HL074175 to B.A.S.); NIH (P20 GM103423 to B.L.K.); NIH (P20 GM104318 to B.L.K.); National Science Foundation (NSF) grant (DEB-1120512 to A.W. and J.R.S.); NSF (DBI-0445967 to R.H.G.); and NSF (EF-0723771 to A.W. and F.G.), and the METACyt Initiative of IU, through a grant from the Lilly Endowment, Inc. to the CGB at IU. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the DOD, NIH, NIEHS, and NSF. Our work benefits from and contributes to the Fundulus Genomics Consortium (http://www.fundulus.org, last accessed August 25, 2014).
References
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:U101–U115. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Bers DM, Despa S. Na/K-ATPase—an integral player in the adrenergic fight-or-flight response. Trends Cardiovasc Med. 2009;19:111–118. doi: 10.1016/j.tcm.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland DE. Adaptive behavior of the chloride cell in the gill of Fundulus heteroclitus. J Morphol. 1950;87:369–379. doi: 10.1002/jmor.1050870208. [DOI] [PubMed] [Google Scholar]
- Daborn K, Cozzi RR, Marshall WS. Dynamics of pavement cell-chloride cell interactions during abrupt salinity change in Fundulus heteroclitus. J Exp Biol. 2001;204:1889–1899. doi: 10.1242/jeb.204.11.1889. [DOI] [PubMed] [Google Scholar]
- Dawson TP, Jackson ST, House JI, Prentice IC, Mace GM. Beyond predictions: biodiversity conservation in a changing climate. Science. 2011;332:53–58. doi: 10.1126/science.1200303. [DOI] [PubMed] [Google Scholar]
- Debat V, David P. Mapping phenotypes: canalization, plasticity and developmental stability. Trends Ecol Evol. 2001;16:555–561. [Google Scholar]
- Dodson SI. The ecological role of chemical stimuli for the zooplankton—predator-induced morphology in daphnia. Oecologia. 1989;78:361–367. doi: 10.1007/BF00379110. [DOI] [PubMed] [Google Scholar]
- Evans DH. Teleost fish osmoregulation: what have we learned since August Krogh, Homer Smith, and Ancel Keys. Am J Physiol Regul Integr Comp Physiol. 2008;295:R704–R713. doi: 10.1152/ajpregu.90337.2008. [DOI] [PubMed] [Google Scholar]
- Evans DH, Piermarini PM, Choe KP. The multifunctional fish gill: dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol Rev. 2005;85:97–177. doi: 10.1152/physrev.00050.2003. [DOI] [PubMed] [Google Scholar]
- Gibson G, Wagner G. Canalization in evolutionary genetics: a stabilizing theory? Bioessays. 2000;22:372–380. doi: 10.1002/(SICI)1521-1878(200004)22:4<372::AID-BIES7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Guo L, Lobenhofer EK, Wang C, Shippy R, Harris SC, Zhang L, Mei N, Chen T, Herman D, Goodsaid FM, et al. Rat toxicogenomic study reveals analytical consistency across microarray platforms. Nat Biotechnol. 2006;24:1162–1169. doi: 10.1038/nbt1238. [DOI] [PubMed] [Google Scholar]
- Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet. 2006;38(Suppl):S20–S24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- Jeyasingh PD, Ragavendran A, Paland S, Lopez JA, Sterner RW, Colbourne JK. How do consumers deal with stoichiometric constraints? Lessons from functional genomics using Daphnia pulex. Mol Ecol. 2011;20:2341–2352. doi: 10.1111/j.1365-294X.2011.05102.x. [DOI] [PubMed] [Google Scholar]
- Katoh F, Kaneko T. Short-term transformation and long-term replacement of branchial chloride cells in killifish transferred from seawater to freshwater, revealed by morphofunctional observations and a newly established ‘time-differential double fluorescent staining' technique. J Exp Biol. 2003;206:4113–4123. doi: 10.1242/jeb.00659. [DOI] [PubMed] [Google Scholar]
- King BL, Davis AP, Rosenstein MC, Wiegers TC, Mattingly CJ. Ranking transitive chemical-disease inferences using local network topology in the comparative toxicogenomics database. Plos One. 2012;7:e46524. doi: 10.1371/journal.pone.0046524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. Genetic properties influencing the evolvability of gene expression. Science. 2007;317:118–121. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- Levine MT, Eckert ML, Begun DJ. Whole-genome expression plasticity across tropical and temperate Drosophila melanogaster populations from Eastern Australia. Mol Biol Evol. 2011;28:249–256. doi: 10.1093/molbev/msq197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manu, Surkova S, Spirov AV, Gursky VV, Janssens H, Kim AR, Radulescu O, Vanario-Alonso CE, Sharp DH, Samsonova M, et al. Canalization of gene expression in the Drosophila blastoderm by gap gene cross regulation. PLoS Biol. 2009;7:591–603. doi: 10.1371/journal.pbio.1000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E, Aglyamova GV, Wang S, Buchanan-Carter J, Abrego D, Colbourne JK, Willis BL, Matz MV. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 2009;10:219. doi: 10.1186/1471-2164-10-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nat Genet. 2002;32:261–266. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- Oleksiak MF, Crawford DL. The relationship between phenotypic and environmental variation: do physiological responses reduce interindividual differences? Physiol Biochem Zool. 2012;85:572–584. doi: 10.1086/666904. [DOI] [PubMed] [Google Scholar]
- Oleksiak MF, Roach JL, Crawford DL. Natural variation in cardiac metabolism and gene expression in Fundulus heteroclitus. Nat Genet. 2005;37:67–72. doi: 10.1038/ng1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons KJ, Sheets HD, Skulason S, Ferguson MM. Phenotypic plasticity, heterochrony and ontogenetic repatterning during juvenile development of divergent Arctic charr (Salvelinus alpinus) J Evol Biol. 2011;24:1640–1652. doi: 10.1111/j.1420-9101.2011.02301.x. [DOI] [PubMed] [Google Scholar]
- Philpott CW, Copeland DE. Fine structure of chloride cells from three species of fundulus. J Cell Biol. 1963;18:389–404. doi: 10.1083/jcb.18.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler FB, Laurenson S, Williams LC, Dodd A, Copp BR, Love DR. Chemical discovery and global gene expression analysis in zebrafish. Nat Biotechnol. 2003;21:879–883. doi: 10.1038/nbt852. [DOI] [PubMed] [Google Scholar]
- Scott GR, Richards JG, Forbush B, Isenring P, Schulte PM. Changes in gene expression in gills of the euryhaline killifish Fundulus heteroclitus after abrupt salinity transfer. Am J Physiol Cell Physiol. 2004;287:C300–C309. doi: 10.1152/ajpcell.00054.2004. [DOI] [PubMed] [Google Scholar]
- Shaw JR, Bomberger JM, VanderHeide J, LaCasse T, Stanton S, Coutermarsh B, Barnaby R, Stanton BA. Arsenic inhibits SGK1 activation of CFTR Cl- channels in the gill of killifish, Fundulus heteroclitus. Aquat Toxicol. 2010;98:157–164. doi: 10.1016/j.aquatox.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JR, Gabor K, Hand E, Lankowski A, Durant L, Thibodeau R, Stanton CR, Barnaby R, Coutermarsh B, Karlson KH, et al. Role of glucocorticoid receptor in acclimation of killifish (Fundulus heteroclitus) to seawater and effects of arsenic. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1052–R1060. doi: 10.1152/ajpregu.00328.2006. [DOI] [PubMed] [Google Scholar]
- Shaw JR, Jackson B, Gabor K, Stanton S, Hamilton JW, Stanton BA. The influence of exposure history on arsenic accumulation and toxicity in the killifish, Fundulus heteroclitus. Environ Toxicol Chem. 2007;26:2704–2709. doi: 10.1897/07-032.1. [DOI] [PubMed] [Google Scholar]
- Shaw JR, Sato JD, VanderHeide J, LaCasse T, Stanton CR, Lankowski A, Stanton SE, Chapline C, Coutermarsh B, Barnaby R, et al. The role of SGK and CFTR in acute adaptation to seawater in Fundulus heteroclitus. Cell Physiol Biochem. 2008;22:69–78. doi: 10.1159/000149784. [DOI] [PubMed] [Google Scholar]
- Shi LM, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollid J, Nilsson GE. Plasticity of respiratory structures—adaptive remodeling of fish gills induced by ambient oxygen and temperature. Respir Physiol Neurobiol. 2006;154:241–251. doi: 10.1016/j.resp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Stanton CR, Thibodeau R, Lankowski A, Shaw JR, Hamilton JW, Stanton BA. Arsenic inhibits CFTR-mediated chloride secretion by killifish (Fundulus heteroclitus) opercular membrane. Cell Physiol Biochem. 2006;17:269–278. doi: 10.1159/000094139. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RDC. R: a language and environment for statisitcal computing. Vienna (Austria): R Foundation for Statistical Computing; 2008. [Google Scholar]
- USEPA. Ambient water quality criteria for arsenic. Washington (DC): Office of Water Regulations and Standards; 1980. p. 211. [Google Scholar]
- Valladares F, Balaguer L, Martinez-Ferri E, Perez-Corona E, Manrique E. Plasticity, instability and canalization: is the phenotypic variation in seedlings of sclerophyll oaks consistent with the environmental unpredictability of Mediterranean ecosystems? New Phytol. 2002;156:457–467. doi: 10.1046/j.1469-8137.2002.00525.x. [DOI] [PubMed] [Google Scholar]
- von Dassow G, Meir E, Munro EM, Odell GM. The segment polarity network is a robust developmental module. Nature. 2000;406:188–192. doi: 10.1038/35018085. [DOI] [PubMed] [Google Scholar]
- Waddington CH. Canalization of development and the inheritance of acquired characters. Nature. 1942;150:563–565. doi: 10.1038/1831654a0. [DOI] [PubMed] [Google Scholar]
- Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 2011;39:D283–D288. doi: 10.1093/nar/gkq930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt WB. Adaptive significance of pigment polymorphisms in colias butterflies .2. Thermoregulation and photoperiodically controlled melanin variation in colias eurytheme. Proc Natl Acad Sci U S A. 1969;63:767. doi: 10.1073/pnas.63.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead A. The Evolutionary radiation of diverse osmotolerant physiologies in killifish (Fundulus Sp.) Evolution. 2010;64:2070–2085. doi: 10.1111/j.1558-5646.2010.00957.x. [DOI] [PubMed] [Google Scholar]
- Whitehead A, Crawford DL. Neutral and adaptive variation in gene expression. Proc Natl Acad Sci U S A. 2006;103:5425–5430. doi: 10.1073/pnas.0507648103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead A, Roach JL, Zhang SJ, Galvez F. Genomic mechanisms of evolved physiological plasticity in killifish distributed along an environmental salinity gradient. Proc Natl Acad Sci U S A. 2011;108:6193–6198. doi: 10.1073/pnas.1017542108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Laurent P. Fish gill morphology: inside out. J Exp Zool, 2002;293:192–213. doi: 10.1002/jez.10124. [DOI] [PubMed] [Google Scholar]
- Wood CM, Grosell M. A critical analysis of transepithelial potential in intact killifish (Fundulus heteroclitus) subjected to acute and chronic changes in salinity. J Comp Physiol B. 2008;178:713–727. doi: 10.1007/s00360-008-0260-1. [DOI] [PubMed] [Google Scholar]
- Wooley DW. Some new aspects of the relationship of chemical structure to biological activity. Science. 1944;100:579–583. doi: 10.1126/science.100.2609.579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.