

Abstract
Background and Purpose
Ulinastatin (UTI), a serine protease inhibitor, was recently found to have an anti-inflammatory action. However, the mechanisms mediating this anti-inflammatory effect are not well understood. This study tested the hypothesis that UTI suppresses allergic inflammation by inducing the expression of haem oxygenase 1 (HO1).
Experimental Approach
Control mice and mice sensitized (on days 1, 9 and 14) and challenged (on days 21 to 27) with ovalbumin (OVA) were treated with UTI. The effects of UTI on basal expression of HO1 and that induced by OVA challenge were examined. The involvement of UTI-induced HO1 expression in anti-inflammatory and antioxidant effects of UTI was also evaluated.
Key Results
UTI markedly increased basal HO1 protein expression in lungs of control mice in a time- and dose-dependent manner, and augmented HO1 protein expression induced by OVA. The up-regulation of HO1 mediated by UTI in sensitized and OVA-challenged mice was associated with reduced airway inflammation, alleviated tissue injury, reduced oxidant stress and enhanced antioxidant enzyme activities. Inhibition of HO1 activity using HO1 inhibitor, zinc protoporphyrin, attenuated inhibitory effects of UTI on inflammation and oxidant stress, and its stimulant effects on antioxidant enzyme activities. Mechanistic analysis showed that UTI increased nuclear translocation of nuclear factor erythroid 2-related factor 2 (Nrf2), stimulated Nrf2 DNA binding activity and concomitantly up-regulated HO1 mRNA expression.
Conclusions and Implications
UTI is a potent and naturally occurring inducer of HO1 expression. HO1 up-regulation contributes significantly to the anti-inflammatory and organ-protective effects of UTI, which has important research and therapeutic implications.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| haem oxygenase 1 | dexamethasone |
| Evans blue dye |
This Table lists protein targets and ligands which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Ulinastatin was originally purified as a serine protease inhibitor (Muramatu et al., 1980) and is also known as urinary trypsin inhibitor (UTI). Recently, UTI was found to have anti-inflammatory and cytoprotective actions. UTI gene deletion exacerbated systemic inflammation and organ injury in endotoxaemic mice (Inoue et al., 2005a,b). Exogenous administration of UTI to endotoxaemic animals inhibited inflammatory mediator production, reversed systemic hypotension (Molor-Erdene et al., 2005), prevented intravascular coagulation (Takano et al., 2009) and ameliorated multiple organ injury (Takano et al., 2009; Zhang et al., 2011). UTI protected against ischaemia-reperfusion (Yano et al., 2003; Koga et al., 2010), haemorrhage (Masuda et al., 2003), smoke inhalation (Qiu et al., 2012) or organ injury induced by sea water inhalation (Rui et al., 2012). UTI has also been shown to have beneficial effects in patients with severe sepsis, traumatic brain injury and acute Kawasaki disease (Chen et al., 2009; Kanai et al., 2011; Tu et al., 2012).
Although anticoagulant and organ-protective effects of UTI may be partially explained by inhibition of protease activities, the anti-inflammatory action of UTI cannot be fully explained by protease inhibition. Mechanisms mediating the anti-inflammatory action of UTI remain to be elucidated.
Investigation into the effects of UTI on inflammation and organ injury has thus far been focused primarily on neutrophil-mediated responses. The effects of UTI on other types of inflammation, including allergic inflammation, have not been studied. Allergic airway inflammation differs significantly from sepsis-associated inflammation in cause, nature, extent of inflammation, as well as cell types and mediators involved. Allergic inflammation is generally associated with eosinophil infiltration (Barnes, 1996; Agrawal and Shao, 2010), whereas endotoxaemic inflammation is characterized by neutrophil infiltration. Studying the effect of UTI on allergic inflammation may gain new insights into other mechanisms underlying UTI’s anti-inflammatory action.
The haem oxygenase 1 (HO1)/carbon monoxide (CO) pathway is an important antioxidant and anti-inflammatory defense mechanism (Ferrándiz and Devesa, 2008; Gozzelino et al., 2010; Raval and Lee, 2010). Increased HO1 expression has been consistently demonstrated to be associated with beneficial effects in a variety of pathological conditions (Ferrándiz and Devesa, 2008; Gozzelino et al., 2010; Raval and Lee, 2010). UTI may exert its anti-inflammatory and organ-protective actions by up-regulating HO1 expression. However, UTI as a new HO1 inducer remains to be proven. The role of the HO1/CO pathway in mediating the anti-inflammatory and organ-protective actions of UTI is unclear.
The purpose of this study was threefold: firstly, to clarify if UTI is anti-inflammatory and tissue-protective in allergic inflammation in an ovalbumin (OVA)-sensitized and challenged mouse model; secondly, to demonstrate that UTI is a novel HO1 inducer; and thirdly, to define the role of HO1 up-regulation in mediating UTI’s anti-inflammatory and antioxidant effects. We demonstrated for the first time that UTI is a potent and naturally occurring inducer of HO1. HO1 up-regulation contributes significantly to the anti-inflammatory and organ-protective effects of UTI. Our study provides new insights into the mechanisms of action of UTI.
Methods
Animal studies
All animal study protocols were approved by Institutional Animal Care and Use Committee, and complied with the regulations of the ministry of health of China and the US National Institute of Health guidelines. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). FVB mice (8–10 weeks-old) were obtained from Vital River Laboratory Animal Technology (Beijing, China), housed five mice per cage, and kept under a 12 h light/dark cycle and constant temperature 21 ± 2oC, humidity, 30–70%, with food and water provided ad libitum. The total number of mice used was 233. After 1 week of acclimatization, mice were randomly divided into study groups. Mice in the non-sensitized control group were sham-sensitized by injection with PBS and sham-challenged with PBS inhalation. Mice in the other groups were sensitized by i.p. injection of OVA (emulsified in 2.5 mg aluminum hydroxide in PBS) 25 μg·per mouse, on days 1, 9 and 14, and challenged by inhalation of 1% OVA in PBS for 20 min day-1 on days 21 to 27. Mice in OVA + UTI or OVA + Dex groups were administered UTI (100 000 U·kg−1·day−1, i.p.) or Dex (1 mg·kg−1·day−1, i.p.) on days 21 to 27. Mice in OVA + UTI + ZnPP group were injected with ZnPP (20 mg·kg−1·day−1, i.p.) 1 h before each UTI administration. On day 28, airway reactivity or lung endothelial permeability was measured, or bronchoalveolar lavage (BAL) was performed followed by the collection of blood and tissue samples.
For OVA inhalation, mice were placed in a plexiglas inhalation chamber maintained under normoxic and normocapnic conditions. Aerosolized OVA or PBS was delivered using an ultrasonic nebulizer (NE-U12, Omron Corp., Beijing, China)
Measurement of airway responsiveness
Mice were anaesthetized with nembutal (60 mg·kg−1, i.p.); absence of withdrawal reflex to toe pinch in combination with assessment of jaw “tone”, heart rate and respiratory rate were used to assess depth of anaesthesia. The trachea was cannulated and the mice were mechanically ventilated and then placed in a sealed whole body plethysmograph connected to an AniRes2005 data collection and analysis system (BestLab, Beijing, China) for monitoring respiratory mechanics. Transpulmonary resistance was recorded at baseline, and after aerosolized PBS or three doses of aerosolized methacholine (3, 9 and 27 mg·mL−1). Airway reactivity was measured as changes in transpulmonary resistance provoked by each dose of methacholine.
Counting cells in the bronchoalveolar lavage fluid (BALF)
BAL was performed by delivering 0.8 mL of prewarmed PBS into the trachea followed by gentle suction. Following removal of debris, pooled BALF from three BALs was centrifuged. The pellet was resuspended in PBS, and total cell number was counted with haemocytometer after cell viability determination by trypan blue exclusion test. For differential cell counts, cells were centrifuged onto cytospin slides, differentially stained with Diff-Quik® stain reagent (Beyotime Institute of Biotechnology, Nanjing, China) and counted.
Histology and immunofluorescence staining
Mouse nasal cavity and lungs were fixed in 10% formalin and processed or were embedded in optimal cutting temperature compound and snap-frozen. For histological analysis, paraffin-embedded sections (4 μm) were stained with haematoxylin and eosin. For HO1 or nuclear factor erythroid 2-related factor 2 (Nrf2) immunofluorescence staining, lung cryosections (6 μm) were fixed with paraformaldehyde, permeabilized and stained with HO1 or Nrf2 antibody (Abcam, Cambridge, MA, USA) followed by staining with fluorescein isothiocyanate-conjugated secondary antibody. Nuclei were counterstained with DAPI.
Measurement of lung endothelial permeability
Lung microvascular endothelial permeability was assessed using EBD leakage index as a marker, as previously described (Wang le et al., 2002). Tissue EBD content (ng EBD·mg−1 dry tissue) was calculated by comparing tissue supernatant A620 readings with an EBD standard curve.
Measurements of serum level of IgE, BALF levels of cytokine, oxidant stress markers and antioxidant activities, and lung tissue levels of protein carbonyl content and malondialdehyde (MDA)
Serum levels of total and OVA-specific IgE, BALF levels of IL-4, IL-5 and IFN-γ, or BALF level of eotaxin-1 were measured using mouse IgE, IL-4, IL-5 and IFN-γ ELISA kits (Bethyl Laboratories, Montgomery, AL, USA) or eotaxin-1 ELISA kit (Yuanye, Shanghai, China). BALF level of total antioxidant capacity (TAOC), glutathione (GSH), superoxide dismutase (SOD) and catalase activities were determined using assay kits (Beyotime Institute of Biotechnology, Nanjing, China).
BALF leukocyte reactive oxygen species activity was measured using a 2′, 7′-dichlorofluorescein diacetate (DCFDA) fluorescence probe (Beyotime Company, Shanghai, China). One millilitre of BALF containing 1 × 105 leukocytes was mixed and incubated with 20 μM DCFDA at 37°C for 20 min. Fluorescence intensity of the oxidized DCFDA was measured using a spectrophotometer (Perkin-Elmer, Waltham, MD, USA).
Lung tissue levels of protein carbonyl content or MDA were determined using a protein carbonyl content assay kit (Abcam) or a MDA assay kit (Beyotime Institute of Biotechnology).
Western blot
Total, cytoplasmic or nuclear protein was extracted from lungs. Equal amounts of proteins (25 μg) were separated on 10% SDS-PAGE gels and transferred to a PVDF membrane. Western blot was performed as previously described (Ye et al., 2008) using eosinophil peroxidase, HO1, Nrf2, β-actin or lamin specific antibodies (Abcam; Zymed, Carlsbad, San Diego, CA, USA or Bioss Inc., Beijing, China).
Real-time PCR
RNA was isolated from lungs using the RNeasy mini kit (Qiagene, Germantown, MD, USA). Reverse transcription was performed using Sensiscript RT kit (Promega, Madison, WI, USA). Real-time PCR was performed in an ABI Prism 7900 sequence detection system (Applied Biosystems, Foster City, CA, USA) using SYBR Green PCR master mix following a standard protocol. The primer sequences are provided in Supporting information Table S1. Results were analysed using the 2−ΔCt method. HO1, IL-4, IL-5 or IFN-γ mRNA was normalized to GAPDH mRNA.
Measurements of Nrf2 and NF-κB binding activity
Nuclear protein was prepared from lungs. Nrf2 or NF-κB DNA binding activity was measured using Nrf2 (antioxidant-response element) or NF-κB electrophoretic mobility shift assay Kit (Signosis, Santa Clara, CA, USA).
Statistical analysis
Data are expressed as means ± SD and analysed by one-way anova followed by post hoc analysis using Holm-Sidak method, or were analysed by Kruskal–Wallis test followed by Mann–Whitney U-test. A P-value less than 0.05 was considered significant.
Chemicals
Dexamethasone (Dex), Evans blue dye (EBD), OVA, methacholine, UTI and zinc protoporphyrin (ZnPP) were purchased from Sigma-Aldrich (St Louis, MO, USA).
Results
UTI inhibits allergic airway inflammation
We examined the anti-inflammatory effect of UTI on allergic inflammation in comparison with the effect of Dex, which is a well-known anti-inflammatory agent and known up-regulator of HO1 expression (Xu et al., 2008). Consistent with the characteristics of allergic inflammation, OVA-sensitized and challenged mice displayed elevated serum levels of total and OVA-specific IgE (Supporting information Figure S1), higher numbers of BALF eosinophils and a higher lung tissue level of eosinophil peroxidase (Figure 1A–C), higher BALF levels of IL-4, IL-5 and eotaxin, and lower BALF level of IFN-γ (Figure 1D–G). These mice also had higher levels of IL-4 and IL-5, and a lower level of IFN-γ mRNA expression in lungs (Supporting information Figure S2). Treatment of OVA-sensitized and challenged mice with UTI reduced serum IgE level (Supporting information Figure S1), decreased airway and lung eosinophil infiltration (Figure 1A, B) and reversed the changes in BALF levels of cytokines and lung tissue levels of cytokine mRNAs (Figure 1 and Supporting information Figure S2). Dex had similar effects (Figure 1 and Supporting information Figures S1 and S2).
Figure 1.

UTI suppresses allergic inflammation. At 24 h after final OVA challenge, numbers of neutrophils, eosinophils and lymphocytes in bronchoalveolar lavage fluid (BALF) were counted, BALF levels of IL-4, IL-5, eotaxin-1 and IFN-γ proteins were measured, and lung tissue levels of eosinophil peroxidase (EPO) were determined in mice of non-sensitized control (Con), ovalbumin sensitization and challenge (OVA), OVA + UTI or OVA plus dexamethasone (OVA + Dex) group. (A) UTI or Dex treatment inhibits OVA sensitization and challenge-induced increase in BALF leukocyte count. Means ± SD of 10 mice per group. (B) Western blot photographs show that UTI or Dex treatment inhibits OVA sensitization and challenge-induced lung tissue level of EPO. (C) The Western blot EPO bands were quantified using densitometry and expressed as fold change over control group. Means ± SD of five mice per group. (D to G) UTI or Dex treatment reverses OVA challenge-induced increase in BALF eoxaxin-1, IL-4 and IL-5, and decrease in BALF IFN-γ. Means ± SD of 10 mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group.
UTI ameliorates OVA sensitization and challenge-induced airway and lung pathologies
Because allergic inflammation affects both the nose and airway (Jeffery and Haahtela, 2006), we examined the effects of UTI on OVA sensitization and challenge-induced pathologies in nose, bronchi and lungs. In nasal mucosa, epithelial cilia loss, epithelial derangement, goblet cell hypertrophy and mucosal inflammatory cell infiltration were observed (Figure 2A). In lungs, inflammatory cell infiltration in bronchial and alveolar walls (Figure 2B), which was associated with increased BALF cell counts, and increased endothelial permeability (Figure 2C) were found. UTI treatment ameliorated these histological changes and prevented the increase in endothelial permeability (Figure 2). Dex treatment provided similar protective effects (Figure 2).
Figure 2.
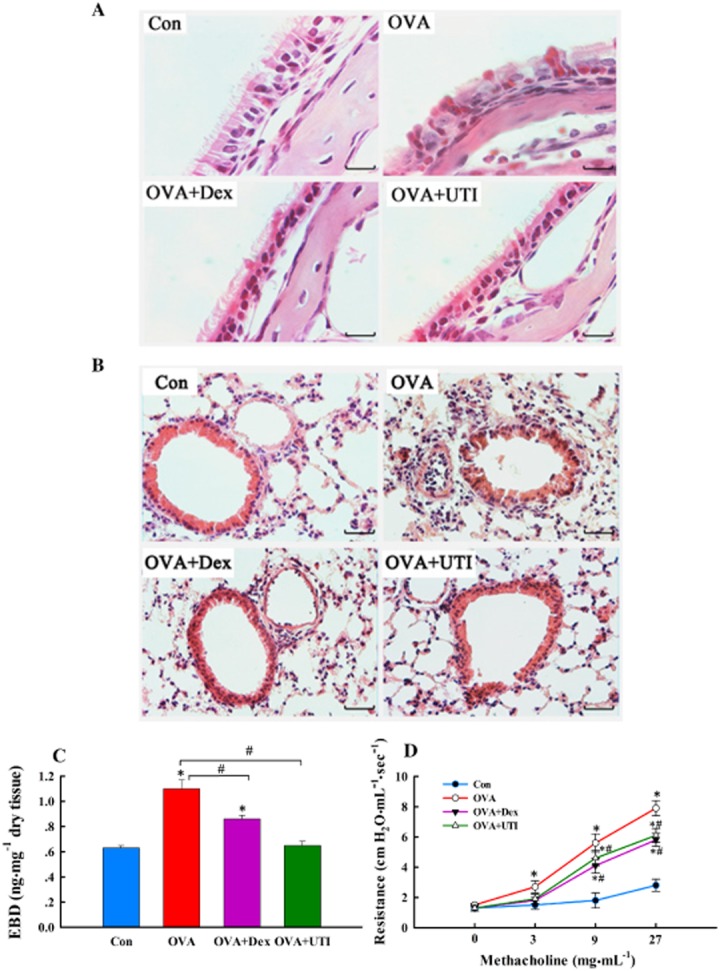
UTI ameliorates OVA sensitization and challenge-induced pathologies and improves lung function. At 24 h after final OVA challenge, paraffin-embedded sections of nasal mucosa and lungs were stained with haematoxylin and eosin (H&E); lung endothelial permeability measured and airway reactivity to methacholine were also assessed. (A) Representative H&E staining images of nasal mucosa sections. Section from sensitized and challenged mice (OVA) exhibited increased inflammatory cell infiltration, epithelial derangement, epithelial deciliation and goblet cell hypertrophy. These histological changes were not obvious in sections from OVA + UTI or OVA + Dex groups of mice. Scale bar = 40 μm. (B) Representative H&E staining images of lung sections. Section from OVA-sensitized and challenged mice displayed inflammatory cell infiltration in bronchial and alveolar walls. These histological changes were ameliorated in sections from OVA + UTI or OVA + Dex groups of mice. Scale bar = 20 μm. (C) UTI reduces OVA sensitization and challenge-induced lung endothelial permeability. Lung endothelial permeability, measured using EBD leakage index as an indicator, increased significantly in OVA alone group, which was prevented in OVA + UTI and OVA + Dex group. Means ± SD of 10 mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group. (D) UTI reduces OVA sensitization and challenge-induced airway hyperactivity. Airway reactivity to aerosolized methacholine increased significantly in OVA alone group, which was prevented in OVA + UTI and OVA + Dex group. Means ± SD of five mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group.
UTI attenuates OVA sensitization and challenge-induced airway hyperreactivity
Suppression of allergic inflammation and pathologies resulted in an improved lung function. OVA-sensitized and challenged mice displayed an increased airway reactivity to methacholine inhalation, which was attenuated in UTI- or Dex-treated mice (Figure 2D).
UTI attenuates OVA sensitization and challenge-induced oxidant stress
We examined the possibility that UTI exerts its anti-inflammatory effect by regulating tissue oxidant and antioxidant balance. We measured markers of tissue oxidative stress, BALF leukocyte reactive oxygen species activity, lung tissue MDA level and lung protein carbonyl content. OVA-sensitized and challenged mice showed elevated levels of the three oxidant stress markers, which were suppressed by treatment with UTI or with Dex to a lesser extent (Figure 3).
Figure 3.
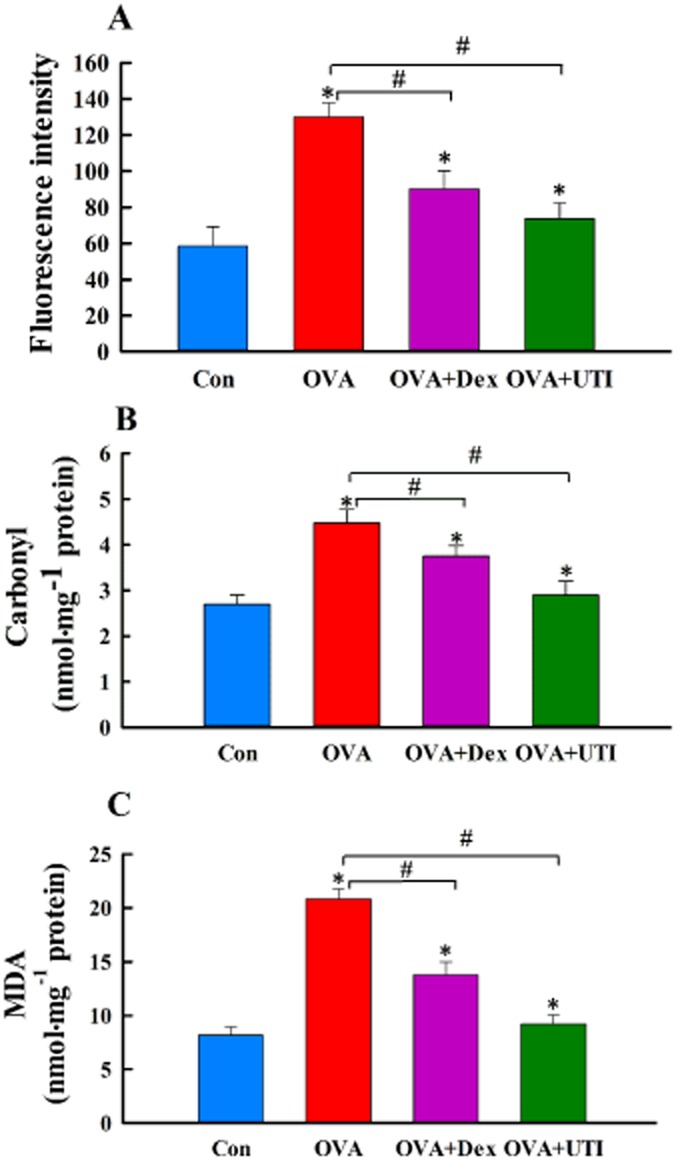
UTI inhibits OVA sensitization and challenge-induced oxidative stress. At 24 h after final OVA challenge, BALF leukocyte reactive oxygen species (ROS) activity (A) and lung tissue protein carbonyl content (B) and malondialdehyde (MDA) level (C) were measured using DCFDA fluorescence probe and expressed as fluorescence intensity, and using protein carbonyl content and MDA assay kits. Means ± SD of 10 mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group.
UTI enhances antioxidant capacity
UTI also enhances endogenous antioxidant capacity. Mice in the OVA-sensitized and challenged group showed significantly reduced BALF levels of GSH, TAOC, SOD and catalase activities, which were prevented by UTI treatment (Figure 4). More importantly, UTI treatment increased BALF GSH, TAOC, SOD or catalase activity to a level that was higher than control (Figure 4), suggesting that UTI stimulates endogenous antioxidant capacity. Dex had similar effects. Thus, UTI treatment reduces tissue oxidant stress and enhances endogenous antioxidant capacity.
Figure 4.

UTI enhances endogenous antioxidant capacity. At 24 h after final OVA challenge, BALF levels of total superoxide dismutase (SOD) actvity (A), catalase activity (CAT, B), glutathione (GSH, C) and total antioxidant capacity (TAOC, D) were measured. Means ± SD of 10 mice per group. *P < 0.05, compared with control (Con) group. #P < 0.05, compared with OVA alone group.
UTI induces HO1 protein expression
We explored the possibility that UTI exerts its antioxidant and anti-inflammatory effects by inducing HO1 expression. Treatment of control mice with UTI increased the basal level of HO1 protein expression in lungs in a time- and dose-dependent manner (Figure 5). OVA sensitization and challenge increased, to a significantly lesser extent, the lung tissue level of HO1 protein. Treatment of OVA-sensitized and challenged mice with UTI increased HO1 protein expression by approximately twofold (Figure 6A, B). Immunofluorescence staining showed that UTI treatment of sensitized and challenged mice increased the number of HO1-positive cells in the lungs (Figure 6C). Stimulation of HO1 expression was not mediated by the excipients of UTI. Excipients at the dose equivalent to the highest concentration of UTI had no effects on HO1 expression and on OVA-induced oxidative stress (Supporting information Figure S3). Thus, UTI stimulates basal and augments OVA-induced HO1 expression, and is an inducer of HO1. Treatment of OVA-sensitized and challenged mice with Dex also increased HO1 expression but to a lesser extent than HO1 (Figure 6).
Figure 5.
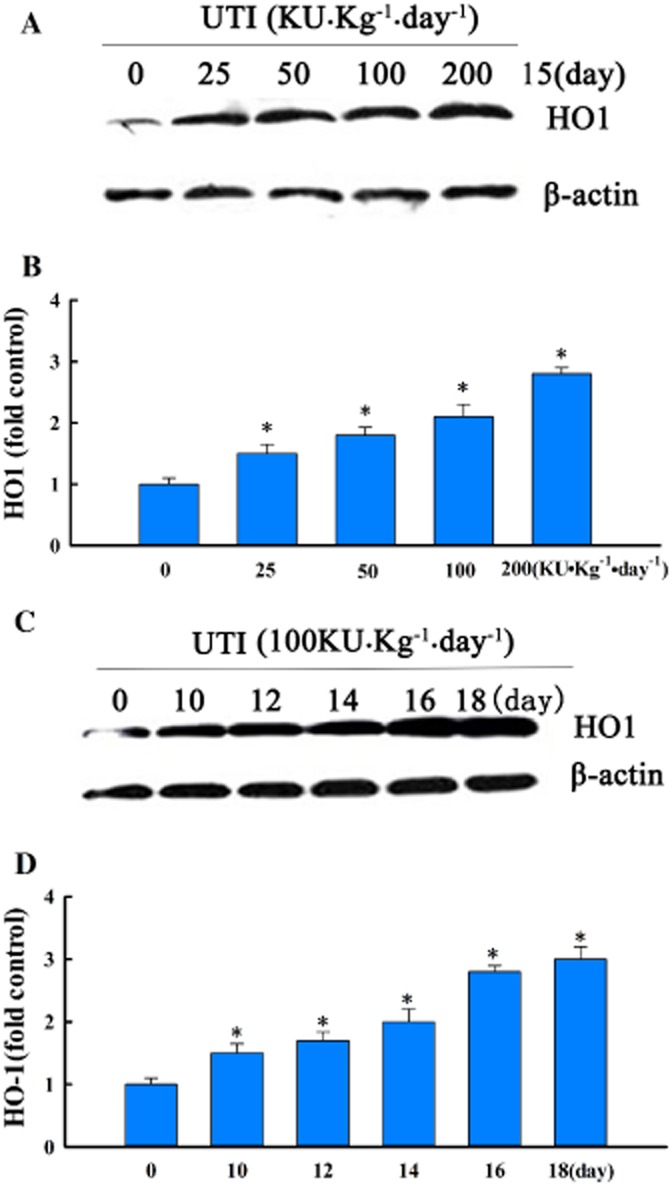
UTI stimulates basal HO1 protein expression. (A and C) Western blot photographs showing that UTI dose- and time-dependently increased basal HO1 protein expression in control lungs. (B and D) Western blot HO1 bands were quantified using densitometry and expressed as fold increase over control group. Means ± SD of four mice per group. *P < 0.05, compared with control group.
Figure 6.
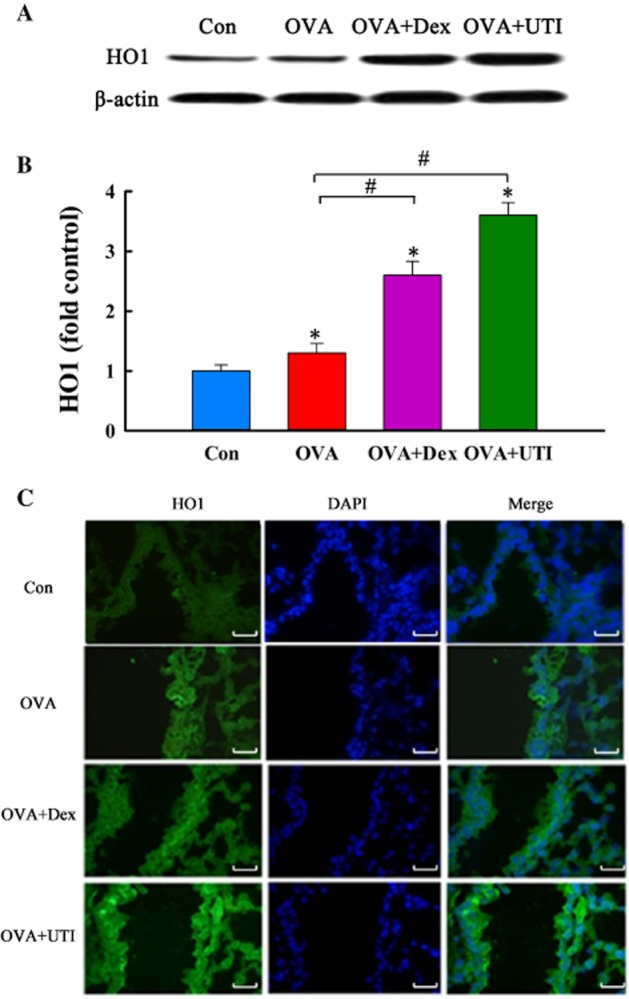
UTI augments OVA-induced HO1 protein expression. At 24 h after final OVA challenge, lung tissue level of HO1 protein was determined. (A) Western blot photographs show that UTI or Dex augments OVA-induced HO1 protein expression. (B) Western blot HO1 bands were quantified using densitometry and expressed as fold increase over control group. Means ± SD of five mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group. (C) Representative photographs of immunofluorescence staining show UTI increases HO1 expressing cells. Lung cryosections were prepared 24 h after final OVA challenge, and immunofluorescence stained with HO1 antibody and nuclear counterstained with DAPI. Specificity of HO1 staining was confirmed using isotype control antibody. OVA sensitization and challenge caused an increase, and OVA + UTI caused a further increase in number of HO1 positive cells. Scale bar = 20 μm.
Inhibition of HO1 activity attenuates the anti-inflammatory and antioxidant activities of UTI
To verify the link between UTI-induced HO1 up-regulation and UTI-mediated anti-inflammatory and antioxidant effects, we examined the effect of inhibiting HO1 activity using ZnPP, a widely used HO1 inhibitor (Bonkovsky et al., 1990; Lee and Chau, 2002), on the anti-inflammatory and antioxidant activities of UTI. UTI treatment reversed OVA-induced increases in BALF IL-4 and lung tissue MDA levels, and decreases in BALF IFN-γ level and SOD activity (Figure 7). Co-treatment of sensitized and challenged mice with UTI plus ZnPP attenuated the inhibitory effects of UTI on IL-4 and MDA production and its stimulant effects on IFN-γ and SOD activity (Figure 7). UTI inhibited OVA-induced decreases in BALF levels of GSH, TAOC and catalase activity, effects that were reversed by co-treatment with ZnPP (Supporting information Figure S4). These results suggest a link between UTI-induced up-regulation HO1 and its anti-inflammatory and antioxidant effects.
Figure 7.

Inhibition of HO1 activity attenuates UTI’s anti-inflammatory and antioxidant activities. Mice in OVA + UTI + ZnPP group were injected with ZnPP (20 mg·kg−1·day−1, i.p.) 1 h before each UTI injection. At 24 h after final OVA challenge, BALF levels of IL-4 (A), IFN-γ (C) and total SOD activity (D), and lung tissue level of MDA (B) were measured. Means ± SD of 10 mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group. $P < 0.05, compared with OVA + UTI group.
UTI stimulates HO1 mRNA expression and Nrf2 DNA binding activity
UTI treatment increased basal HO1 mRNA expression in control mice and augmented OVA-induced HO1 mRNA expression in sensitized and challenged mice (Figure 8A), suggesting that UTI stimulates HO1 gene transcription.
Figure 8.

UTI stimulates Nrf2 activity and up-regulates HO1 mRNA expression. At 24 h after final OVA challenge, cytoplasmic and nuclear levels of Nrf2 protein, Nrf2 DNA binding activity and tissue level of HO1 mRNA in lungs were determined. (A) Q-RT-PCR shows that UTI up-regulates HO1 mRNA expression in lungs. HO1 mRNA was normalized to GAPDH mRNA. Means ± SD of five mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group. (B) Western blot photographs show that UTI increases nuclear and decreases cytoplasmic Nrf2 content, indicating Nrf2 nuclear translocation. (C and D) Western blot Nrf2 bands were quantified using densitometry and expressed as fold increase or decrease over control group. Means ± SD of four mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group. (E) EMSA photograph shows that UTI stimulates Nfr2 DNA binding activity in lungs. Representative of three independent experiments.
HO1 gene transcription is mediated by Nrf2, which, upon stimulation, is translocated from the cytoplasm into the nucleus, inducing HO1 gene transcription (Kobayashi and Yamamoto, 2005). We therefore examined the effect of UTI on Nrf2 activation. Western blot analysis demonstrated that UTI treatment of control or OVA-challenged mice increased nuclear, and decreased cytoplasmic levels of Nrf2 in parallel in lungs (Figure 8B–D). Immunofluorescence staining showed that lung sections from OVA + UTI mice had increased numbers of Nrf2-positive nuclei (Supporting information Figure S5), indicating an increased nuclear translocation of Nrf2. EMSA revealed an increased Nrf2 DNA binding activity in lungs from UTI and OVA + UTI groups of mice (Figure 8E).
UTI inhibits NF-κB DNA binding activity
The NF-κB pathway is a major inflammatory way (Liu and Malik, 2006). We also examined the effect of UTI on NF-κB activity. UTI attenuated the increased NF-κB DNA binding activity induced by OVA sensitization and challenge (Supporting information Figure S6).
Discussion and conclusions
A major finding of this study is that UTI is a potent inducer of HO1 expression. UTI markedly increased basal HO1 protein expression in control lungs in a time- and dose-dependent manner. UTI augmented the increased expression of HO1 induced by OVA sensitization and challenge. UTI, at the highest dose tested, increased basal HO1 protein level by approximately 3.5-fold. Thus, we demonstrated that UTI is a novel and naturally occurring inducer of HO1 expression.
Two previous reports have examined the effect of UTI on HO1 expression. One study claimed that treatment of rats with UTI for 4 h augmented oleic acid-induced HO1 expression. However, in that study, lung HO1 level was analysed by immunohistochemical staining, which is quantifiable (Jin et al., 2007). The other study found that UTI had no effect on basal and hemin-induced HO1 expression in a septic shock model (Yu and Yao, 2008). Differences in the duration and dose of UTI treatment may explain the discrepancies between the previous and our current studies. In the previous studies, rats were treated with UTI for a few hours (Jin et al., 2007; Yu and Yao, 2008), whereas we found that 48 h of UTI treatment was needed to induce HO1 expression in vivo.
Our findings could have therapeutic applications. HO1 is the rate-limiting enzyme that degrades haem into CO and biliverdin. The HO1/CO pathway is an important antioxidant and anti-inflammatory defense mechanism (Ferrándiz and Devesa, 2008; Gozzelino et al., 2010; Raval and Lee, 2010). The induction of HO1 has consistently been shown to be associated with beneficial effects in a variety of pathological conditions (Ferrándiz and Devesa, 2008; Gozzelino et al., 2010; Raval and Lee, 2010). However, HO1 is an inducible enzyme whose expression is minimal in most tissues under physiological conditions. Controlled up-regulation of this enzyme is a major means of activating the HO1/CO pathway and achieving potential therapeutic goals. Although several chemical HO1 inducers have been reported (Ferrándiz and Devesa, 2008), UTI has an advantage over these chemicals as it is an endogenous peptide that has been in clinical use for over 30 years with no significant side effects reported.
The mechanisms by which UTI induces HO1 expression remain to be elucidated. HO1 gene transcription is mainly regulated by Nrf2 (Kobayashi and Yamamoto, 2005). Nrf2 is retained in the cytoplasm by binding to Kelch-like ECH associating protein 1, which facilitates Nrf2 ubiquitination and degradation by ubiquitin proteasome system. Upon stimulation, Nrf2 is dissociated from Kelch-like ECH associating protein 1, translocated into the nucleus and binds to the antioxidant-response element on the HO1 gene promoter, leading to HO1 gene transcription (Kobayashi and Yamamoto, 2005; Kensler et al., 2007). We demonstrated that UTI increased the translocation of Nrf2 into the nucleus, stimulated Nrf2 DNA binding activity and concomitantly up-regulated HO1 mRNA expression. This result suggests that UTI may stimulate Nrf2-mediated HO1 mRNA expression. There are several possible ways that UTI could activate Nrf2. Many signalling molecules, including Nrf2 and HO1, are degraded by the ubiquitin proteasome system. UTI is a serine protease inhibitor and might inhibit Nrf2 degradation. UTI may directly interact with Nrf2 protein to alter its function through an, as yet, unknown mechanism. Alternatively, UTI might directly inhibit proteases involved in the degradation of HO1 or upstream signalling molecules that mediate HO1 expression. These potential mechanisms can be important and warrant further investigation.
The second finding of this study is that UTI is also an effective anti-inflammatory agent against allergic inflammation. Previous studies have reported that UTI protected against neutrophil-mediated inflammation and organ injury (Masuda et al., 2003; Inoue et al., 2005a,b; Molor-Erdene et al., 2005; Takano et al., 2009; Koga et al., 2010; Qiu et al., 2012; Rui et al., 2012). However, the effect of UTI on allergic inflammation has not been studied. Allergic airway inflammation is type 2 T cell-mediated eosinophilic inflammation (Barnes, 1996; Agrawal and Shao, 2010), which differs from neutrophil-mediated inflammation in both cellular mechanisms and mediators involved. We demonstrated here that UTI treatment of OVA-sensitized and challenged mice decreased the serum level of OVA-specific IgE, reduced eosinophil infiltration, decreased BALF and lung tissue levels of cytokines and ameliorated many of the pathological signs of inflammation. Our findings extend those from previous studies by demonstrating that UTI acts as an anti-inflammatory in allergic inflammation.
Up-regulation of HO1 expression may contribute significantly to the antioxidant and anti-inflammatory effects of UTI. We demonstrated here that UTI treatment up-regulated HO1 expression, and concomitantly inhibited inflammation, alleviated tissue oxidant stress and mitigated tissue damage in airway and lungs of OVA-sensitized and challenged mice. The HO1/CO pathway is a major antioxidant defense pathway (Ferrándiz and Devesa, 2008; Gozzelino et al., 2010; Raval and Lee, 2010). Numerous previous studies have demonstrated anti-inflammatory and antioxidant effects of HO1 in allergic inflammation (Xia et al., 2006; Ferrándiz and Devesa, 2008; Gozzelino et al., 2010; Raval and Lee, 2010; Sheikh et al., 2011) and in systemic inflammatory syndrome (Tanaka et al., 2010). More importantly, we demonstrated that inhibition of HO1 activity attenuated the inhibitory effect of UTI on inflammation and tissue oxidant stress, and the stimulant effects of UTI on endogenous antioxidant capacity. Taken together, our results demonstrate that UTI may exert its anti-inflammatory effect by inducing HO1 expression. We also demonstrated that UTI inhibited OVA-induced NF-κB activity, which may also contribute to its anti-inflammatory effects. However, this effect was rather weak.
A previous study reported that UTI augments hydrogen sulfide-induced Nrf2 mRNA expression (Ge et al., 2012). We found no evidence that UTI increases Nrf2 protein expression, even though UTI stimulated Nrf2 nuclear translocation and Nrf2 DNA binding activity. It is possible that the antioxidant and anti-inflammatory effects of UTI are attributable to increased Nrf2 activity, but not to increased Nrf2 expression. In addition to HO1, Nrf2 mediates the transcription of many antioxidant genes. The contributions of these gene products to the antioxidant and anti-inflammatory effects of UTI warrant further investigation.
Our observation that Dex up-regulated HO1 is consistent with results from previous studies, where it was shown that Dex up-regulates HO1 in a rat lungs (Xu et al., 2008) and in human monocytes (Yamazaki et al., 2007). However, in contrast, Dex has been shown to repress IL-6-mediated HO1 up-regulation in cultured endothelial cells (Deramaudt et al., 1999). Differences in cell types, models, experimental conditions and mechanisms of action of Dex may all contribute to these discrepancies.
In conclusion, UTI is a potent and naturally occurring inducer of HO1 expression. Up-regulation of HO1 by UTI may involve Nrf2 activation. UTI inhibits allergic inflammation in an OVA-sensitized and challenged mouse model. Activation of the HO1 antioxidant pathway may contribute to the anti-inflammatory and tissue-protective effects of UTI.
Acknowledgments
This work was supported by Hebei Province Natural Science Foundation of China (H2012206004).
Glossary
- BALF
bronchoalveolar lavage fluids
- CO
carbon monoxide
- DCFDA
2′, 7′-dichlorofluorescein diacetate
- Dex
dexamethasone
- EBD
Evans blue dye
- HO1
haem oxygenase 1
- MDA
malondialdehyde
- Nrf2
nuclear factor erythroid 2-related factor 2
- OVA
ovalbumin
- SOD
superoxide dismutase
- TAOC
total antioxidant capacity
- UTI
ulinastatin (urinary trypsin inhibitor)
- ZnPP
zinc protoporphyrin
Author contributions
B. W. and D. S. proposed the hypothesis; G. S., B. W., D. S. and W. S. designed the experiments; D. S., Y. N., J. W., L. Y., J. Y. and X. L. performed the experiments; D. S., Y. N. and W. S. analysed the data; S. F. L. and D. S. wrote the manuscript; and H. S. significantly edited the manuscript.
Conflicts of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site: http://dx.doi.org/10.1111/bph.12780
Figure S1 UTI reduces serum levels of total and OVA-specific IgE. Mice in sham sensitized control group (Con) were sham sensitized and challenged with PBS. Mice in ovalbumin sensitization and challenge group (OVA), OVA plus UTI (OVA + UTI) or OVA plus dexamethasone (OVA + Dex) groups were sensitized with OVA (25 μg·mouse−1·day−1, i.p.) on days 1, 9 and 14, and challenged by inhalation of 1% OVA for 20 min per day on days 21 to 27. Mice in OVA + UTI or OVA + Dex group were administered UTI (100 000 Ukg·day−1, i.p.) or dexamethasone (1 mg·kg−1·day−1, i.p.) on days 21 to 27. At 24 h after final OVA challenge, serum levels of total (A) and OVA-specific IgE (B) were quantified using ELISA kit. Means ± SD of 10 mice per group. *P < 0.05, compared with Con group. #P < 0.05, compared with OVA alone group.
Figure S2 UTI prevents OVA-induced up-regulation or down-regulation of cytokine mRNAs. At 24 h after final OVA challenge, lungs were harvested. Lung tissue levels of IL-4, IL-5 and IFN-γ mRNA were determined using qRT-PCR and normalized to GAPDH mRNA. UTI or Dex treatment prevented IL-4 (A) and IL-5 (B) mRNA up-regulation, and IFN-γ mRNA (C) down-regulation in OVA-sensitized and challenged mice. Means ± SD of five mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group.
Figure S3 Excipients of UTI had no effects on HO1 expression and on OVA-induced oxidative stress. Control mice were injected with saline (Con) or excipients of UTI (Excipient) at the dose equivalent to 200 KU·kg−1 of UTI once daily for 15 days and lung tissue harvested. OVA-sensitized and challenged mice were untreated (OVA), or treated with UTI (OVA + UTI) or equivalent dose of excipients on days 21 to 27. At 24 h after final OVA challenge, lung tissue level of HO1 protein was determined or bronchoalveolar lavage fluid level (BALF) leukocyte reactive oxygen species (ROS) activity measured using DCF-DA fluorescence probe and expressed as fluorescence intensity. (A) Western blot photographs show that excipients of UTI had no effect on basal and OVA-induced HO1 protein expression. (B) The western blot HO1 bands were quantified using densitometry and expressed as fold increase over control. Means ± SD of five mice per group. *P < 0.05, compared with Con or Excipient group. #P < 0.05, compared with OVA or OVA + Excipient group. (C) Bar graph shows that the excipients of UTI had no effect on basal and OVA-induced BALF leukocyte ROS activity. Means ± SD of five mice per group. *P < 0.05, compared with Con or Excipient group. #P < 0.05, compared with OVA or OVA + Excipient group.
Figure S4 Inhibition of HO1 activity abrogates the stimulatory effects of UTI on antioxidant capacities. Mice in Con, OVA, OVA + UTI or OVA + UTI + ZnPP group were sham or OVA-sensitized and challenged, and treated with UTI as described above. Mice in OVA + UTI + ZnPP group were injected with ZnPP (20 mg·kg−1·day−1, i.p.) 1 h before each UTI administration. At 24 h after final OVA challenge, BALF levels of glutathione (GSH, A), total antioxidant capacity (TAOC, B) and catalase activity (CAT, C) were measured. Means ± SD of 10 mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group. $P < 0.05, compared with OVA + UTI group.
Figure S5 Representative immunofluorescence staining shows that UTI stimulates Nrf2 nuclear translocation. At 24 h after final OVA challenge, lung cryosections were prepared and IF stained with Nrf2 antibody. The specificity of Nrf2 antibody staining was confirmed using isotype control antibody. Lung sections from OVA-sensitized and challenged mice (OVA) have an increased, and lung sections from OVA + UTI group mice have further increased number of Nrf2/DAPI positive nuclei (bright blue dots), indicating that UTI augments OVA-induced Nrf2 nuclear translocation. Scale bar = 20 μm.
Figure S6 EMSA photograph shows that UTI inhibits NF-κB DNA binding activity. Mice were sensitized, challenged and treated with UTI as described above. At 24 h after final OVA challenge, lungs were harvested and NF-κB DNA binding activity determined. Representative of three independent experiments.
Table S1 Primer sequences for real time PCR.
References
- Agrawal DK, Shao Z. Pathogenesis of allergic airway inflammation. Curr Allergy Asthma Rep. 2010;10:39–48. doi: 10.1007/s11882-009-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Pathophysiology of asthma. Br J Clin Pharmacol. 1996;42:3–10. doi: 10.1046/j.1365-2125.1996.03721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkovsky HL, Healey JF, Pohl J. Purification and characterization of heme oxygenase from chick liver. Comparison of the avian and mammalian enzymes. Eur J Biochem. 1990;189:155–166. doi: 10.1111/j.1432-1033.1990.tb15472.x. [DOI] [PubMed] [Google Scholar]
- Chen H, He MY, Li YM. Treatment of patients with severe sepsis using ulinastatin and thymosin alpha1: a prospective, randomized, controlled pilot study. Chin Med J (Engl) 2009;122:883–888. [PubMed] [Google Scholar]
- Deramaudt TB, da Silva JL, Remy P, Kappas A, Abraham NG. Negative regulation of human heme oxygenase in microvessel endothelial cells by dexamethasone. Proc Soc Exp Biol Med. 1999;222:185–193. doi: 10.1046/j.1525-1373.1999.d01-130.x. [DOI] [PubMed] [Google Scholar]
- Ferrándiz ML, Devesa I. Inducers of heme oxygenase-1. Curr Pharm Des. 2008;14:473–486. doi: 10.2174/138161208783597399. [DOI] [PubMed] [Google Scholar]
- Ge Y, Sun W, Wu ZS, Jiang XZ, Qiu QM, Hong GL, et al. Effect of ulinastatin on oxidative stress and nuclear factor E2-related factor 2 expression in the lung tissues of acute hydrogen sulfide intoxicated rats. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2012;30:27–32. [PubMed] [Google Scholar]
- Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- Inoue K, Takano H, Shimada A, Yanagisawa R, Sakurai M, Yoshino S, et al. Urinary trypsin inhibitor protects against systemic inflammation induced by lipopolysaccharide. Mol Pharmacol. 2005a;67:673–680. doi: 10.1124/mol.104.005967. [DOI] [PubMed] [Google Scholar]
- Inoue K, Takano H, Yanagisawa R, Sakurai M, Shimada A, Yoshino S, et al. Protective role of urinary trypsin inhibitor in acute lung injury induced by lipopolysaccharide. Exp Biol Med (Maywood) 2005b;230:281–287. doi: 10.1177/153537020523000408. [DOI] [PubMed] [Google Scholar]
- Jeffery PK, Haahtela T. Allergic rhinitis and asthma: inflammation in a one-airway condition. BMC Pulm Med. 2006;30(Suppl. 1):S5. doi: 10.1186/1471-2466-6-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LY, Xu JM, He ZB, Ruan WY, Chai XP. Effect of ulinastain on the expression of hemeoxygenase-1 in oleic acid-induced acute lung injury in rats. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2007;32:675–678. [PubMed] [Google Scholar]
- Kanai T, Ishiwata T, Kobayashi T, Sato H, Takizawa M, Kawamura Y, et al. Ulinastatin, a urinary trypsin inhibitor, for the initial treatment of patients with Kawasaki disease: a retrospective study. Circulation. 2011;124:2822–2828. doi: 10.1161/CIRCULATIONAHA.111.028423. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Koga Y, Fujita M, Tsuruta R, Koda Y, Nakahara T, Yagi T, et al. Urinary trypsin inhibitor suppresses excessive superoxide anion radical generation in blood, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats. Neurol Res. 2010;32:925–932. doi: 10.1179/016164110X12645013515133. [DOI] [PubMed] [Google Scholar]
- Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- Liu SF, Malik AB. NF-κB activation as a pathologic mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- Masuda T, Sato K, Noda C, Ikeda KM, Matsunaga A, Ogura MN, et al. Protective effect of urinary trypsin inhibitor on myocardial mitochondria during hemorrhagic shock and reperfusion. Crit Care Med. 2003;31:1987–1992. doi: 10.1097/01.CCM.0000057037.44171.BA. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molor-Erdene P, Okajima K, Isobe H, Uchiba M, Harada N, Okabe H. Urinary trypsin inhibitor reduces LPS-induced hypotension by suppressing tumor necrosis factor-alpha production through inhibition of Egr-1 expression. Am J Physiol Heart Circ Physiol. 2005;288:H1265–H1271. doi: 10.1152/ajpheart.00885.2004. [DOI] [PubMed] [Google Scholar]
- Muramatu M, Mori S, Matsuzawa Y, Horiguchi Y, Nakanishi Y, Tanaka M. Purification and characterization of urinary trypsin inhibitor, UT168, from normal human urine, and its cleavage by human uropepsin. J Biochem. 1980;88:1317–1329. doi: 10.1093/oxfordjournals.jbchem.a133100. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Ji S, Wang J, Li H, Xia T, Pan B, et al. The therapeutic efficacy of ulinastatin for rats with smoking inhalation injury. Int Immunopharmacol. 2012;14:289–295. doi: 10.1016/j.intimp.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Raval CM, Lee PJ. Heme oxygenase-1 in lung disease. Curr Drug Targets. 2010;11:1532–1540. doi: 10.2174/1389450111009011532. [DOI] [PubMed] [Google Scholar]
- Rui M, Duan YY, Zhang XH, Wang HL, Wang DP. Urinary trypsin inhibitor attenuates seawater induced acute lung injury by influencing the activities of nuclear factor-κB and its related inflammatory mediators. Respiration. 2012;83:335–343. doi: 10.1159/000333378. [DOI] [PubMed] [Google Scholar]
- Sheikh SZ, Hegazi RA, Kobayashi T, Onyiah JC, Russo SM, Matsuoka K, et al. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol. 2011;186:5506–5513. doi: 10.4049/jimmunol.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano H, Inoue K, Shimada A, Sato H, Yanagisawa R, Yoshikawa T. Urinary trypsin inhibitor protects against liver injury and coagulation pathway dysregulation induced by lipopolysaccharide/D-alactosamine in mice. Lab Invest. 2009;89:833–839. doi: 10.1038/labinvest.2009.35. [DOI] [PubMed] [Google Scholar]
- Tanaka R, Fujita M, Tsuruta R, Fujimoto K, Aki HS, Kumagai K, et al. Urinary trypsin inhibitor suppresses excessive generation of superoxide anion radical, systemic inflammation, oxidative stress, and endothelial injury in endotoxemic rats. Inflamm Res. 2010;59:597–606. doi: 10.1007/s00011-010-0166-8. [DOI] [PubMed] [Google Scholar]
- Tu Y, Diao YF, Yang XP, Sun HT, Zhang S. Effect of ulinastatin in traumatic brain injury with multiple injuries. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2012;24:677–679. [PubMed] [Google Scholar]
- Wang le F, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, et al. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med. 2002;165:1634–1639. doi: 10.1164/rccm.2110017. [DOI] [PubMed] [Google Scholar]
- Xia ZW, Zhong WW, Xu LQ, Sun JL, Shen QX, Wang JG, et al. Heme oxygenase-1-mediated CD4 + CD25high regulatory T cells suppress allergic airway inflammation. J Immunol. 2006;177:5936–5945. doi: 10.4049/jimmunol.177.9.5936. [DOI] [PubMed] [Google Scholar]
- Xu X, Zhong LL, Jiao SM, Liu SS, Li Y, Zhang B. Effect of budesonide on the heme oxygenase-1 expression in lung tissues of rats with asthma. Zhongguo Dang Dai Er Ke Za Zhi. 2008;10:376–380. [PubMed] [Google Scholar]
- Yamazaki H, Ohta K, Tsukiji H, Toma T, Hashida Y, Ishizaki A, et al. Corticosteroid enhances heme oxygenase-1 production by circulating monocytes by up-regulating hemoglobin scavenger receptor and amplifying the receptor-mediated uptake of hemoglobin-haptoglobin complex. Biochem Biophys Res Commun. 2007;358:506–512. doi: 10.1016/j.bbrc.2007.04.136. [DOI] [PubMed] [Google Scholar]
- Yano T, Anraku S, Nakayama R, Ushijima K. Neuroprotective effect of urinary trypsin inhibitor against focal cerebral ischemia-reperfusion injury in rats. Anesthesiology. 2003;98:465–473. doi: 10.1097/00000542-200302000-00028. [DOI] [PubMed] [Google Scholar]
- Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-κB in multiple organ injury and bacterial clearance in murine models of sepsis. J Exp Med. 2008;205:1303–1315. doi: 10.1084/jem.20071393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JB, Yao SL. Protective effects of hemin pretreatment combined with ulinastatin on septic shock in rats. Chin Med J (Engl) 2008;121:49–55. [PubMed] [Google Scholar]
- Zhang X, Liu F, Liu H, Cheng H, Wang W, Wen Q, et al. Urinary trypsin inhibitor attenuates lipopolysaccharide-induced acute lung injury by blocking the activation of p38 mitogen-activated protein kinase. Inflamm Res. 2011;60:569–575. doi: 10.1007/s00011-010-0305-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 UTI reduces serum levels of total and OVA-specific IgE. Mice in sham sensitized control group (Con) were sham sensitized and challenged with PBS. Mice in ovalbumin sensitization and challenge group (OVA), OVA plus UTI (OVA + UTI) or OVA plus dexamethasone (OVA + Dex) groups were sensitized with OVA (25 μg·mouse−1·day−1, i.p.) on days 1, 9 and 14, and challenged by inhalation of 1% OVA for 20 min per day on days 21 to 27. Mice in OVA + UTI or OVA + Dex group were administered UTI (100 000 Ukg·day−1, i.p.) or dexamethasone (1 mg·kg−1·day−1, i.p.) on days 21 to 27. At 24 h after final OVA challenge, serum levels of total (A) and OVA-specific IgE (B) were quantified using ELISA kit. Means ± SD of 10 mice per group. *P < 0.05, compared with Con group. #P < 0.05, compared with OVA alone group.
Figure S2 UTI prevents OVA-induced up-regulation or down-regulation of cytokine mRNAs. At 24 h after final OVA challenge, lungs were harvested. Lung tissue levels of IL-4, IL-5 and IFN-γ mRNA were determined using qRT-PCR and normalized to GAPDH mRNA. UTI or Dex treatment prevented IL-4 (A) and IL-5 (B) mRNA up-regulation, and IFN-γ mRNA (C) down-regulation in OVA-sensitized and challenged mice. Means ± SD of five mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group.
Figure S3 Excipients of UTI had no effects on HO1 expression and on OVA-induced oxidative stress. Control mice were injected with saline (Con) or excipients of UTI (Excipient) at the dose equivalent to 200 KU·kg−1 of UTI once daily for 15 days and lung tissue harvested. OVA-sensitized and challenged mice were untreated (OVA), or treated with UTI (OVA + UTI) or equivalent dose of excipients on days 21 to 27. At 24 h after final OVA challenge, lung tissue level of HO1 protein was determined or bronchoalveolar lavage fluid level (BALF) leukocyte reactive oxygen species (ROS) activity measured using DCF-DA fluorescence probe and expressed as fluorescence intensity. (A) Western blot photographs show that excipients of UTI had no effect on basal and OVA-induced HO1 protein expression. (B) The western blot HO1 bands were quantified using densitometry and expressed as fold increase over control. Means ± SD of five mice per group. *P < 0.05, compared with Con or Excipient group. #P < 0.05, compared with OVA or OVA + Excipient group. (C) Bar graph shows that the excipients of UTI had no effect on basal and OVA-induced BALF leukocyte ROS activity. Means ± SD of five mice per group. *P < 0.05, compared with Con or Excipient group. #P < 0.05, compared with OVA or OVA + Excipient group.
Figure S4 Inhibition of HO1 activity abrogates the stimulatory effects of UTI on antioxidant capacities. Mice in Con, OVA, OVA + UTI or OVA + UTI + ZnPP group were sham or OVA-sensitized and challenged, and treated with UTI as described above. Mice in OVA + UTI + ZnPP group were injected with ZnPP (20 mg·kg−1·day−1, i.p.) 1 h before each UTI administration. At 24 h after final OVA challenge, BALF levels of glutathione (GSH, A), total antioxidant capacity (TAOC, B) and catalase activity (CAT, C) were measured. Means ± SD of 10 mice per group. *P < 0.05, compared with control group. #P < 0.05, compared with OVA alone group. $P < 0.05, compared with OVA + UTI group.
Figure S5 Representative immunofluorescence staining shows that UTI stimulates Nrf2 nuclear translocation. At 24 h after final OVA challenge, lung cryosections were prepared and IF stained with Nrf2 antibody. The specificity of Nrf2 antibody staining was confirmed using isotype control antibody. Lung sections from OVA-sensitized and challenged mice (OVA) have an increased, and lung sections from OVA + UTI group mice have further increased number of Nrf2/DAPI positive nuclei (bright blue dots), indicating that UTI augments OVA-induced Nrf2 nuclear translocation. Scale bar = 20 μm.
Figure S6 EMSA photograph shows that UTI inhibits NF-κB DNA binding activity. Mice were sensitized, challenged and treated with UTI as described above. At 24 h after final OVA challenge, lungs were harvested and NF-κB DNA binding activity determined. Representative of three independent experiments.
Table S1 Primer sequences for real time PCR.