

Abstract
Background and Purpose
The KCNQ-encoded voltage-gated potassium channel family (Kv7.1-Kv7.5) are established regulators of smooth muscle contractility, where Kv7.4 and Kv7.5 predominate. Various Kv7.2–7.5 channel enhancers have been developed that have been shown to cause a vasorelaxation in both rodent and human blood vessels. Recently, two novel Kv7 channel enhancers have been identified, ML213 and NS15370, that show increased potency, particularly on Kv7.4 channels. The aim of this study was to characterize the effects of these novel enhancers in different rat blood vessels and compare them with Kv7 enhancers (S-1, BMS204352, retigabine) described previously. We also sought to determine the binding sites of the new Kv7 enhancers.
Key Results
Both ML213 and NS15370 relaxed segments of rat thoracic aorta, renal artery and mesenteric artery in a concentration-dependent manner. In the mesenteric artery ML213 and NS15370 displayed EC50s that were far lower than other Kv7 enhancers tested. Current-clamp experiments revealed that both novel enhancers, at low concentrations, caused significant hyperpolarization in mesenteric artery smooth muscle cells. In addition, we determined that the stimulatory effect of these enhancers relied on a tryptophan residue located in the S5 domain, which is the same binding site for the other Kv7 enhancers tested in this study.
Conclusions and Implications
This study has identified and characterized ML213 and NS15370 as potent vasorelaxants in different blood vessels, thereby highlighting these new compounds as potential therapeutics for various smooth muscle disorders.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| Kv7.1 channels | Methoxamine |
| Kv7.2 channels | Linopirdine |
| Kv7.3 channels | Nicardipine |
| Kv7.4 channels | Paxilline |
| Kv7.5 channels | Retigabine |
| BMS204352 |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Five members of the KCNQ gene family have been identified (KCNQ1–5) that encode the voltage-gated potassium channels (Kv7.1–7.5). Expression of the different KCNQ isoforms varies among mammalian cell types (Soldovieri et al., 2011), although the majority of research into these channels has focussed on their expression in the heart (KCNQ1) and nervous system (KCNQ2-5). KCNQ-encoded channels have also been identified in various mammalian smooth muscle tissues. In smooth muscles, the expression of KCNQ1, KCNQ4 and KCNQ5 dominates (Ohya et al., 2003; Yeung et al., 2007; Ng et al., 2011; Chadha et al., 2012a,b; Khanamiri et al., 2013) with recent evidence that a Kv7.4/Kv7.5 heteromer is the main molecular species in vascular smooth muscle (Brueggemann et al., 2014; Chadha et al., 2014). In both rodent and human blood vessels, pharmacological modulators of KCNQ-encoded channels have been used to identify a crucial role for these channels in regulating smooth muscle contractility (Jepps et al., 2013; Stott et al., 2014). Kv7 channel enhancers, such as retigabine, S-1 and BMS204352, which enhance Kv7.2–Kv7.5, have been used to elucidate the importance of these channels in smooth muscle contractility and are effective vasorelaxants. Retigabine, S-1 and BMS204352 act on Kv7.2–7.5, but not Kv7.1 (Schenzer et al., 2005; Wuttke et al., 2005), and shift both the activation threshold and voltage for half-activation in the hyperpolarizing direction, leading to an increase in current amplitude at more negative potentials. The mechanism of enhancement for these compounds occurs through direct binding to a tryptophan residue (Trp236 in Kv7.2; Trp265 in Kv7.3) in a hydrophobic pocket of the cytoplasmic part of S5, which stabilizes the channel in the open state (Schenzer et al., 2005; Wuttke et al., 2005; Bentzen et al., 2006; Lange et al., 2013).
Recently two new activators have been described, ML213 and NS15370, which enhance the activity of Kv7.2 and Kv7.4, and Kv7.2–7.5 channels, respectively, with increased potency in overexpression systems compared with the Kv7 channel enhancers described previously. ML213 was identified by a high throughput fluorescent screen of the Molecular Libraries Small Molecule Repository and structure activity relationship studies, and has been shown to enhance Kv7.2 and Kv7.4 channels with EC50s of 230 and 510 nM respectively (Yu et al., 2011); whereas NS15370 was developed as a chemical analogue of retigabine and can enhance Kv7.2–7.5 channels with EC50s ranging between 40 and 150 nM (Dalby-Brown et al., 2013).
However, the role of Kv7 channels in controlling neuronal excitability has led to widespread interest in the development of novel pharmacological enhancers of these channels. Pharmacological enhancement of the ‘neuronal’ Kv7 channels suppresses neuronal activity and prevents the generation of seizures in various animal models of epilepsy (Dailey et al., 1995; Rostock et al., 1996; Tober et al., 1996). Retigabine (Trobalt®) is effective in clinical trials as an anti-epileptic (Brodie et al., 2010; French et al., 2011) and is now on the market in both the United States and Europe. While the development of Kv7 channel enhancers is aimed predominantly at treating neurological disorders, the aim of this study was to determine whether the new Kv7 channel enhancers, ML213 and NS15370, were effective at suppressing contractile activity in vascular smooth muscle, thus highlighting these enhancers as possible therapeutics for various vascular disorders. Therefore, this study compared the effects of three established enhancers of Kv7.2–7.5 with those of the two novel agents in various rat blood vessels, and also determined the effects of the novel enhancers on the resting membrane potential of rat mesenteric artery smooth muscle cells. In addition, we determine whether these new activators rely on the same tryptophan residue essential for the binding of retigabine and the other Kv7 enhancers.
Methods
All experiments were performed on arteries from male, 12–16-week-old Wistar rats (Charles River UK, Ltd., Margate, UK) killed by cervical dislocation. A total of 30 animals were used in this study.
Quantitative PCR (QPCR)
QPCR analysis of KCNQ isoform expression in the rat thoracic aorta, renal artery and mesenteric artery was performed as described previously in Khanamiri et al. (2013). Briefly, RNA was extracted using the RNEasy Micro Extraction Kit (Qiagen, Manchester, UK) and reverse transcribed using Oligo(dT)12–18 primers and Moloney murine leukemia virus (Life Technologies). Quantitative analysis of specific genes of interest within our cDNA samples was determined using Precision-iC SYBR green mastermix (PrimerDesign, Ltd., Southampton, UK) with the CFX96 Real-Time PCR Detection System (Bio-Rad, Hertfordshire, UK). Duplicate reactions were performed in 10 μL volumes containing 5 μL Precision-iC SYBR green master mix (PrimerDesign, Ltd.), 300 nM primer (PrimerDesign, Ltd.), 5–10 ng cDNA and made up to 10 μL with nuclease-free water. The following cycling conditions were used: initial activation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min, and data were collected during each cycling phase. Melt curve analysis, to ensure each primer set amplified a single, specific product, completed the protocol. Reverse transcription-negative samples and no-template controls were run alongside all reactions to assess contamination. Quantification cycle (Cq) values were determined using Bio-Rad CFX96 Manager 3.0 software and the single threshold mode.
The geNorm reference gene selection kit (PrimerDesign, Ltd.) was used to identify the most stable reference genes and to determine optimal number of reference genes required for reliable normalization of the genes of interest in our samples (Vandesompele et al., 2002). All reference genes in the rat geNorm reference gene selection kit and the KCNQ1–5 assays (Table 1) were designed and optimized by PrimerDesign, Ltd.
Table 1.
KCNQ 1–5 assays used for QPCR analysis of rat blood vessels
| Gene | Primer sequence (+) sense, (−) antisense | GenBank accession number | Amplicon (bp) | Region spanned |
|---|---|---|---|---|
| KCNQ1 | (+) 5′-CCATCTTTGTTCATCCCCATCT-3′ | NM_032073 | 100 | 1797–1896 |
| (−) 5′- CCAGTTGTGTCACCTTGTCTT -3′ | ||||
| KCNQ2 | (+) 5′-GGTGTCTCATTCTTCGCTCTT-3′ | NM_133322 | 100 | 1023–1122 |
| (−) 5′-TCCGCCGTTTCTCAAAGTG-3′ | ||||
| KCNQ3 | (+) 5′-ATACACATTTATCTGCTCTTCCTTTTA-3′ | NM_031597 | 122 | 3299–3420 |
| (−) 5′-TGCTCTCAGTTTATCCGAATCAA-3′ | ||||
| KCNQ4 | (+) 5′-GCTCATCTTCGCCTCTTTCC-3′ | XM_233477 | 112 | 861–972 |
| (−) 5′-GCCAATGGTCGTCAGTGTAAT-3′ | ||||
| KCNQ5 | (+) 5′-CCTGGCGTACACGAGAGTAT-3′ | XM_001071249 | 80 | 2383–2462 |
| (−) 5′-TTTGACTGGGCGAACTGAAC-3′ |
The analysis was designed and optimized by PrimerDesign, Ltd.
Myography
Arteries were cleaned of adherent tissue and segments (∼2 mm) were mounted in a myograph (Danish Myo Technology, Aarhus, Denmark) for isometric tension recording. The composition of the physiological salt solution (PSS) in the chambers was 125 mM NaCl, 4.6 mM KCl, 2.5 mM CaCl2, 25.4 mM NaHCO3, 1 mM Na2HPO4, 0.6 mM MgSO4 and 10 mM glucose, maintained at 37°C and aerated with 95% O2/5% CO2. The vessels were allowed to equilibrate for 30 min before undergoing a passive force normalization procedure (Mulvany & Halpern, 2011). Before the application of the Kv7 enhancers, the arterial segments were contracted with the α1-adrenoceptor agonist methoxamine (3 μM in the thoracic aorta and renal artery, 10 μM in the mesenteric artery), which was determined previously to produce a sub-maximal contraction (80–90% of the maximal contraction) in the respective vessels (data not shown).
Mesenteric artery myocyte isolation and electrophysiology
Following dissection, mesenteric arteries were bathed for 10 min in nominally Ca2+-free PSS before incubation at 37°C in Ca2+-free PSS containing collagenase type IA (2 mg·mL−1), protease type X (1 mg·mL−1) and DL-dithiothreitol (1 mg·mL−1) for 15 min followed by a 10 min wash in Ca2+-free PSS solution at room temperature. Single cells were liberated by gentle mechanical agitation with a wide bore Pasteur pipette and the suspension was transferred to experimental chambers and kept for at least 30 min at room temperature to allow adherence. Cells were used in patch-clamp experiments within 6 h of isolation. Membrane potential recordings were made using standard amphotericin B (300 μg·mL−1) perforated-patch technique in current clamp mode. Patch pipettes were fire-polished and had a resistance of 4–8 MΩ when filled with the pipette solution of the following composition: 126 mM KCl, 1.2 mM MgCl2, 10 mM HEPES, 0.5 mM EGTA, pH was adjusted to 7.2 with KOH and PSS was used as an external solution. All experiments were performed at room temperature. The electrical signals were recorded using an Axopatch 200B patch-clamp amplifier (Molecular Devices, Sunnyvale, CA, USA). Electrical signals were generated and digitized at 1 kHz using a Digidata 1322A hosted by a PC running pClamp 9.0 software (Axon Instruments, Sunnyvale, CA, USA). Data were analysed and plotted using pClamp and MicroCal Origin software. All data are presented as mean ± SEM. Comparative analysis of the data was performed using Student’s t-test.
HEK cell transfection and electrophysiology
Monoclonal HEK293 cells stably expressing human Kv7.4 were grown in DMEM, supplemented with 10% FBS (Th Geyer, Renningen, Germany) supplemented with Glutamax (Substrate Department, the Panum Institute, Copenhagen, Denmark) and incubated at 37°C in 5% CO2. In addition, HEK293 cells were transiently transfected with hKv7.4-W242L mutant channels using siLentFect™ Lipid (Bio-Rad), according to manufacturer’s instructions. The point mutation W242L of human Kv7.4 was constructed as described previously (Bentzen et al., 2006).
Potassium currents were recorded from HEK cells stably expressing human Kv7.4 using a QPatch 16 HT automated patch-clamp system, (Sophion-Bioscience, Ballerup, Denmark), with disposable single-hole QPlates (Sophion-Bioscience). On the day of the experiments, HEK293 cells were rinsed with PBS, detached from T175 bottles with 5 mL Detachin (Th Geyer) and re-suspended in serum-free medium supplemented with 100 U·mL−1 penicillin/streptomycin, 0.04 mg·mL−1 Soy bean trypsin inhibitor and 25 mM HEPES (Sigma-Aldrich, Brondby, Denmark) at a density of 1–5 million cells mL−1. Cells were transferred to a QPatch stirring station, and allowed to recover for 10 min before the experiment was initiated. Conventional whole-cell patch-clamp electrophysiology was used to record currents from HEK cells expressing hKv7.4-W242L. In all experiments, the extracellular solution contained the following: 145 mM NaCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 4 mM KCl and 10 mM glucose. The pH was adjusted to 7.4 with NaOH and the osmolarity to 305 mOsm with sucrose. The intracellular solution contained: 1.75 mM MgCl2, 10 mM EGTA, 110 mM KCl, 5.4 mM CaCl2 and 4 mM Na2-ATP, pH adjusted to 7.4 with KOH and osmolarity to 295 mOsm. The intracellular free calcium concentration was 100 nM. Kv7.4 currents were elicited every 5 s by depolarizing the membrane potential to −10 mV for 500 ms from a holding potential of −80 mV. After 2 min of stabilization, 6 periods of increasing concentrations of ML213 or NS15370 were added for 1 min to determine the stimulatory effects of the enhancers on the Kv7.4 current at −10 mV. A double step protocol from −100 mV to +60 mV (20 mV increments, 500 ms duration) returning to −30 mV to measure the tail current was applied before addition of the Kv7 enhancer and after the highest concentration.
Drugs
Retigabine, S-1, BMS204352 and NS15370 were synthesized at NeuroSearch A/S (Ballerup, Denmark). Linopirdine was purchased from Sigma-Aldrich, and ML213 was purchased from Tocris (Bristol, U.K.).
Results
Relative expression of KCNQ isoforms in rat blood vessels
Figure 1 shows the relative abundance of KCNQ gene expression in the cDNA from the rat thoracic aorta (n = 4), renal artery (n = 6) and mesenteric artery (n = 4). In the thoracic aorta and renal artery the relative expression of KCNQ1 predominated with KCNQ4 and KCNQ5 also being present. In the mesenteric artery KCNQ4 expression dominated relative to KCNQ1 and KCNQ5, which were also present. KCNQ3 was observed in the renal artery, but its expression was variable among samples, whereas in the thoracic aorta and mesenteric artery, KCNQ3 expression was negligible. KCNQ2 was never detected in these arteries (Figure 1).
Figure 1.
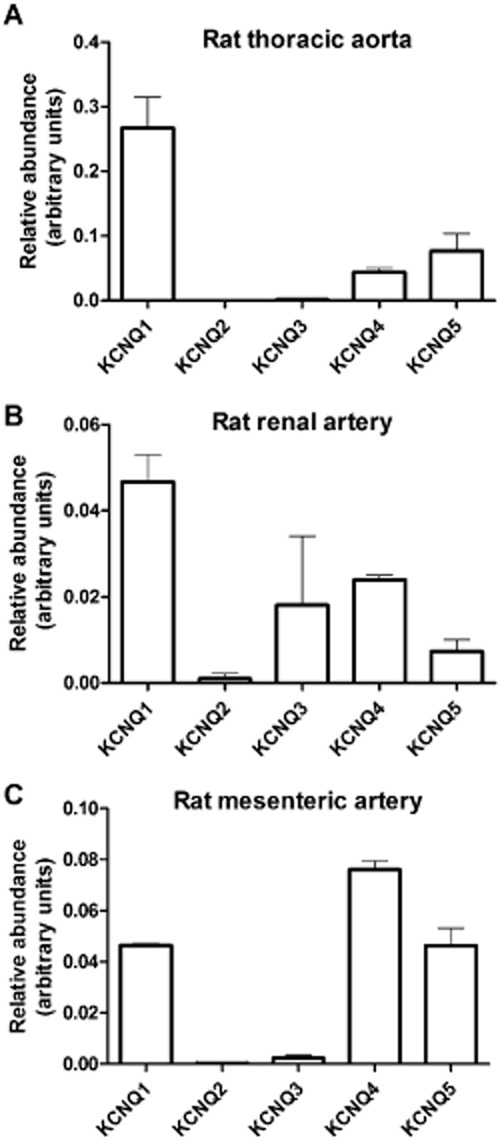
QPCR analysis of relative abundance of KCNQ genes in (A) the rat thoracic aorta (n = 4); (B) the rat renal artery (n = 6); (C) the rat mesenteric artery (n = 4), normalized to the reference genes. The relative abundance of each gene was calculated using the 2−ΔCq method. Data represent the mean ± SEM.
ML213 and NS15370 are potent vasorelaxants of rat blood vessels
Following contraction with the α1-adrenoceptor agonist methoxamine, application of increasing concentrations of ML213 and NS15370 to segments of rat thoracic aorta, renal artery and mesenteric artery resulted in concentration-dependent relaxations, which were considerably different from the minimal loss of tone seen when vehicle alone was applied (Figure 2A). When comparing the three arteries, ML213 and NS15370 were more potent at relaxing precontracted segments of mesenteric artery than the thoracic aorta and renal artery (n = 7–10; Table 2). In the mesenteric artery, pre-application of the Kv7 blocker linopirdine (10 μM) prevented ML213 from relaxing the artery (n = 6; Figure 3). The effect of NS15370 was also blunted significantly in the presence of 10 μM linopirdine (n = 6; Figure 3). Similar effects were observed in the aorta with 15 μM ML213 producing a 100 ± 2% and 10 ± 2% relaxation in the absence and presence of 10 μM linopirdine (P < 0.001; n = 5; data not shown). The lack of response to the Kv7 activators in the presence of linopirdine was not due to a reduced ability of the vessels to relax as application of 1 μM nicardipine caused a vasorelaxation in all vessels tested (Figure 3). Neither agent was able to relax arteries contracted with 60 mM KCl (e.g. tone with 15 μM ML213 was 100 ± 4% of initial, n = 6; data not shown). These data reveal ML213 and NS15370 to be highly potent relaxants of precontracted rat arteries.
Figure 2.
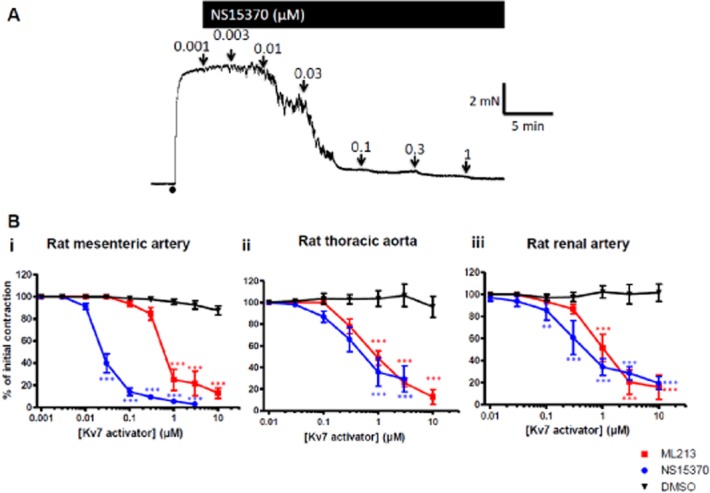
NS15370 and ML213 cause a vasorelaxation in different precontracted rat blood vessels. (A) Representative effect of increasing concentrations of NS15370 on a segment of small mesenteric artery mounted in a wire myograph precontracted with 10 μM methoxamine. (B) Concentration–effect curves for ML213 and NS15370 on (i) small mesenteric arteries, (ii) thoracic aorta and (iii) renal arteries. Each point is the mean of 7–10 animals ± SEM. A Bonferroni post hoc test was performed following a two-way anova and *** P < 0.001.
Table 2.
Comparison of the EC50 values of different Kv7 activators when applied to the rat thoracic aorta, renal artery and mesenteric artery under isometric conditions
| Kv7 channel enhancer | EC50s (μM) | ||
|---|---|---|---|
| Thoracic aorta (n) | Renal artery (n) | Mesenteric artery (n) | |
| NS15370 | 0.55 ± 0.13 (7) | 0.72 ± 0.21 (7) | 0.03 ± 0.00 (8) |
| ML213 | 1.03 ± 0.09 (10) | 1.03 ± 0.16 (7) | 0.75 ± 0.09 (9) |
| S-1 | 8.96 ± 0.8 (11) | 0.76 ± 0.16 (6) | 2.75 ± 0.26 (11) |
| BMS204352 | 10.08 ± 1.15 (7) | 3.31 ± 0.51 (6) | 2.06 ± 0.21 (9) |
| Retigabine | 8.75 ± 1.22 (8) | 9.21 ± 2.76 (6) | 10.59 ± 1.47 (7) |
| Bonferroni’s multiple comparison test | |||
| NS15370 vs. ML213 | ns | ns | ns |
| NS15370 vs. S-1 | *** | ns | ** |
| NS15370 vs. BMS204352 | *** | ns | ns |
| NS15370 vs. Retigabine | *** | *** | *** |
| ML213 vs. S-1 | *** | ns | ns |
| ML213 vs. BMS204352 | *** | ns | ns |
| ML213 vs. Retigabine | *** | *** | *** |
The EC50 values were analysed statistically using a Bonferroni multiple comparison test, where **P < 0.01 and ***P < 0.005; ns, not significant.
Figure 3.
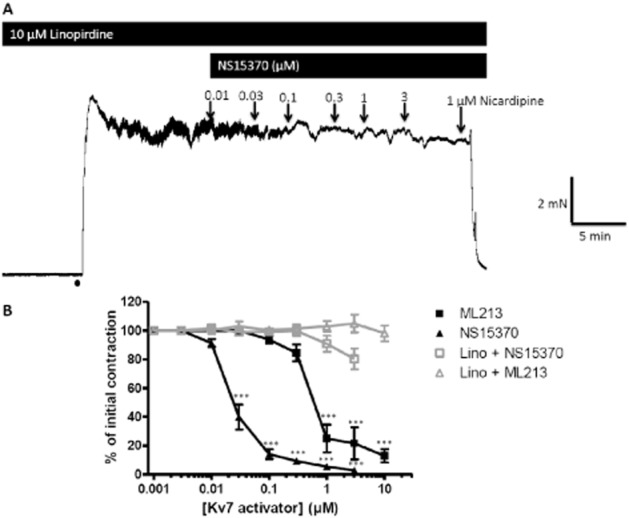
The effects of the Kv7 channel enhancers in the presence of a Kv7 channel blocker. (A) Representative isometric recording traces of the effect of NS15370 on precontracted (10 μM methoxamine) segments of mesenteric artery in the presence of the Kv7 blocker linopirdine (10 μM). At the end of the experiment 1 μM nicardipine was applied to show the vessel was still able to fully relax. (B) Mean data summerizing the effect of ML213 and NS15370 in the presence and absence of 10 μM linopirdine on segments of mesenteric artery (n = 6). Data represent the mean ± SEM. A Bonferroni post hoc test was performed following a two-way anova, and **P < 0.01 and ***P < 0.001.
ML213 and NS15370 are more potent in the vasculature than other Kv7 enhancers
Previously, other structurally different enhancers of Kv7.2–7.5 have been used to study the role of Kv7 channels in numerous blood vessels (Yeung et al., 2007; 2008; Mackie et al., 2008; Joshi et al., 2009; Zhong et al., 2010; Jepps et al., 2011; Mani et al., 2011; Ng et al., 2011; Chadha et al., 2012a; 2014; Khanamiri et al., 2013). We therefore undertook a series of experiments to compare the effects of S-1, BMS204352 and retigabine with the relaxant ability of the two novel compounds. Under isometric conditions, all the Kv7 channel enhancers tested relaxed precontracted segments of thoracic aorta, renal artery and mesenteric artery with differing potencies (see Table 2). All relaxations were prevented by prior application of linopirdine. Statistical analysis of the EC50 values in Table 2 (one-way anova followed by a Bonferroni multiple comparisons test) showed that in the thoracic aorta NS15370 and ML213 were significantly more potent at causing a vasorelaxation compared with S-1, BMS204352 and retigabine. In the renal and mesenteric arteries, NS15370 and ML213 were significantly more potent compared with retigabine, and NS15370 showed significant differences compared with S-1 in the mesenteric artery (Figure 4 and Table 2).
Figure 4.
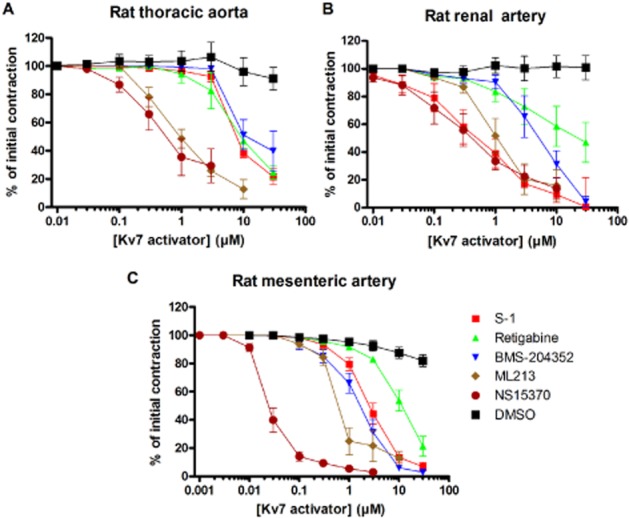
Comparison of the vasorelaxant effects of different Kv7 channel enhancers in the (A) thoracic aorta, (B) renal artery and (C) mesenteric artery. Data represent the mean ± SEM taken from 6 to 11 animals.
ML213 and NS15370 hyperpolarize mesenteric artery smooth muscle cells
Current-clamp recordings were performed to determine the action of ML213 and NS15370 on the resting membrane potential of single smooth muscle cells freshly isolated from rat mesenteric arteries. As shown in Figure 5, the membrane potential in mesenteric artery smooth muscle cells was often superimposed by transient hyperpolarizations. These were sensitive to paxilline and, therefore, reflect the sporadic activation of calcium-activated potassium channels. We did not investigate this aspect further. Importantly, application of 10 μM ML213 produced a significant hyperpolarization within a few minutes of application (P < 0.001; n = 11 cells). Application of NS15370 also hyperpolarized the membrane potential significantly at lower concentrations than ML213 (Figure 5B).
Figure 5.
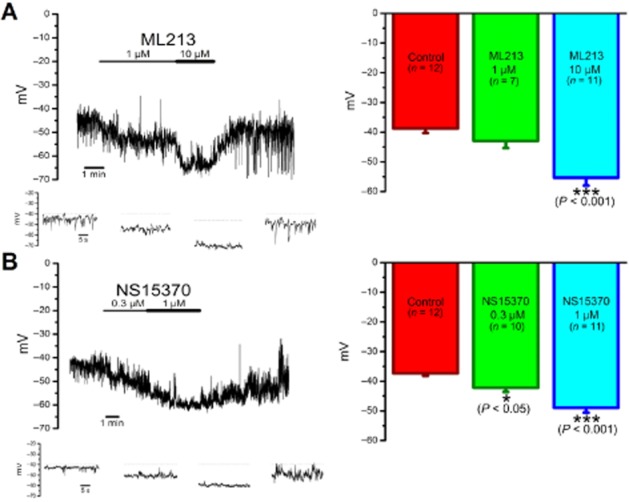
(A) ML213 and (B) NS15370 hyperpolarize the resting membrane potential of mesenteric artery smooth muscle cells. (Left panel) Representative traces of current-clamp recordings performed on smooth muscle cells isolated from rat mesenteric arteries to show the hyperpolarization caused by application of ML213 and NS15370. Expanded examples of the recordings are shown in four insets beneath each of the main current-clamp recordings. (Right panel) Mean effect on the membrane potential of different concentrations of the Kv7 channel enhancers (n = 7–12 cells) ± SEM. According to a one-way anova followed by a Bonferroni post hoc test, *P < 0.05 and ***P < 0.001 significantly different from control.
Effect of the novel enhancers relies on a tryptophan residue in the S5 transmembrane domain of Kv7.4
In QPatch experiments, application of increasing concentrations of ML213 and NS15370 stimulated Kv7.4 currents at −10 mV with EC50 values of 1.8 ± 0.3 μM and 42 ± 8 nM, respectively (n = 6 and 8 cells). As a tryptophan residue in the S5 pore domain is crucial for the stimulatory effects of retigabine, S-1 and BMS204352 (Schenzer et al., 2005; Wuttke et al., 2005; Bentzen et al., 2006), but is not involved with the enhancing effects of another Kv7.2–7.5 enhancer, ICA27243 (Padilla et al., 2009; Blom et al., 2010); we investigated whether NS15370 and ML213 activities relied on this tryptophan residue. In the HEK cells overexpressing KCNQ4, ML213 (10 μM) caused a significant increase in steady-state current (Figure 6A), which was associated with a significant leftward shift in the voltage-dependence of channel activation with the voltage at half-maximal activation (V½) of activation changing by −33.0 ± 3.9 mV (n = 7 cells). In contrast, in cells overexpressing a KCNQ4 mutant with the tryptophan-242 residue changed to a leucine (KCNQ4-W242L), ML213 had no effect on the steady-state currents and produced a 1.6 ± 4.4 mV (n = 7 cells) change in the V½ of activation, which was not significant according to a two-way anova (Figure 6B). NS15370 (1 μM) had more complex effects on the currents produced by the stable expression of hKv7.4, with enhancement (not significant according to a two-way anova and Bonferroni’s post hoc test) seen at potentials negative to 0 mV and current depression at more positive membrane potentials (Figure 7A). However, 1 μM NS15370 produced a leftward shift in the voltage-dependence of activation with a decrease in V½ of −39.9 ± 1.7 mV (n = 8 cells). These effects were not observed in cells expressing the Kv7.4 mutant (n = 7 cells; Figure 7B).
Figure 6.
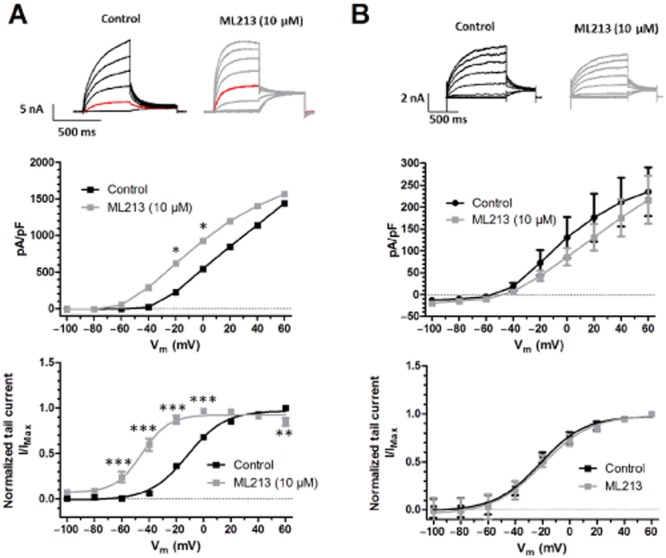
Effect of ML213 on (A) hKv7.4 channels and (B) on mutated hKv7.4-W242L channels. (Top panel) Representative current traces recorded from HEK293 cells either (A) stably expressing hKv7.4 or (B) transiently transfected with hKv7.4-W242L before and after application of ML213. Red traces represent the current elicited at −20 mV. (Middle panel) Current–voltage relationship comparing the effect of ML213 on hKv7.4 and hKv7.4-W242L. Current values are plotted against test potential before and after application of the drug. Currents were elicited by depolarizing steps from −100 to +60 mV (20 mV increments, 500 ms duration for the hKv7.4 currents and 1 s for the hKv7.4-W242L currents) from a holding potential of −80 mV. A Bonferroni post hoc test was performed following a two-way anova and *P < 0.05. (Bottom panel) Voltage-dependence of activation. Curves were obtained by normalizing the tail current at −30 mV against the maximal tail current measured in each experiment and plotted as a function of the preceding step potential (n = 6). Finally, a Boltzmann fit was performed to determine the V½ values. A two-way anova followed by a Bonferroni multiple comparisons test was performed, and **P < 0.01 and ***P < 0.001.
Figure 7.
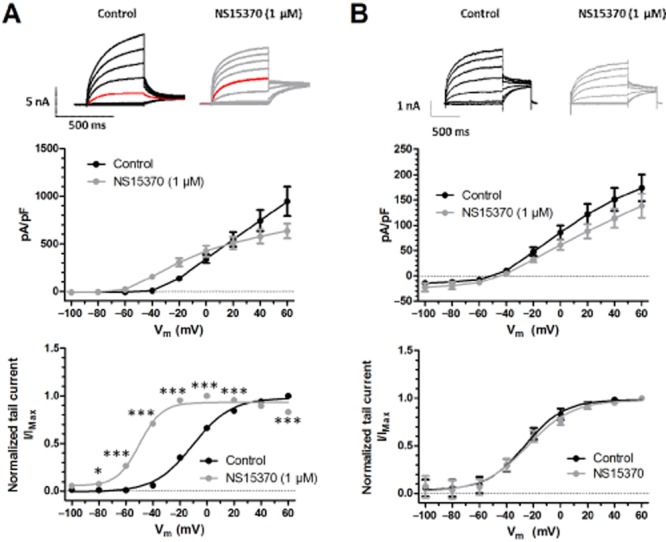
Effect of NS15370 on (A) hKv7.4 channels and (B) on mutated hKv7.4-W242L channels. (Top panel) Representative current traces recorded from HEK293 cells either (A) stably expressing hKv7.4 or (B) transiently transfected with hKv7.4-W242L before and after application of NS15370. (Middle panel) Current–voltage relationship comparing the effect of NS15370 on hKv7.4 and hKv7.4-W242L. Current values are plotted against test potential before and after application of drug. Currents were elicited by depolarizing steps from −100 to +60 mV (20 mV increments, 500 ms duration for the hKv7.4 currents and 1 s for the hKv7.4-W242L currents) from a holding potential of −80 mV. A Bonferroni post hoc test was performed following a two-way anova and no points were found to be significantly different from their respective control. (Bottom panel) Voltage-dependence of activation. Tail current–voltage relationship was obtained by plotting the normalized tail current amplitude at −30 mV against the maximal tail current and plotted as a function of the preceding step potential (n = 6–8). Finally a Boltzmann fit was performed to determine the V½ values. A Bonferroni post hoc test was performed following a two-way anova, and *P < 0.05 and ***P < 0.001.
Discussion
The results of this study show that two newly identified compounds, ML213 and NS15730, are highly potent vasorelaxants in a range of blood vessels. Moreover, these agents are more potent vasorelaxants than retigabine, S-1 and BMS204352, which have been used along with flupirtine to investigate vascular effects of Kv7 channels previously (Yeung et al., 2007; 2008; Mackie et al., 2008; Joshi et al., 2009; Zhong et al., 2010; Jepps et al., 2011; Mani et al., 2011; Chadha et al., 2012a; 2014; Khanamiri et al., 2013). In addition, ML213 and NS15730 produced marked hyperpolarization of the membrane in freshly isolated smooth muscle cells. These effects could be sufficient to counter any depolarizing surges that would be likely to lead to increased opening of voltage-dependent calcium channels and contraction. All effects on smooth muscle cells were attenuated by either high-potassium bathing solutions or specific Kv7 blockers, such as linopirdine, consistent with both agents producing arterial relaxation through specific enhancement of Kv7 channel activity and subsequent hyperpolarization.
This study also determined the relative expression of different KCNQ isoforms in the rat thoracic aorta, renal artery and mesenteric artery. In these vessels, KCNQ1, KCNQ4 and KCNQ5 expression was detected, KCNQ3 expression was variable and KCNQ2 message was never detected, suggesting that this gene is not expressed in these arteries or the copy number is considerably lower than detectable levels for this assay. These data support previous findings that KCNQ1, KCNQ4 and KCNQ5 dominate in vascular smooth muscle. Furthermore, the present study shows that the relative levels of KCNQ isoform expression vary considerably between different vessels, with KCNQ1 expression predominating in the aorta and renal artery, whereas in third-order mesenteric arteries, KCNQ4 expression predominated.
A particularly interesting observation in this study was the increased potency of ML213 and NS15730 in the mesenteric artery compared with the aorta or renal artery. ML213 and NS15730 affects Kv7.2 and 7.4 and Kv7.2–7.5, respectively, without affecting Kv7.1 (Yu et al., 2011; Dalby-Brown et al., 2013). The expression profiles of the KCNQ isoforms in these vessels suggest that the relative contributions of KCNQ4 and KCNQ5 are greater in the mesenteric artery than the other vessels tested. In the thoracic aorta and renal artery, KCNQ1 expression predominates, which if translated in these ratios to a protein level, might explain the reduced potency of the Kv7 enhancers in these vessels compared with the mesenteric artery. Considering the pharmacological profiles of the Kv7 enhancers and given that KCNQ3 expression is rarely detected in the vasculature and KCNQ2 is never detected (Ohya et al., 2003; Yeung et al., 2007; Ng et al., 2011; Chadha et al., 2012a,b; Chadha et al., 2014; Brueggemann et al., 2014; this study), the effect of ML213 at the concentrations reported in the present study are likely to be due to the enhancement of native Kv7.4 channels, whereas the effects of NS15730 are probably due the enhancement of both Kv7.4 and 7.5. Recently, proximity ligation assays in cerebral and mesenteric arteries (Brueggemann et al., 2014; Chadha et al., 2014) have shown that, in these vessels, Kv7.4 subunits are likely to exist as either homomeric Kv7.4 channels or a Kv7.4/Kv7.5 heteromultimer. Therefore, the increased potency of ML213 and NS15730 in the mesenteric artery might reflect a dominance of the Kv7.4 or Kv7.4/Kv.7.5 channels compared with in the aorta or renal artery. However, linopirdine and XE991, which block all Kv7 channels, rarely contract mesenteric arteries especially in the absence of any vasoconstrictor pre-tone, whereas aorta and renal arteries contract readily to these agents (Chadha et al., 2012a,b). Future studies need to focus on the various factors that are responsible for the regional effectiveness of Kv7 modulators.
In the mesenteric artery, this study found the relaxant EC50 values of the previously reported enhancers (S-1, BMS204352 and retigabine) to range between 2 and 11 μM, whereas ML213 displayed an EC50 of 0.74 μM and NS15370 was particularly potent with an EC50 of 0.026 μM. To date, no other Kv7 channel enhancers have been shown to relax blood vessels with such potency. ML213 and NS15370 also relaxed thoracic aorta segments with increased potency compared with the other enhancers tested, and in the renal artery, as well as NS15370 and ML213, S-1 was also equally potent at causing vasorelaxations. The low relaxant EC50s presented in this study for NS15370 are in keeping with EC50 values estimated from concentration–response curves using a Fluorimetric Imaging Plate Reader-based Tl+ influx assay (Dalby-Brown et al., 2013), as well QPatch data presented in this study. Importantly, the vasorelaxant effects were inhibited by the Kv7 channel blocker linopirdine, suggesting the effects of these new enhancers can be attributed specifically to Kv7 channel enhancement.
We tested the effect of the two novel Kv7 channel enhancers on HEK293B cells stably expressing KCNQ4 and used a mutated KCNQ4 to determine the binding site of these novel enhancers. NS15370 and ML213 caused hyperpolarizing shifts in the V½ of activation as previously reported (Dalby-Brown et al., 2013). This is in keeping with the effects other pharmacological enhancers of Kv7 channels that have also been shown to cause a hyperpolarizing shift in the V½ of activation, including BMS204352 (Schrøder et al., 2001), S-1 (Bentzen et al., 2006) and retigabine (Main et al., 2000). In this study, we also observed that NS15370 inhibited currents at positive potentials and the threshold for a net inhibition became more negative with increasing concentration. These findings are in agreement with those of Dalby-Brown et al. (2013), who also observed a current depression at higher concentrations of this enhancer. This effect has also been reported for retigabine and S-1 in overexpression systems (Main et al., 2000; Bentzen et al., 2006). Moreover, remarkably similar bimodal effects were reported for retigabine on native K+ currents in mouse portal vein smooth muscle cells (Yeung et al., 2008), and were observed in voltage clamp studies on mesenteric arteries in the present study (data not shown). Interestingly, both the stimulatory and inhibitory effects of NS15370 were lost in HEK cells overexpressing a KCNQ4 mutant with the tryptophan-242 residue changed to a leucine that has been determined to be crucial for retigabine’s action. This residue is found in a hydrophobic pocket of the S5 domain and only when the channel is in the open state is this residue available for the Kv7 channel enhancers to bind and stabilize the channel in the open configuration (Schenzer et al., 2005; Wuttke et al., 2005; Lange et al., 2013). The finding that ML213 and NS15730 produced marked membrane hyperpolarization and are reliant upon this essential tryptophan residue adds credence to the view that Kv7 channels contribute to the resting membrane conductance in smooth muscle cells.
Recently, the use of retigabine (Trobalt) to pharmacologically enhance Kv7 channels has been shown to prevent seizure generation by suppressing neuronal activity (Brodie et al., 2010; French et al., 2011). However, retigabine enhances Kv7.2–7.5 channels, which leads to various smooth muscle side effects (Jepps et al., 2013); therefore, to treat epilepsy, it is desirable to produce Kv7 enhancers with increased specificity and potency for Kv7.2–7.3 channels. NS15370 and ML213 are two such activators that were identified in an attempt to develop such an anti-epileptic, or possibly to treat other neurological disorders such as mania and psychosis (Dalby-Brown et al., 2013). However, the data presented in this study show that these activators are also potent vasorelaxants in a range of blood vessels and can hyperpolarize the resting membrane potential of mesenteric artery smooth muscle cells. Moreover, ML213 has been shown to inhibit contractions in pig detrusor muscle suggesting that the use of selective Kv7.4 channel modulators could be used in the treatment of detrusor overactivity (Svalø et al., 2013). This study has only focused on vascular smooth muscle; however, it is also known that Kv7 channels are expressed and regulate non-vascular smooth muscle contractility, including the digestive system (Ohya et al., 2002; Jepps et al., 2009; Ipavec et al., 2011), uterus (McCallum et al., 2009; 2011), bladder (Streng et al., 2004; Rode et al., 2010; Svalø et al., 2011; 2013) and airways (Brueggemann et al., 2012), all of which should be considered when trying to characterize a new Kv7 channel enhancer as an anti-epileptic. It is now widely recognized that the ‘neuronal’ Kv7 channels are not exclusively expressed in neurones, and this study highlights the possibility that these new compounds have potential as therapeutics for various smooth muscle disorders.
Acknowledgments
We would like to thank the staff at the Biological Research Facility at St. George’s University of London. J. B. S. and S. P. were funded by the British Heart Foundation and by Medical Research Council grants (PG/12/63/29824 and MR/K019074/1) respectively.
Glossary
- BMS204352
(3S)-(+)-(5-chloro-2-methoxyphenyl)-1,3-dihydro-3-fluoro-6-(trifluoromethyl)-2H-indol-2-one
- Kv
voltage-gated potassium channel
- ML213
N-mesitylbicyclo[2.2.1]heptane-2-carboxamide
- NS15370
(2-(3,5-difluorophenyl)-N-[6-[(4-fluorophenyl)methylamino]-2-morpholino-3-pyridyl]acetamide)hydrochloride
- QPCR
quantitative PCR
- S-1
(S)-N-[1-(3-morpholin-4-yl-phenyl)-ethyl]-3-phenyl-acrilamide
- V½
voltage at half-maximal activation
Author contributions
T. A. J., B. H. B., J. B. S., O. V. P. and K. S. performed experiments and analysed data. T. A. J., B. H. B., J. B. S. and W. D. B. and I. A. G. were involved in the preparation and submission of the paper. I. A. G. also provided funding.
Conflict of interests
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen BH, Schmitt N, Calloe K, Dalby-Brown W, Grunnet M, Olesen SP. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology. 2006;51:1068–1077. doi: 10.1016/j.neuropharm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Blom SM, Schmitt N, Jensen HS. Differential effects of ICA-27243 on cloned Kv7 channels. Pharmacology. 2010;86:174–181. doi: 10.1159/000317525. [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Lerche H, Gil-Nagel A, Elger C, Hall S, Shin P, et al. Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology. 2010;75:1817–1824. doi: 10.1212/WNL.0b013e3181fd6170. [DOI] [PubMed] [Google Scholar]
- Brueggemann LI, Kakad PP, Love RB, Solway J, Dowell ML, Cribbs LL, et al. Kv7 potassium channels in airway smooth muscle cells: signal transduction intermediates and pharmacological targets for bronchodilator therapy. Am J Physiol Lung Cell Mol Physiol. 2012;302:L120–L132. doi: 10.1152/ajplung.00194.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Cribbs LL, Freda J, Tripathi A, Majetschak M, et al. Differential protein kinase C-dependent modulation of Kv7.4 and Kv7.5 subunits of vascular Kv7 channels. J Biol Chem. 2014;289:2099–2111. doi: 10.1074/jbc.M113.527820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha PS, Zunke F, Zhu HL, Davis AJ, Jepps TA, Olesen SP, et al. Reduced KCNQ4-encoded voltage-dependent potassium channel activity underlies impaired β-adrenoceptor-mediated relaxation of renal arteries in hypertension. Hypertension. 2012a;59:877–884. doi: 10.1161/HYPERTENSIONAHA.111.187427. [DOI] [PubMed] [Google Scholar]
- Chadha PS, Zunke F, Davis AJ, Jepps TA, Linders JT, Schwake M, et al. Pharmacological dissection of Kv7.1 channels in systemic and pulmonary arteries. Br J Pharmacol. 2012b;166:1377–1387. doi: 10.1111/j.1476-5381.2012.01863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha PS, Jepps TA, Carr G, Stott JB, Zhu HL, Cole WC, et al. Contribution of Kv7.4/Kv7.5 heteromers to intrinsic and calcitonin gene-related peptide-induced cerebral reactivity. Arterioscler Thromb Vasc Biol. 2014;34:887–893. doi: 10.1161/ATVBAHA.114.303405. [DOI] [PubMed] [Google Scholar]
- Dailey JW, Cheong JH, Ko KH, Adams-Curtis LE, Jobe PC. Anticonvulsant properties of D-20443 in genetically epilepsy-prone rats: prediction of clinical response. Neurosci Lett. 1995;195:77–80. doi: 10.1016/0304-3940(95)11783-s. [DOI] [PubMed] [Google Scholar]
- Dalby-Brown W, Jessen C, Hougaard C, Jensen ML, Jacobsen TA, Nielsen KS, et al. Characterization of a novel high potency positive modulator of Kv7 channels. Eur J Pharmacol. 2013;709:52–63. doi: 10.1016/j.ejphar.2013.03.039. [DOI] [PubMed] [Google Scholar]
- French JA, Abou-Khalil BW, Leroy RF, Yacubian EM, Shin P, Hall S, et al. Randomized, double-blind, placebo-controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology. 2011;76:1555–1563. doi: 10.1212/WNL.0b013e3182194bd3. [DOI] [PubMed] [Google Scholar]
- Ipavec V, Martire M, Barrese V, Taglialatela M, Currò D. Kv7 channels regulate muscle tone and nonadrenergic noncholinergic relaxation of the rat gastric fundus. Pharmacol Res. 2011;64:397–409. doi: 10.1016/j.phrs.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps TA, Greenwood IA, Moffatt JD, Sanders KM, Ohya S. Molecular and functional characterization of Kv7 K+ channel in murine gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol. 2009;297:G107–G115. doi: 10.1152/ajpgi.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps TA, Chadha PS, Davis AJ, Harhun MI, Cockerill GW, Olesen SP, et al. Downregulation of Kv7.4 channel activity in primary and secondary hypertension. Circulation. 2011;124:602–611. doi: 10.1161/CIRCULATIONAHA.111.032136. [DOI] [PubMed] [Google Scholar]
- Jepps TA, Olesen SP, Greenwood IA. One man’s side effect is another man’s therapeutic opportunity: targeting Kv7 channels in smooth muscle disorders. Br J Pharmacol. 2013;168:19–27. doi: 10.1111/j.1476-5381.2012.02133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther. 2009;329:368–376. doi: 10.1124/jpet.108.147785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanamiri S, Soltysinska E, Jepps TA, Bentzen BH, Chadha PS, Schmitt N, et al. Contribution of Kv7 channels to basal coronary flow and active response to ischemia. Hypertension. 2009;62:1090–1097. doi: 10.1161/HYPERTENSIONAHA.113.01244. [DOI] [PubMed] [Google Scholar]
- Lange W, Geissendörfer J, Schenzer A, Grötzinger J, Seebohm G, Friedrich T, et al. Refinement of the binding site and mode of action of the anticonvulsant Retigabine on KCNQ K+ channels. Mol Pharmacol. 2013;75:272–280. doi: 10.1124/mol.108.052282. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- Mani BK, Brueggemann LI, Cribbs LL, Byron KL. Activation of vascular KCNQ (Kv7) potassium channels reverses spasmogen-induced constrictor responses in rat basilar artery. Br J Pharmacol. 2011;164:237–249. doi: 10.1111/j.1476-5381.2011.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum LA, Greenwood IA, Tribe RM. Expression and function of Kv7 channels in murine myometrium throughout oestrous cycle. Pflugers Arch. 2009;457:1111–1120. doi: 10.1007/s00424-008-0567-5. [DOI] [PubMed] [Google Scholar]
- McCallum LA, Pierce SL, England SK, Greenwood IA, Tribe RM. The contribution of Kv7 channels to pregnant mouse and human myometrial contractility. J Cell Mol Med. 1977;15:577–586. doi: 10.1111/j.1582-4934.2010.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 2011;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Ng FL, Davis AJ, Jepps TA, Harhun MI, Yeung SY, Wan A, et al. Expression and function of the K+ channel KCNQ genes in human arteries. Br J Pharmacol. 2011;162:42–53. doi: 10.1111/j.1476-5381.2010.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya S, Asakura K, Muraki K, Watanabe M, Imaizumi Y. Molecular and functional characterization of ERG, KCNQ, and KCNE subtypes in rat stomach smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2002;282:G277–G287. doi: 10.1152/ajpgi.00200.2001. [DOI] [PubMed] [Google Scholar]
- Ohya S, Sergeant GP, Greenwood IA, Horowitz B. Molecular variants of KCNQ channels expressed in murine portal vein myocytes: a role in delayed rectifier current. Circ Res. 2003;92:1016–1023. doi: 10.1161/01.RES.0000070880.20955.F4. [DOI] [PubMed] [Google Scholar]
- Padilla K, Wickenden AD, Gerlach AC, McCormack K. The KCNQ2/3 selective channel opener ICA-27243 binds to a novel voltage-sensor domain site. Neurosci Lett. 2009;465:138–142. doi: 10.1016/j.neulet.2009.08.071. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode F, Svalø J, Sheykhzade M, Rønn LC. Functional effects of the KCNQ modulators retigabine and XE991 in the rat urinary bladder. Eur J Pharmacol. 2010;638:121–127. doi: 10.1016/j.ejphar.2010.03.050. [DOI] [PubMed] [Google Scholar]
- Rostock A, Tober C, Rundfeldt C, Bartsch R, Engel J, Polymeropoulos EE, et al. D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996;23:211–223. doi: 10.1016/0920-1211(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grötzinger J, et al. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrøder RL, Jespersen T, Christophersen P, Strøbaek D, Jensen BS, Olesen SP. KCNQ4 channel activation by BMS-204352 and retigabine. Neuropharmacology. 2001;40:888–898. doi: 10.1016/s0028-3908(01)00029-6. [DOI] [PubMed] [Google Scholar]
- Soldovieri MV, Miceli F, Taglialatela M. Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. Physiology. 2011;26:365–376. doi: 10.1152/physiol.00009.2011. [DOI] [PubMed] [Google Scholar]
- Stott JB, Jepps TA, Greenwood IA. Kv7 potassium channels: a new therapeutic target in smooth muscle disorders. Drug Discov Today. 2014;19:413–424. doi: 10.1016/j.drudis.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Streng T, Christoph T, Andersson KE. Urodynamic effects of the K+ channel (KCNQ) opener retigabine in freely moving, conscious rats. J Urol. 2004;172:2054–2058. doi: 10.1097/01.ju.0000138155.33749.f4. [DOI] [PubMed] [Google Scholar]
- Svalø J, Hansen HH, Rønn LC, Sheykhzade M, Munro G, Rode F. Kv7 positive modulators reduce detrusor overactivity and increase bladder capacity in rats. Basic Clin Pharmacol Toxicol. 2011;110:145–153. doi: 10.1111/j.1742-7843.2011.00765.x. [DOI] [PubMed] [Google Scholar]
- Svalø J, Bille M, Parameswaran Theepakaran N, Sheykhzade M, Nordling J, Bouchelouche P. Bladder contractility is modulated by Kv7 channels in pig detrusor. Eur J Pharmacol. 2013;715:312–320. doi: 10.1016/j.ejphar.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Tober C, Rostock A, Rundfeldt C, Bartsch R. D-23129: a potent anticonvulsant in the amygdala kindling model of complex partial seizures. Eur J Pharmacol. 1996;303:163–169. doi: 10.1016/0014-2999(96)00073-8. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:34. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Yeung S, Schwake M, Pucovský V, Greenwood IA. Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br J Pharmacol. 2008;155:62–72. doi: 10.1038/bjp.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SY, Pucovský V, Moffatt JD, Saldanha L, Schwake M, Ohya S, et al. Molecular expression and pharmacological identification of a role for Kv7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Wu M, Townsend SD, Zou B, Long S, Daniels JS, et al. Discovery, synthesis, and structure activity relationship of a series of N-Aryl-bicyclo[2.2.1]heptane-2-carboxamides: characterization of ML213 as a novel KCNQ2 and KCNQ4 potassium channel opener. ACS Chem Neurosci. 2011;2:572–577. doi: 10.1021/cn200065b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, et al. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]