

Abstract
Background and Purpose
Doxorubicin-based chemotherapy induces cardiotoxicity, which limits its clinical application. We previously reported the protective effects of quercetin against doxorubicin-induced hepatotoxicity. In this study, we tested the effects of quercetin on the expression of Bmi-1, a protein regulating mitochondrial function and ROS generation, as a mechanism underlying quercetin-mediated protection against doxorubicin-induced cardiotoxicity.
Experimental Approach
Effects of quercetin on doxorubicin-induced cardiotoxicity was evaluated using H9c2 cardiomyocytes and C57BL/6 mice. Changes in apoptosis, mitochondrial function, oxidative stress and related signalling were evaluated in H9c2 cells. Cardiac function, serum enzyme activity and reactive oxygen species (ROS) generation were measured in mice after a single injection of doxorubicin with or without quercetin pre-treatment.
Key Results
In H9c2 cells, quercetin reduced doxorubicin-induced apoptosis, mitochondrial dysfunction, ROS generation and DNA double-strand breaks. The quercetin-mediated protection against doxorubicin toxicity was characterized by decreased expression of Bid, p53 and oxidase (p47 and Nox1) and by increased expression of Bcl-2 and Bmi-1. Bmi-1 siRNA abolished the protective effect of quercetin against doxorubicin-induced toxicity in H9c2 cells. Furthermore, quercetin protected mice from doxorubicin-induced cardiac dysfunction that was accompanied by reduced ROS levels and lipid peroxidation, but enhanced the expression of Bmi-1 and anti-oxidative superoxide dismutase.
Conclusions and Implications
Our results demonstrate that quercetin decreased doxorubicin-induced cardiotoxicity in vitro and in vivo by reducing oxidative stress by up-regulation of Bmi-1 expression. The findings presented in this study have potential applications in preventing doxorubicin-induced cardiomyopathy.
Table of Links
| LIGANDS |
|---|
| Doxorubicin |
| Quercetin |
This Table lists the protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Doxorubicin, an anthracycline antibiotic, is commonly used to treat several types of cancer. A limitation of doxorubicin-based chemotherapy is the severe adverse effects to non-tumour tissues such as the heart, liver and kidney (Silber and Barber, 1995) caused by the therapy. The maximum allowable dose of doxorubicin during cancer treatment is limited by the risk of developing congestive heart failure (Steinherz et al., 1991).
Several key studies found that the damaging effects of doxorubicin on cardiomyocytes is through the disruption of mitochondrial function and the generation of reactive oxygen species (ROS), such as hydrogen peroxide, superoxide and hydroxyl radicals (Wallace, 2003; Mukhopadhyay et al., 2009; Pereira et al., 2011). Cardiomyocytes are more susceptible to doxorubicin-induced free radical-mediated damage because these cells have relatively low levels of antioxidant enzymes such as superoxide dismutase (SOD) and catalase (Doroshow et al., 1980; Kalyanaraman et al., 2002), which provide protection by converting hydrogen peroxide into water and oxygen. Thus, doxorubicin-induced cardiotoxicity can be prevented by quenching the ROS generated by doxorubicin in cardiomyocytes (Peng et al., 2005; Kaiserova et al., 2007).
The iron-chelating, antioxidant and carbonyl reductase-inhibitory characteristics of flavonoids make them attractive compounds for mitigating anthracycline-induced cardiotoxicity. Quercetin (3,3′,4′,5,7-pentahydroxyflavone), an important dietary flavonoid present in several fruits and vegetables, exhibits antioxidant, anti-inflammatory and anti-cancer properties (Gibellini et al., 2011). Quercetin scavenges superoxide anion, singlet oxygen and lipid peroxy radicals (Morales et al., 2012; Ertuğ et al., 2013) and inhibits copper-catalysed oxidation (Filipe et al., 2004). Several reports demonstrate that quercetin scavenges ROS and inhibits the activation of ERK and MAP kinase in ROS-induced cardiomyopathy (Kyaw et al., 2004; Angeloni et al., 2007). Furthermore, quercetin protects H9c2 cardiomyocytes from the oxidative stress induced by H2O2 through its antioxidant activity and it modulates the apoptosis signal transduction pathway (Angeloni et al., 2007). Quercetin was more effective than other flavonoids such as naringenin, pycnogenol and trolox, in protecting against daunorubicin-induced cytotoxicity in H9c2 cardiomyocytes (Mojzisová et al., 2009). Quercetin enhances the therapeutic efficacy of doxorubicin in highly invasive breast cancer cells and simultaneously reduces doxorubicin-induced toxic side effects (Staedler et al., 2011). We have previously demonstrated that quercetin protects mice from doxorubicin-induced hepatotoxicity while potentiating the anti-hepatoma activity of doxorubicin (Wang et al., 2012). Similarly, quercetin attenuated doxorubicin-induced cardiotoxicity in vivo (Pei et al., 2007) and protected cardiomyocytes from doxorubicin-induced toxicity by chelating iron, inducing antioxidant activity and inhibiting carbonyl reductase (Kaiserova et al., 2007).
The present study was undertaken to elucidate the molecular mechanisms underlying the protection by quercetin of myocardial cells against doxorubicin-induced toxicity in vitro and in vivo. We found that quercetin attenuated doxorubicin-induced cardiotoxicity by reducing oxidative stress. Our results demonstrated that quercetin decreased doxorubicin-induced ROS levels and DNA damage to maintain cardiac cell viability, through up-regulated Bmi-1 expression.
Methods
Cell culture and treatment
Rat embryonic ventricular myocardial H9c2 cells were purchased from Cell Bank (Chinese Academy of Sciences). Cells were cultured in RPMI-1640 (Gibco, Life Technologies Inc., Grand Island, NY, USA) with 10% FBS in a humidified incubator at 37°C with 5% CO2. Bmi-1 Smartpool si-RNA (siBmi-1), purchased from Dharmacon (Dharmacon, Abgene Ltd., Epsom, UK), was used to transfect sub-confluent H9c2 cells. The cells were transfected using the Lipofectamine RNAiMAX (Invitrogen Corp., Carlsbad, CA, USA) reverse transfection protocol according to the manufacturer’s instructions. Following transfection, the cells were incubated for 48 h prior to treatments. A scrambled pool of siRNA served as the control. Bmi-1 over-expressed vector pGC-FU-GFP-Bmi-1 and pGC-FU-GFP control were purchased from GeneChem (Shanghai, China). Cells were infected with the lentivirus and selected under puromycin, according to the manufacturer’s instructions.
MTT cell viability assay
The cells were plated in triplicate wells in 96-well plates (5 × 103 cells per well) overnight and incubated with doxorubicin (doses: 0–16 μM) with/without quercetin (doses: 50 or 100 μM) containing complete RPMI-1640 medium for 48 h. MTT (0.5 mg/mL) was added to the medium for the last 4 h of incubation. The surviving cells converted MTT into formazan, which generated a blue-purple colour when dissolved in DMSO and was measured at 570 nm using an ELX800 Microplate Reader (Bio-Tek Instruments Inc., Winooski, VT, USA). The relative percentage of cell survival was calculated by dividing the absorbance of treated cells by the control in each experiment.
Apoptosis assay
Apoptosis was determined using the Annexin V-FITC apoptosis detection kit (BD Biosciences), according to the manufacturer’s protocol, to discriminate between live cells (Annexin-V−/PI−), apoptotic cells (Annexin-V+/PI− and Annexin-V+/PI+) and dead cells (Annexin-V−/PI+). The stained cells were analysed by FACSCalibur using CellQuest software (Becton Dickinson, San Jose, CA, USA). Hoechst 33258 staining method was also used to determine apoptosis. After drug treatment, the cells were washed with PBS, the nuclei were stained with 2 μg·mL−1 Hoechst 33258 for 10 min and stained cells were analysed using a Zeiss fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Electron microscopy
Cell samples were harvested by low-speed centrifugation (200× g), washed twice with PBS and fixed in 1.4% glutaraldehyde fixation buffer for 2 h at room temperature. Sample sections (50 nm) were obtained using standard procedures and the sections were briefly stained with lead citrate (2–5 min). Transmission electron microscopic analysis was performed with a Philips 410 (Philips, Amsterdam, the Netherlands) at the acceleration voltage of 60 kV.
Detection of mitochondrial membrane potential (MMP)
The cells were harvested and stained with JC-1 according to the manufacturer’s protocol and analysed with FACSCalibur. Cells on glass coverslips were stained with JC-1, analysed by a Zeiss LSM 710 confocal microscope system (Carl Zeiss) and processed with ZEN software (Carl Zeiss). Moreover, cells were evaluated after staining with TMRM, another MMP-dependent dye, according to the manufacturer’s protocol.
Mitochondrial and intracellular ROS levels
MitoSOX Red and CM-H2DCFDA were used to detect mitochondrial and intracellular peroxides. Cells on glass coverslips were stained with 5 μM MitoSOX Red for 30 min at 37°C; images obtained by a Zeiss LSM 710 confocal microscope system (Carl Zeiss) and MitoSOX dye intensities were normalized to the intensity average of control cells and at least 100 cells were analysed with ZEN LE software. The harvested cells were stained with 10 μM CM-H2DCFDA for 30 min at 37°C, washed twice with PBS and quantified by FACSCalibur using CellQuest software. The blank samples without dye were evaluated to exclude autofluorescence.
Real-time polymerase chain reaction (qPCR)
Total cellular RNA was isolated using the RNeasy Mini kit. Reverse transcription was performed using the SuperScript II Reverse Transcriptase according to the manufacturer’s recommendations. Bmi-1, Nox1, p47 and actin were subjected to qPCR in triplicate for each sample, as previously described (Dong et al., 2011). The primer sequences were obtained from the Universal Probe Library (Roche, Basel, Switzerland). The 2-ΔΔCq value determination method was used to compare the fold differences in expression.
Immunoblotting and immunofluorescence staining
Immunoblotting was performed as described in our previous study (Dong et al., 2011). Immunofluorescence staining was performed on cells plated on coverslips. The cells were fixed in 3.7% formaldehyde for 15 min, washed three times with PBS and permeabilized with 0.25% Triton X-100 in PBS for 10 min. Mouse monoclonal anti-phospho-γ-H2AX and Alex Fluor568 goat anti-mouse IgG were used as primary and secondary antibodies respectively. Images were obtained using a Zeiss HBO-100 fluorescence microscope.
Comet assay
To detect DNA double-strand breaks (DSBs), a neutral comet assay was performed with the CometAssay kit (Trevigen, Gaithersburg, MD, USA), according to the manufacturer’s guidelines. H9c2 cells were harvested after the treatment, separated by electrophoresis in the CometSlides and stained with SYBR Green Gold (Trevigen). Images were obtained with the Zeiss fluorescence microscope. The degree of DNA damage was compared in the groups by measuring the tail lengths with CometAssay IV software.
Ethics statement and animal treatment protocols
All animal care and experimental procedures were in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of Zhejiang University and were approved by the Committee on the Ethics of Animal Experiments of Sir Run Run Shaw Hospital (Permit Number: 20120906). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 174 animals were used in the experiments described here.
Because the daily intake of quercetin in the human diet has been estimated to be in the range of 2–40 mg, we selected a dose of 100 mg·kg−1 of quercetin for the mouse experiments in this study. C57BL/6 female mice aged 8–10 weeks were randomly divided into 4 groups. The first group received saline (i.p., n = 25). Mice in the second group were treated with a single dose of doxorubicin (20 mg·kg−1, i.p., n = 30). Quercetin was administered (100 mg·kg−1, p.o., n = 25) to the mice in the third group once daily for 10 days. Mice in the fourth group were pretreated with quercetin daily for 5 days prior to a single dose of doxorubicin (n = 30). For survival determination, mice mortality was monitored (n = 10 in each group) for 5 weeks after doxorubicin injection. Mice in other groups were killed and weighed 5 days after doxorubicin injection. A blood sample from each animal was collected in tubes, and serum was separated by centrifugation at 200× g/15 min for determination of creatine kinase (CK), LDH, SOD and malondialdehyde (MDA) by commercial enzyme assay kits [CK/LDH assay kit, Biosino Bio-technology and Science Inc., Beijing, China; SOD/MDA assay kit, DiaSys Diagnostic Systems (Shanghai) Co., Ltd, Shanghai, China] according to the manufacturer’s instructions. The measurements were performed with a 7020 chemistry analyzer (Hitachi, Tokyo, Japan). For determination of the histopathological changes, hearts of mice from the various treatment groups were collected, weighed and divided into two parts. The top half of each heart was processed for histological analysis. The bottom half was placed embedded in tissue-freezing medium, frozen at −80°C and processed for DHE staining according to the instructions. Tissue sections were imaged using a Zeiss fluorescent microscope. Part heart tissues were subjected to Western blot analysis for Bmi-1.
Measurement of cardiac function
Because 20 mg·kg−1 doxorubicin treatment induced high mortality (25%) and extremely weakened the mice, a single dose of 10 mg·kg−1 doxorubicin was used to measure cardiac function (n = 6 in each group). Transthoracic echocardiography was measured with a 14 MHz linear array probe (a diagnostic ultrasound system Nemio SSA-550A, Toshiba, Tokyo, Japan) after sodium pentobarbital administration (50 mg·kg−1 of body weight, i.p.) for 10 min.
Histopathology
Tissue samples (hearts and livers) were fixed in 10% formol saline for 24 h and embedded in paraffin, cut in 5 μm sections, stained with Harris haematoxylin & eosin and evaluated for any structural changes under a bright field microscope. Standard immunoperoxidase procedures were used to visualize Bmi-1 in samples. Briefly, sections were deparaffinized, blocked with goat serum, followed by incubation with anti-Bmi-1(1:150) overnight at 4°C. After incubation with HRP-linked secondary antibody for 30 min, the sections were counterstained with Mayer’s haematoxylin. The TUNEL positive cells in heart sections were detected using ApopTag Plus Peroxidase in Situ Apoptosis Detection Kit according to the manufacturer’s protocol.
Data analysis
Resulsst are expressed as means ± SEM. Mean values for more than three groups were compared by one-way anova, followed by Tukey’s multiple comparison test. Differences with P < 0.05 were considered significant.
Materials
Doxorubicin, quercetin, MTT, anti-GAPDH, anti-rabbit or mouse IgG HRP were purchased from Sigma (St. Louis, MO, USA). JC-1, Mitosox Red, TMRM and 7′-dihydroethidium (DHE) were from Molecular Probes (Eugene, OR, USA); Alex Fluor®568 goat anti-mouse IgG, DMSO, CM-H2DCFDA and SuperScript II Reverse Transcriptase were from Invitrogen-Life Technologies (Merelbeke, Belgium); RNeasy Mini kit was from Qiagen (Shanghai, China); apoptosis detection kit, anti-Bcl-2 and anti-Bid antibodies were from BD Biosciences (San Jose, CA, USA); anti-p16, anti-p53 and anti-p19 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-phospho-γ-H2AX, anti-Bmi-1 antibodies and ApopTag® Plus Peroxidase in Situ Apoptosis Detection Kit were from Millipore Inc. (Billerica, MA, USA); anti-Rad51 was from Epitomics Inc. (Burlingame, CA, USA); anti-Ku70, anti-PARP, anti-Bmi-1 and anti-cleaved caspase-3 antibodies were from Cell Signaling Technology (Beverly, CA, USA); Fluo 4-AM was from Dojindo Laboratories (Kumamoto, Japan).
Results
Quercetin protected H9c2 cardiomyocytes from doxorubicin-induced cell death
H9c2 cells were treated with various concentrations of doxorubicin and the cell numbers were evaluated using an MTT assay to investigate the ability of quercetin to protect cardiomyocytes against doxorubicin-induced cytotoxicity. Doxorubicin treatment for 48 h reduced the number of cardiomyocytes in a dose-dependent manner (Figure 1A). However, the doxorubicin-induced reduction in cell numbers was prevented by 2 h of pre-treatment with 50 μM quercetin and 100 μM quercetin (Figure 1A). The peak doxorubicin concentration during anti-cancer therapy is 5 μM in human plasma (Minotti et al., 2004). Quercetin alone (<200 μM) did not affect the cell numbers (data not shown). Thus, for all in vitro studies, the H9c2 cells were incubated for 24 h with 5 μM doxorubicin, with or without 100 μM quercetin pre-treatment, unless otherwise specified.
Figure 1.
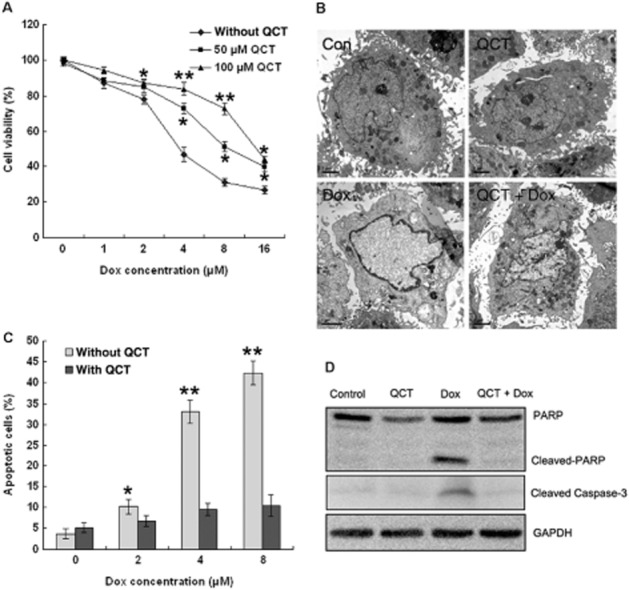
Quercetin (QCT) protected against doxorubicin-induced cell death in H9c2 cardiomyocytes. (A) H9c2 cells were treated with doxorubicin (Dox; 0–16 μM) for 48 h with and without quercetin pre-treatment (50 and 100 μM) and the surviving cells were measured using an MTT assay. Data represent the mean ± SEM (n = 3). *P < 0.05, **P < 0.01 for each group versus doxorubicin without quercetin. (B) Electron microscopic evaluation of apoptosis. H9c2 cells treated with doxorubicin (5 μM) for 24 h with and without quercetin pre-treatment (100 μM) were stained and viewed under EM. Doxorubicin-treated cells showed homogeneous chromatin condensation within the nucleus, and control cells showed normal nuclear morphology. (C) H9c2 cells were treated with doxorubicin (2, 4, 6 and 8 μM) for 24 h with and without quercetin pre-treatment (100 μM), and apoptotic cells were analysed by flow cytometry using Annexin-V/PI staining. Quercetin inhibited doxorubicin-induced apoptosis. n = 3 in each group. *P < 0.05, **P < 0.01 for each group versus Dox. (D) Cleaved PARP and caspase-3 expression were assessed through Western blot analysis. GAPDH served as the loading control. Quercetin inhibited doxorubicin-induced cleaved PARP and caspase-3 expression (data are representative of three independent cell cultures).
Doxorubicin induces cell death through apoptosis. Cardiomyocyte apoptosis was observed by electron microscopy, as illustrated in Figure 1B. Compared with the control, the doxorubicin-treated cells exhibited typical apoptotic morphology, characterized by homogeneous condensation of chromatin to one side or to the periphery of the nuclei and apoptotic body formation with nuclear fragmentation. However, the majority of the cells were normal under doxorubicin treatment with quercetin pre-treatment. Flow cytometric analysis (Figure 1C) showed that the proportion of apoptotic cells was increased concentration-dependently by doxorubicin (2, 4 and 8 μM) and that quercetin pre-treatment clearly mitigated the doxorubicin-induced apoptosis. Cardiomyocyte apoptosis was also studied in terms of PARP cleavage and caspase-3 activation. Treating the cardiomyocytes with doxorubicin dramatically increased the levels of cleaved PARP and caspase-3, which was inhibited by quercetin pre-treatment (Figure 1D).
Quercetin attenuated doxorubicin-induced mitochondrial dysfunction in H9C2 cells
Myocardial mitochondria are the target organelles of doxorubicin-induced toxicity in cardiomyocytes. The inner matrix of some mitochondria in the doxorubicin-treated cells showed increased electron density but quercetin pre-treatment maintained the normal mitochondrial morphology (Figure 2A). Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis (Green and Leeuwenburgh 2002). To determine whether doxorubicin induced apoptosis through disrupting MMP and how quercetin affected this process, changes in MMP were observed using the confocal ratio imaging of JC-1 fluorescence. The red/green ratio in doxorubicin-treated cells decreased, which indicates disruption of the MMP (Figure 2B) and quercetin pre-treatment increased MMP to near normal levels, thereby confirming the protective effect of quercetin on MMP integrity (Figure 2C). To confirm the above results, mitochondrial depolarization was also evaluated by TMRM staining. Quercetin effectively prevented doxorubicin-induced mitochondrial depolarization (data not shown). Doxorubicin induces apoptosis through the mitochondrial pathway in cardiomyocytes. The expression levels of Bcl-2 family proteins were assessed to elucidate the molecular mechanisms involved in the anti-apoptotic effect of quercetin. Doxorubicin down-regulated the expression of the anti-apoptotic Bcl-2 and up-regulated expression of the pro-apoptotic Bid in H9c2 cells, whereas quercetin maintained Bcl-2 expression and repressed Bid expression (Figure 2D). p53 is involved in doxorubicin-induced cardiomyocyte apoptosis; hence, we checked the p53 expression levels. doxorubicin dramatically increased p53 expression, whereas quercetin partially reduced doxorubicin-induced p53 expression (Figure 2D).
Figure 2.
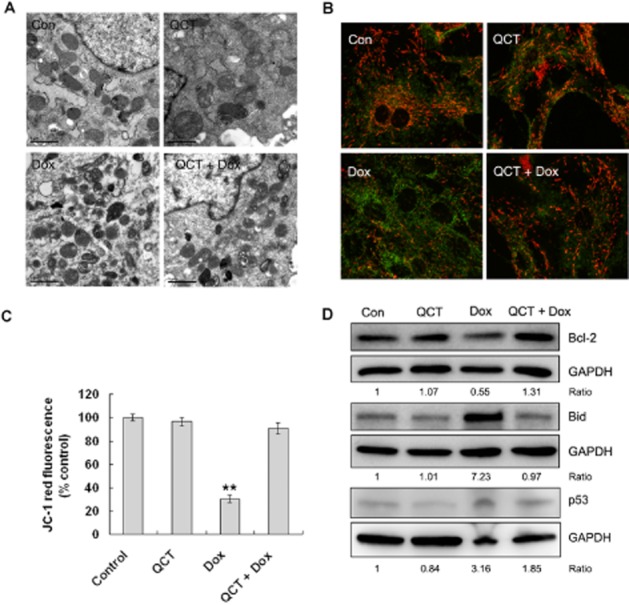
Quercetin (QCT) maintained mitochondrial function in H9c2 cells. (A) Electron microscopic evaluation of mitochondrial morphology. H9c2 cells treated with doxorubicin (Dox; 5 μM) for 24 h with and without 100 μM quercetin pre-treatment were stained. The doxorubicin-treated cells showed increased electron density, whereas the control cells showed normal mitochondrial morphology. (B) Effect of quercetin (100 μM) on the collapse of MMP in H9c2 cells treated with doxorubicin (5 μM) for 24 h. JC-1 was observed as green fluorescent monomers in the cytosol or as red fluorescent aggregates in intact mitochondria. The reduction in red fluorescence intensity indicates the collapse of the mitochondria with intact membrane potential. The micrographs were recorded under confocal microscopy (Zeiss) at 40× magnification. (C) The red JC-1 fluorescence intensity was quantified by FACS and the relative fluorescence changes are shown. JC-1 red was reduced to 30% of the control levels in the doxorubicin-treated cells (24 h). Quercetin effectively prevented the breakdown of the MMP. n = 3 in each group. **P < 0.01 each group versus Dox. (D) Bcl-2, Bid and p53 expression were assessed through Western blot analysis. GAPDH served as the loading control. Quercetin inhibited doxorubicin-induced Bid and p53 expression and maintained Bcl-2 expression. Data are calculated as ratios of target protein levels to those of GAPDH. Ratios are compared with the ratios in control samples set at 1 (data are representative of three independent cell cultures).
Quercetin inhibited doxorubicin-induced oxidative stress in H9C2 cells
Doxorubicin causes cellular and genetic damage by increasing intracellular ROS levels. Hence, we assayed the effect of quercetin on doxorubicin-induced mitochondrial ROS and the total cellular ROS levels in H9c2 cells. Mitochondrial superoxide production was measured with MitoSOX Red under a confocal microscope. MitoSOX Red fluorescence in doxorubicin-treated cells increased by 5.6-fold compared with the untreated control and the quercetin-treated cells, which suggested a significant increase in mitochondrial ROS production by doxorubicin (Figure 3A). Interestingly, quercetin pre-treatment dramatically inhibited mitochondrial superoxide production, as indicated by reduced fluorescence. The total cellular ROS levels were measured via FACS-based quantification of fluorescent CM-H2DCF. As shown in Figure 3B, H9c2 cells displayed marked total cellular ROS accumulation after 24 h of treatment with 5 μM doxorubicin, whereas cellular ROS accumulation was clearly inhibited in the cells pre-treated with quercetin. The mean fluorescence density increased above that of the control in the doxorubicin-treated cells (2 -8 μΜ). However, pre-treatment with quercetin blocked this increased fluorescence (Figure 3C). The mRNA levels of NADHP oxidase involved in ROS production (Nox1 and p47-phox) were compared through reverse transcription quantitative PCR (Figure 3D). The doxorubicin treatment increased Nox1 expression by 80-fold and increased p47-phox by 2.2-fold, but the quercetin pre-treatment prevented this increased expression of these genes (Figure 3E).
Figure 3.
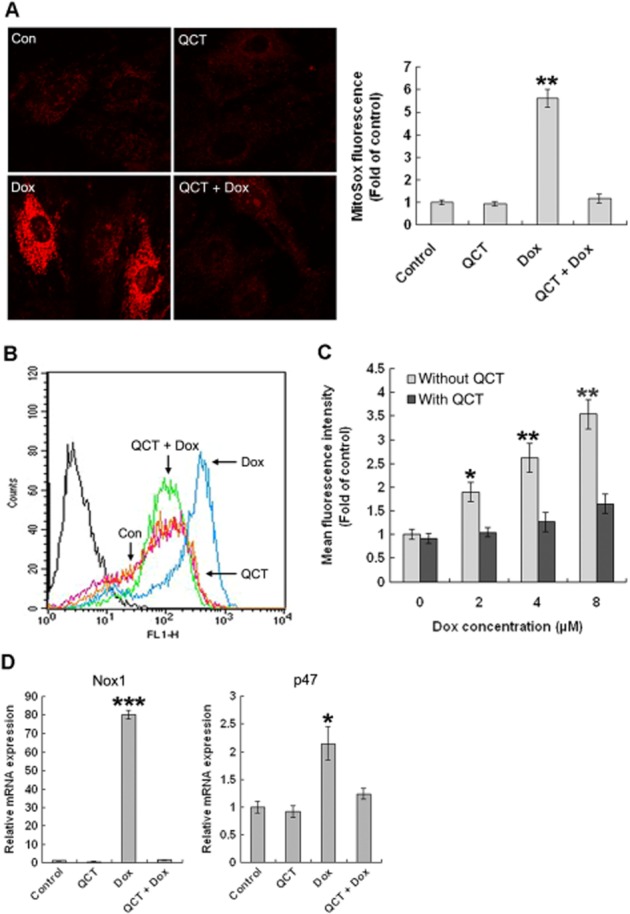
Quercetin (QCT) reduced doxorubicin(Dox)-induced ROS generation in H9c2 cells. (A) Assessment of mitochondrial superoxide production. MitoSOX Red staining revealed increased superoxide anion in doxorubicin-treated cells, but quercetin reduced the red staining to almost normal levels, as in the control cells. **P < 0.01 each group versus Dox. (B) Cells were stained for intracellular ROS using CM-H2DCFDA, a fluorescent marker of ROS, and were assayed via flow cytometry. (C) Fluorescence intensity was measured through flow cytometry in individual cells and plotted. Quercetin effectively attenuated doxorubicin-induced total ROS generation. n = 3 in each group. *P < 0.05, **P < 0.01 each group versus Dox. (D) The mRNA levels of p47 and Nox1 were measured through qPCR and normalized against actin. Quercetin significantly inhibited doxorubicin-induced Nox1 and p47 mRNA expression. n = 3 in each group. *P < 0.05, ***P < 0.001 each group versus Dox (data are representative of three independent cell cultures).
H9c2 cells were stained to identify the γ-H2AX foci, which co-localized with DNA double stranded breaks (DSB) in situ to determine DNA damage. Doxorubicin markedly increased γ-H2AX foci formation and this was greatly diminished in the quercetin-pre-treated cells (Figure 4A). A similar result was obtained via a neutral comet assay. The doxorubicin-treated cells exhibited much longer comet tails than control cells, indicating that doxorubicin-treated cells contained more DSBs (Figure 4B) and quercetin pre-treatment effectively inhibited tail formation. We examined the effect of quercetin treatment on the expression of the proteins involved in the repair of doxorubicin-induced DSBs. The level of Rad51 increased following quercetin treatment in H9c2 cells, but the level of Ku70 expression did not change (Figure 4C). These results indicate that quercetin mitigates the genotoxic effects of doxorubicin.
Figure 4.
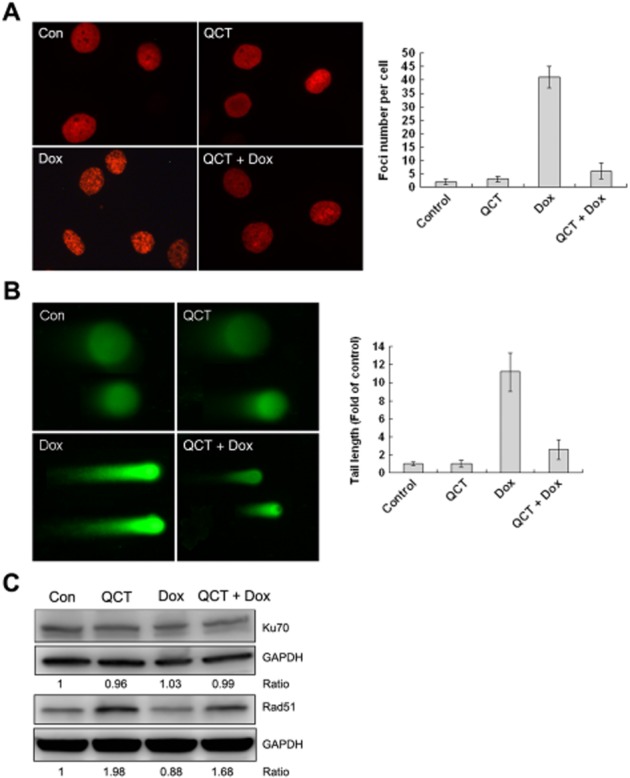
Quercetin (QCT) reduced doxorubicin(Dox)-induced DNA damage in H9c2 cells. (A) Representative staining of the γ-H2AX in cells. The percentage of cells that exhibited γ-H2AX intranuclear foci was determined by counting at least 50 cells. Quercetin attenuated doxorubicin-induced γ-H2AX foci formation. **P < 0.01 each group versus Dox. (B) DNA double-strand breaks measured using a neutral comet assay. Quercetin also effectively attenuated doxorubicin-induced comet tail formation. **P < 0.01 each group versus Dox. (C) Western blot analysis was performed for Ku70, Rad51 and GAPDH served as the loading control. Data are calculated as ratios of target protein levels to those of GAPDH. Ratios are compared with the ratios in control samples set at 1. Doxorubicin had no effect on Ku70 expression, but it reduced Rad51 expression. Quercetin maintained Rad51 expression (data are representative of 3 independent cell cultures).
Quercetin exerted its protective effect by up-regulating Bmi-1 expression in H9C2 cells
Bmi-1 has an important role in the response to DNA damage (Ismail et al., 2010; Gieni et al., 2011), in mitochondrial function and in ROS homeostasis (Chatoo et al., 2009). We first determined the effects of quercetin on Bmi-1 expression levels in H9c2 cells. The mRNA expression level of Bmi-1 was increased by 2.3-fold that of control cells, and longer incubation times (8 and 24 h) with quercetin did not increase the Bmi-1 expression further. Western blot analysis confirmed the gene expression results (Figure 5A). Quercetin also increased Bmi-1 expression in primary cardiomyocytes (data not shown). In H9c2 cells (Figure 5B), doxorubicin alone decreased Bmi-1 and markedly raised the expression of the Bmi-1 targeting proteins, p16INK4a and p19ARF, compared with control and those treated with quercetin alone. Quercetin pre-treatment repressed the increased p16INK4a and p19ARF expression induced by doxorubicin and restored Bmi-1 expression to near normal levels (Figure 5B). Smartpool (Bmi-1 siRNA) was used to knockdown Bmi-1 to determine the role of Bmi-1 on the effects of quercetin in doxorubicin-treated H9c2 cells. Bmi-1 knockdown in H9c2 cells increased the number of apoptotic cells in the control and the cells treated with quercetin alone and now quercetin pre-treatment did not attenuate doxorubicin-induced apoptosis (Figure 5C). Similarly, Bmi-1 knockdown in H9c2 cells increased ROS generation in the control and the cells treated with quercetin alone and quercetin pre-treatment did not attenuate doxorubicin-induced ROS generation (Figure 5D). Furthermore, Bmi-1 overexpression in H9c2 cells significantly decreased doxorubicin-induced apoptosis from 25 to 10% (Figure 5E). quercetin pre-treatment further decreased the number to 6% in control cells and 2% in Bmi-1 overexpressed cells respectively. Thus, up-regulation of Bmi-1 expression accounted for the protective effects of quercetin against doxorubicin-induced cardiotoxicity in H9c2 cells.
Figure 5.
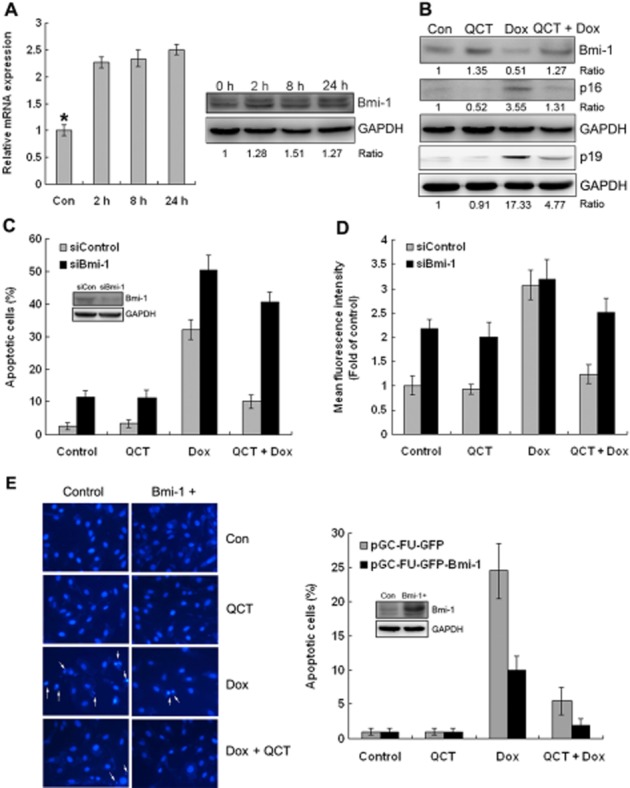
Quercetin (QCT) up-regulated Bmi-1 expression in H9c2 cells. (A) Effect of quercetin on Bmi-1mRNA and protein expression levels as a function of time. Quercetin treatment for 2 h increased Bmi-1 expression. *P < 0.05 each group versus the control. (B) H9c2 cells treated with doxorubicin (Dox; 5 μM) for 24 h with and without 100 μM quercetin pre-treatment and Western blot analysis was performed for Bmi-1, p16Ink4a, p19Arf and GAPDH served as the loading control. Quercetin inhibited doxorubicin-induced p16Ink4a and p19Arf expression and maintained Bmi-1 expression. (C, D) H9c2 cells were transfected with scrambled siRNA (siCon) or siBmi-1. After 2 days, the expressions of Bmi-1 and GAPDH (loading control) were determined using Western blot analysis. The cells were then treated with doxorubicin (5 μM) for 24 h with and without 100 μM quercetin pre-treatment. Apoptotic cells (C) and ROS generation (D) were analysed through flow cytometry using Annexin-V/PI and CM-H2DCFDA staining. n = 3 in each group. Quercetin had no protective effect in Bmi-1-depleted H9c2 cells. (E) Nuclear staining of H9c2 cells with Hoechst 33258. After infection with pGC-FU-GFP control or pGC-FU-GFP-Bmi-1, cells treated with doxorubicin (5 μM) for 24 h with and without 100 μM quercetin pre-treatment, then stained with Hoechst 33258 and visualized using a fluorescent microscope. The percentage of apoptotic cells was calculated by counting the number of the cells with nuclear condensation relative to the total number of the cells. At least 300 cells were counted in each group (data are representative of three independent cell cultures).
Quercetin attenuated doxorubicin-induced acute cardiotoxicity in mice
The general appearance of mice in all four groups was observed daily after the 20 mg·kg−1 doxorubicin treatment. Mice in the doxorubicin-treated group appeared weak, lethargic and lost weight, and 90% had died by 10 days after treatment (Figure 6A). However, these signs were less severe in the mice treated with doxorubicin + quercetin and mortality was reduced to 30% by 10 days. Quercetin alone caused no deaths over the observation period (30 days). Doxorubicin treatment significantly decreased the heart weight (34%) and the heart-to-body weight ratio (Figure 6B) 5 days after doxorubicin treatment compared with the control, but the heart-to-body weight ratio in the quercetin + doxorubicin group was similar to those of the control and the quercetin-treated groups. In accordance with these findings, serum CK, a marker of myocarditis, was significantly increased (P < 0.01) in the doxorubicin group compared with the control and the quercetin treatment groups (Figure 6C). The CK level was significantly decreased in the quercetin + doxorubicin group (P < 0.01) compared with the doxorubicin group. Furthermore, to determine the level of apoptosis in doxorubicin-treated hearts, a TUNEL assay was performed. As shown in Figure 6D, a significant larger number of TUNEL positive nuclei (about sixfold) were detected in doxorubicin-treated hearts compared with other groups, which showed very low level of TUNEL positive nuclei.
Figure 6.

Quercetin (QCT) attenuated doxorubicin-induced acute cardiac functional loss. (A) Survival curve for mice after doxorubicin (Dox) injection. Results were examined by a Kaplan–Meier survival curve analysis. n = 10 in each group. (B) Mice were treated with doxorubicin (20 mg·kg−1, i.p.) with and without quercetin pre-treatment (100 mg·kg−1, p.o.). Body weight and heart weight were recorded. HW, heart weight; BW, body weight; HW/BW, heart weight to body weight ratio. Quercetin attenuated doxorubicin-induced heart and body weight loss. n = 10–15 in each group, *P < 0.05 each group versus Dox. (C) Serum CK activity was measured in mice treated as (A). Quercetin reduced doxorubicin-induced CK activation. n = 5–10 in each group, **P < 0.01 each group versus Dox. (D) Heart sections were subjected to analysis for TUNEL. Quercetin reduced the number of TUNEL positive cardiomyocytes induced by doxorubicin treatment. n = 4–5 sets of hearts. (E) Mice were treated with a doxorubicin (10 mg·kg−1, i.p.) with or without pre-treatment of quercetin (100 mg·kg−1, p.o.). Representative echocardiographic images were taken after 5 days. (F) Cardiac function was measured after 5 days. LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension. Quercetin prevented the doxorubicin-induced loss of cardiac function. n = 5 in each group, * P < 0.05 each group versus Dox.
We evaluated the effects of quercetin on acute cardiac dysfunction using 10 mg·kg−1 doxorubicin because treatment with 20 mg·kg−1 induced high morbidity. As shown in Figure 6E and F, mice treated with quercetin alone did not exhibit abnormal cardiac function compared with the vehicle controls. Doxorubicin alone significantly caused cardiac dysfunction, which was characterized by decreased left ventricular ejection fraction, left ventricular fraction shortening, as well as increased left ventricular end-systolic dimension. Quercetin pre-treatment prevented doxorubicin-induced loss of cardiac function. Taken together, these results indicate that quercetin pre-treatment significantly attenuated doxorubicin-induced acute cardiotoxicity.
Quercetin reduced doxorubicin-induced oxidative stress in mice
ROS overproduction is involved in the pathogenesis of doxorubicin-induced cardiomyopathy (Sag et al., 2013). Therefore, we measured superoxide production via DHE staining of the frozen heart sections of mice. DHE is a fluorescent dye that specifically reacts with intracellular O2●− and is converted into the red fluorescent compound ethidium, which then binds irreversibly to double-stranded DNA and appears as punctate nuclear staining (Miller et al., 1998). O2●− was detected throughout the heart sections (Figure 7A). DHE fluorescence was more intense in the doxorubicin-treated hearts, but quercetin pre-treatment attenuated the fluorescence to normal levels, similar to those in the control. We then examined the effects of quercetin on lipid antioxidative mechanisms, as SOD activity and lipid peroxidation activity, as MDA and LDH in serum. As shown in Figure 7B, both MDA (+164%; P < 0.01) and LDH activities (+468%; P < 0.001) were markedly increased in the doxorubicin group compared with the control and quercetin-treated groups. Quercetin pre-treatment reduced MDA activity (by 31%; P < 0.05) and LDH activity (by 57%; P < 0.01). SOD activity was significantly down-regulated in the doxorubicin group compared with the placebo. By contrast, SOD expression was significantly increased (+18%; P < 0.05) in the quercetin + doxorubicin group compared with those in the doxorubicin-treated mice. These results demonstrate that quercetin depresses the high oxidative stress caused by doxorubicin treatment.
Figure 7.

Quercetin (QCT) reduced doxorubicin-induced oxidative stress in mice. (A) Mice were treated with doxorubicin (Dox; 20 mg·kg−1, i.p.) with and without quercetin pre-treatment (100 mg·kg−1, p.o.). Oxidative stress in heart sections was measured by DHE staining. These micrographs are representative images of DHE-stained heart sections from control, QCT, Dox, quercetin + doxorubicin mice. The relative fluorescence changes are shown in the adjacent panel. n = 5 in each group, **P < 0.01 each group versus Dox. (B) LDH, MDA and SOD activity in the serum were measured. quercetin attenuated doxorubicin-induced LDH and MDA activity and maintained SOD activity. n = 5–10 in each group, *P < 0.05, **P < 0.01 each group versus Dox.
Quercetin up-regulated Bmi-1 expression in mice
Mouse heart sections were assessed via immunohistochemistry (IHC) to investigate the effects of the various treatments on Bmi-1 expression (Figure 8A). Treatment with quercetin alone did not change the the proportion of Bmi-1 positive cells, compared with the vehicle control, whereas doxorubicin treatment decreased the proportion of Bmi-1 positive cells. Quercetin pre-treatment partly blocked this effect of doxorubicin. Western blot results confirmed that quercetin maintained Bmi-1 expression in heart tissues (Figure 8B).
Figure 8.
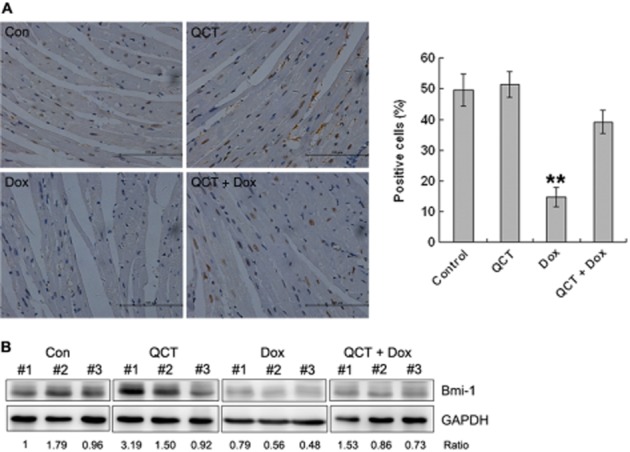
Quercetin (QCT) up-regulated Bmi-1 expression in mice. (A) Heart sections were subjected to immunohistochemical analysis for Bmi-1. Quercetin maintained Bmi-1 expression in the heart. n = 4–5 sets of hearts, **P < 0.01 each group versus Dox. (B) Heart tissues were subjected to Western blot analysis for Bmi-1. Quercetin attenuated doxorubicin-induced Bmi-1 reduction in hearts. Data are calculated as ratios of target protein levels to those of GAPDH. Ratios are compared with the ratios in control samples set at 1. n = 3 in each group.
Additional IHC analyses were performed on the liver sections. We showed in a previous study that quercetin attenuates doxorubicin-induced hepatotoxicity in mice. Similar trends in Bmi-1 expression in the liver were observed, with almost all the cells (>90%) being Bmi-1 positive in the control, QCT and the quercetin + doxorubicin groups, whereas fewer (<20%) Bmi-1 positive cells were present in the livers of doxorubicin-treated mice (Supporting Information Fig. S1). These results are consistent with our in vitro findings.
Discussion
Several mechanisms have been suggested to underlie doxorubicin-induced cardiotoxicity, such as mitochondrial dysfunction, enhanced ROS production and apoptosis caused by doxorubicin-induced DNA damage (Arola et al., 2000; Yoshida et al., 2009).
Cellular aerobic respiration requires functioning mitochondria, which are the main source of intracellular ROS. The potential sources of ROS in cardiomyocytes include mitochondrial electron transport and NADPH oxidase (Tsutsui et al., 2006). Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis (Green and Leeuwenburgh, 2002). Doxorubicin-induced mitochondrial dysfunction in cardiac tissue is characterized by inhibition of oxidative phosphorylation, decreased calcium-loading capacity and increased ROS production (Wallace, 2003; Oliveira et al., 2006; Kuznetsov et al., 2011; Pereira et al., 2011). Our results also showed doxorubicin-induced changes in mitochondrial morphology and MMP. Quercetin helped H9c2 cells maintain normal mitochondrial morphology and MMP and it regulated a series of Bcl-2 family genes (Bcl-2 and Bid) upstream of MMP. In this study, doxorubicin increased the mitochondrial superoxide and total cellular ROS levels in H9c2 cells and doxorubicin-treated mice hearts, and quercetin pre-treatment significantly reduced ROS generation in vitro and in vivo. Previous studies have shown that doxorubicin increased the expression and activity of NADHP oxidases, such as Nox1, Nox2, Nox4 and p47phox (Li et al., 2012; Park et al., 2012). Consistent with these earlier findings, doxorubicin increased Nox1 and p47phox mRNA expression, especially Nox1 expression in H9c2 cells and quercetin dramatically reduced Nox1 expression. A previous study showed that mitochondrial ROS is essential but not sufficient to promote cell death, which requires sustained ROS accumulation through the subsequent action of Nox1 (Lee et al., 2006). Thus, the protective effect of quercetin may be related to inhibition of NADHP oxidase, especially Nox1 expression and ROS generation.
DNA damage plays an important role in doxorubicin-induced lethal cardiomyocyte injury through a pathway involving p53 and the mitochondrial ROS generation (L’Ecuyer et al., 2006). Once activated by doxorubicin-induced oxidative stress, p53 translocates to the nucleus and induces the expression of genes that prevent cell division and causes apoptosis (e.g., Bid; Sax et al., 2002). Subsequently, this promotes mitochondrial ROS production and DNA damage, resulting in increased cell death (Lebrecht and Walker, 2007). p53 overexpression represses glutathione S-transferase α1, NAD(P)H, quinine oxidoreductase and cystine/glutamate transporter expression, and interferes with the antioxidant defences activated by Nrf2 (Faraonio et al., 2006). A recent study indicated that the selective loss of p53 in cardiomyocytes was insufficient to prevent doxorubicin-induced ROS generation and apoptosis (Feridooni et al., 2011), which suggests that additional p53-independent pathways are involved in doxorubicin-induced myocardial damage.
Our study demonstrated that quercetin also has potent antioxidant effects against doxorubicin-induced oxidative stress in vivo, as evidenced by the inhibition of ROS generation, reduction in lipid peroxidation activity (MDA and LDH) and restoration of SOD activity. As an important superoxide radical scavenger, SOD dismutates superoxide anion to hydrogen peroxide, which then can be detoxified by GSH/GSH-Px to yield reduced GSH. In addition to the down-regulated NADHP oxidase expression, we found that quercetin up-regulated Bmi-1 expression in vitro and in vivo, which participated in the cardioprotective effect of quercetin against doxrubicin.
Bmi-1, a polycomb group (PcG) protein, is important for the self-renewal of stem cells (Park et al., 2003). Bmi-1 regulates mitochondrial function by regulating the expression of genes related to mitochondrial function and ROS generation. Bmi-1-deficient cells have impaired mitochondrial function and increased intracellular ROS levels because of the dysregulation of the expression of genes involved in mitochondrial function. This result suggests that Bmi-1 directly regulates oxidative stress levels (Liu et al., 2009). Our previous study showed that Bmi-1 reduced ROS generation by irradiation and repressed the expression of Nox subunit gene in human keratinocytes (Dong et al., 2011). Bmi-1 regulates antioxidant defences in neurons by repressing p53 pro-oxidant activity (Chatoo et al., 2009). Bmi-1 negatively regulates the Ink4a/Arf locus, which encodes the tumour suppressor proteins p16INK4a and p19ARF (Molofsky et al., 2005). ROS signalling activates p38 and eventually induces the transcriptional repression of p16Ink4a and p19Arf (Ito et al., 2006). In the current study, quercetin enhanced Bmi-1 mRNA and protein expression in H9c2 and primary cardiomyocytes within 2 h, and the increased expression was maintained for at least 1 day. Quercetin also attenuated doxorubicin-induced decrease in Bmi-1 and inhibited the expression of p16INK4a and p19ARF. Thus, Bmi-1 up-regulation could partly explain the quercetin-induced inhibition of Nox gene expression (Nox1 and p47phox) and ROS generation.
Bmi-1 also functions in the DNA damage response pathway. Bmi-1 is a very early DNA damage response protein that accumulates at DNA DSB foci and promotes DSB repair (Ismail et al., 2010). Bmi-1 is required for DNA damage-induced ubiquitination of histone H2A at lysine 119, and the loss of Bmi-1 impairs the repair of DNA DSBs through homologous recombination (Ginjala et al., 2011). We found that doxorubicin induced DNA DSB, which was shown as increased γ-H2AX foci formation and comet tails. Quercetin pre-treatment significantly reduced DSB. Thus, up-regulated Bmi-1 expression also explains the quercetin-induced reduction in DSBs and the maintenance of DNA repair capacity of cardiomyocytes.
Bmi-1 overexpression in H9c2 cells significantly decreased doxorubicin-induced apoptosis; meanwhile, quercetin pre-treatment further decreased the number. This result confirmed that up-regulation of Bmi-1 by quercetin played an important role and suggested that there may be other mechanisms also involved in its cardioprotective effects. Quercetin pre-treatment significantly reduced the G/GO-induced apoptosis of mice thymocytes and suppressed DNA binding activity of redox state-sensitive transcription factors, such as NF-κB, AP-1 and p53 (Lee et al., 2003). We found that quercetin reduced doxorubicin-induced p53 expression, which could partly explain quercetin attenuation of doxorubicin-induced apoptosis. Studies showed that doxorubicin also induces inflammatory effects and increases pro-inflammatory cytokines (TNF-α, IL-1β and IL-2) in cardiomyocytes, and anti-inflammatory agents can reduce doxorubicin-induced cardiotoxicity (Bruynzeel et al., 2007). Quercetin reduced neuronal apoptosis induced by IL-1β through inhibition of inflammatory mediators, including IL-6, IL-8 and the chemokine CCL2 (MCP-1), and the increase in the expression of SOD and thioredoxin mediators (Sharma et al., 2007). Recently, quercetin reduced doxorubicin-induced TNF-α and protected rat cardiomyocytes (Matouk et al., 2013). Thus, the anti-inflammation effect of quercetin may, in part, be attributed to its cardioprotective effect against doxorubicin treatment.
Also, doxorubicin-mediated alteration of Ca2+ homeostasis may be another possible mechanism of cardiotoxicity. Doxorubicin-induced Ca2+ overload of cardiac cells causes mitochondrial calcium overloading, resulting in alteration of energy metabolism and promotion of ROS generation (Kim et al., 2006). Quercetin is known to inhibit Ca2+-dependent ATP hydrolysis, ATP-dependent Ca2+ uptake and chelator-induced Ca2+ release (Shoshan et al., 1980). We found that quercetin reduced doxorubicin-induced Ca2+ overload (Supporting Information Fig. S2). This may also be one of the possible mechanisms of cardioprotective effect of quercetin.
In conclusion, the protective role of quercetin against doxorubicin cardiotoxicity could be ascribed mainly to its antioxidant activity. The ability to decrease ROS levels and DNA damage and maintain cardiac cell viability could be due to quercetin-induced up-regulation of Bmi-1 expression. Thus, quercetin intake may counteract and prevent doxorubicin-induced cardiac stress.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81272493), the Science Technology Department of Zhejiang Province (Nos. 2012R10047 and 2012C33116), the 45th Scientific Research Foundation for Returned Scholars by Ministry of Education of China, the Zhejiang Educational Committee (No. Y201224108), the Health Bureau of Zhejiang Province (No. 2012RCB027) and the Fundamental Research Funds for the Central Universities, HUST (No. 2013TS084).
Glossary
- CK
creatine kinase
- DSB
double-strand break
- MDA
malondialdehyde
- MMP
mitochondrial membrane potential
- ROS
reactive oxygen species
- SOD
superoxide dismutase
Author contributions
Q. D., Q. L. and G. W. conceived and designed the experiment. Q. D., L. C., Q. L., L. L., S. M. and G. W. performed the experiments. Q. D., Q. L., S. S. and G. W. wrote the paper. G. W., Q. D. and S. S. analysed the data.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site: http://dx.doi.org/10.1111/bph.12795
Figure S1 Liver sections were subjected to immunohistochemical analysis for Bmi-1. Quercetin maintained Bmi-1 expression in the liver. n = 4–5 sets of livers.
Figure S2 Intracellular Ca2+ concentration was detected by Fluo 4-AM staining 2 h after doxorubicin treatment with and without 100 μM quercetin pre-treatment. Quercetin inhibited doxorubicin-induced Ca2+ overload.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1458. [Google Scholar]
- Angeloni C, Spencer JP, Leoncini E, Biagi PL, Hrelia S. Role of quercetin and its in vivo metabolites in protecting H9c2 cells against oxidative stress. Biochimie. 2007;89:73–82. doi: 10.1016/j.biochi.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–1792. [PubMed] [Google Scholar]
- Bruynzeel AM, Abou El Hassan MA, Schalkwijk C, Berkhof J, Bast A, Niessen HW, et al. Anti-inflammatory agents and monoHER protect against doxorubicin-induced cardiotoxicity and accumulation of CML in mice. Br J Cancer. 2007;96:937–943. doi: 10.1038/sj.bjc.6603640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatoo W, Abdouh M, David J, Champagne MP, Ferreira J, Rodier F, et al. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J Neurosci. 2009;29:529–542. doi: 10.1523/JNEUROSCI.5303-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Q, Oh JE, Chen W, Kim R, Kim RH, Shin KH, et al. Radioprotective effects of Bmi-1 involve epigenetic silencing of its target genes and enhanced DNA repair in normal human keratinocytes. J Invest Dermatol. 2011;131:1216–1225. doi: 10.1038/jid.2011.11. [DOI] [PubMed] [Google Scholar]
- Doroshow JH, Locker GY, Myers CE. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J Clin Invest. 1980;65:128–135. doi: 10.1172/JCI109642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertuğ PU, Aydinoglu F, Goruroglu Ozturk O, Singirik E, Ögülener N. Comparative study of the quercetin, ascorbic acid, glutathione and superoxide dismutase for nitric oxide protecting effects in mouse gastric fundus. Eur J Pharmacol. 2013;698:379–387. doi: 10.1016/j.ejphar.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Faraonio R, Vergara P, Di Marzo D, Pierantoni MG, Napolitano M, Russo T, et al. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem. 2006;281:39776–39784. doi: 10.1074/jbc.M605707200. [DOI] [PubMed] [Google Scholar]
- Feridooni T, Hotchkiss A, Remley-Carr S, Saga Y, Pasumarthi KB. Cardiomyocyte specific ablation of p53 is not sufficient to block doxorubicin induced cardiac fibrosis and associated cytoskeletal changes. PLoS ONE. 2011;6:e22801. doi: 10.1371/journal.pone.0022801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe P, Morlière P, Patterson LK, Hug GL, Mazière JC, Freitas JP. Oxygen-copper (II) interplay in the repair of semi-oxidized urate by quercetin bound to human serum albumin. Free Radic Res. 2004;38:295–301. doi: 10.1080/10715760310001626257. [DOI] [PubMed] [Google Scholar]
- Gibellini L, Pinti M, Nasi M, Montagna JP, De Biasi S, Roat E, et al. Quercetin and cancer chemoprevention. Evid Based Complement Alternat Med. 2011;2011:591356. doi: 10.1093/ecam/neq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieni RS, Ismail IH, Campbell S, Hendzel MJ. Polycomb group proteins in the DNA damage response: a link between radiation resistance and ‘stemness’. Cell Cycle. 2011;10:883–894. doi: 10.4161/cc.10.6.14907. [DOI] [PubMed] [Google Scholar]
- Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M, et al. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol. 2011;31:1972–1982. doi: 10.1128/MCB.00981-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PS, Leeuwenburgh C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochim Biophys Acta. 2002;1588:94–101. doi: 10.1016/s0925-4439(02)00144-8. [DOI] [PubMed] [Google Scholar]
- Ismail IH, Andrin C, McDonald D, Hendzel MJ. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol. 2010;191:45–60. doi: 10.1083/jcb.201003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- Kaiserova H, Simunek T, van der Vijgh WJ, Bast A, Kvasnickova E. Flavonoids as protectors against doxorubicin cardiotoxicity: role of iron chelation, antioxidant activity and inhibition of carbonyl reductase. Biochim Biophys Acta. 2007;1772:1065–1074. doi: 10.1016/j.bbadis.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem. 2002;234–235:119–124. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Kim SJ, Kim BJ, Rah SY, Chung SM, Im MJ, et al. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp Mol Med. 2006;38:535–545. doi: 10.1038/emm.2006.63. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Margreiter R, Amberger A, Saks V, Grimm M. Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochim Biophys Acta. 2011;1813:1144–1152. doi: 10.1016/j.bbamcr.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Kyaw M, Yoshizumi M, Tsuchiya K, Izawa Y, Kanematsu Y, Tamaki T. Atheroprotective effects of antioxidants through inhibition of mitogen-activated protein kinases. Acta Pharmacol Sin. 2004;25:977–985. [PubMed] [Google Scholar]
- Lebrecht D, Walker UA. Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7:108–113. doi: 10.1007/s12012-007-0009-1. [DOI] [PubMed] [Google Scholar]
- Lee JC, Kim J, Park JK, Chung GH, Jang YS. The antioxidant, rather than prooxidant, activities of quercetin on normal cells: quercetin protects mousethymocytes from glucose oxidase-mediated apoptosis. Exp Cell Res. 2003;291:386–397. doi: 10.1016/s0014-4827(03)00410-5. [DOI] [PubMed] [Google Scholar]
- Lee SB, Bae IH, Bae YS, Um HD. Link between mitochondria and NADPH oxidase 1 isozyme for the sustained production of reactive oxygen species and cell death. J Biol Chem. 2006;281:36228–36235. doi: 10.1074/jbc.M606702200. [DOI] [PubMed] [Google Scholar]
- L’Ecuyer T, Sanjeev S, Thomas R, Novak R, Das L, Campbell W, et al. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am J Physiol Heart Circ Physiol. 2006;291:H1273–H1280. doi: 10.1152/ajpheart.00738.2005. [DOI] [PubMed] [Google Scholar]
- Li JZ, Yu SY, Wu JH, Shao QR, Dong XM. Paeoniflorin protects myocardial cell from doxorubicin-induced apoptosis through inhibition of NADPH oxidase. Can J Physiol Pharmacol. 2012;90:1569–1575. doi: 10.1139/y2012-140. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu C, Chen J, Song S, Lee IH, Quijano C, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matouk AI, Taye A, Heeba GH, El-Moselhy MA. Quercetin augments the protective effect of losartan against chronic doxorubicin cardiotoxicity in rats. Environ Toxicol Pharmacol. 2013;36:443–450. doi: 10.1016/j.etap.2013.05.006. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FJ, Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- Mojzisová G, Sarisský M, Mirossay L, Martinka P, Mojzis J. Effect of flavonoids on daunorubicin-induced toxicity in H9c2 cardiomyoblasts. Phytother Res. 2009;23:136–139. doi: 10.1002/ptr.2566. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–1437. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J, Günther G, Zanocco AL, Lemp E. Singlet oxygen reactions with flavonoids. A theoretical-experimental study. PLoS ONE. 2012;7:e40548. doi: 10.1371/journal.pone.0040548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Ba’tkai S, Kashiwaya Y, Hasko G, Liaudet L, et al. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol. 2009;296:1466–1483. doi: 10.1152/ajpheart.00795.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira PJ, Santos MS, Wallace KB. Doxorubicin-induced thiol-dependent alteration of cardiac mitochondrial permeability transition and respiration. Biochemistry (Mosc) 2006;71:194–199. doi: 10.1134/s000629790602012x. [DOI] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- Park J, Park E, Ahn BH, Kim HJ, Park JH, Koo SY, et al. NecroX-7 prevents oxidative stress-induced cardiomyopathy by inhibition of NADPH oxidase activity in rats. Toxicol Appl Pharmacol. 2012;263:1–6. doi: 10.1016/j.taap.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Research. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei TX, Xu CQ, Li B, Zhang ZR, Gao XX, Yu J, et al. Protective effect of quercetin against adriamycin-induced cardiotoxicity and its mechanism in mice. Yao Xue Xue Bao. 2007;42:1029–1033. [PubMed] [Google Scholar]
- Peng X, Chen B, Lim CC, Sawyer DB. The cardiotoxicology of anthracycline chemotherapeutics: translating molecular mechanism into preventative medicine. Mol Interv. 2005;5:163–171. doi: 10.1124/mi.5.3.6. [DOI] [PubMed] [Google Scholar]
- Pereira GC, Silva AM, Diogo CV, Carvalho FS, Monteiro P, Oliveira PJ. Drug induced cardiac mitochondrial toxicity and protection: from doxorubicin to carvedilol. Curr Pharm Des. 2011;17:2113–2129. doi: 10.2174/138161211796904812. [DOI] [PubMed] [Google Scholar]
- Sag CM, Wagner S, Maier LS. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic Biol Med. 2013;63:338–349. doi: 10.1016/j.freeradbiomed.2013.05.035. [DOI] [PubMed] [Google Scholar]
- Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–849. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- Sharma V, Mishra M, Ghosh S, Tewari R, Basu A, Seth P, et al. Modulation of interleukin-1 beta mediated inflammatory response in human astrocytes by flavonoids: implications in neuroprotection. Brain Res Bull. 2007;3:55–63. doi: 10.1016/j.brainresbull.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Shoshan V, Campbell KP, MacLennan DH, Frodis W, Britt BA. Quercetin inhibits Ca2+ uptake but not Ca2+ release by sarcoplasmic reticulum in skinned muscle fibers. Proc Natl Acad Sci USA. 1980;77:4435–4438. doi: 10.1073/pnas.77.8.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber JH, Barber G. Doxorubicin-induced cardiotoxicity. N Engl J Med. 1995;333:1359–1360. [PubMed] [Google Scholar]
- Staedler D, Idrizi E, Kenzaoui BH, Juillerat-Jeanneret L. Drug combinations with quercetin: doxorubicin plus quercetin in human breast cancer cells. Cancer Chemother Pharmacol. 2011;68:1161–1172. doi: 10.1007/s00280-011-1596-x. [DOI] [PubMed] [Google Scholar]
- Steinherz LJ, Steinherz PG, Tan CT, Heller G, Murphy ML. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA. 1991;266:1672–1677. [PubMed] [Google Scholar]
- Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006;8:1737–1744. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003;93:105–115. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- Wang G, Zhang J, Liu L, Sharma S, Dong Q. Quercetin potentiates doxorubicin mediated antitumor effects against liver cancer through p53/Bcl-xl. PLoS ONE. 2012;12:e51764. doi: 10.1371/journal.pone.0051764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Shiojima I, Ikeda H, Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol. 2009;47:698–705. doi: 10.1016/j.yjmcc.2009.07.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Liver sections were subjected to immunohistochemical analysis for Bmi-1. Quercetin maintained Bmi-1 expression in the liver. n = 4–5 sets of livers.
Figure S2 Intracellular Ca2+ concentration was detected by Fluo 4-AM staining 2 h after doxorubicin treatment with and without 100 μM quercetin pre-treatment. Quercetin inhibited doxorubicin-induced Ca2+ overload.