

Abstract
Background and Purpose
Finding new indications for existing drugs, also known as drug repositioning or repurposing, is a powerful approach to accelerate drug discovery and development. The unfolded protein response pathways have been proposed to be a viable target for developing new anticancer drugs.
Experimental Approach
We screened the Johns Hopkins Drug Library for inhibitors of prostate cancer cell proliferation to identify new antiprostate cancer treatments among known drugs. We systematically investigated the mechanism underlying the anticancer activity of a hit and assessed its efficacy in blocking prostate tumour growth in a mouse model.
Key Results
The antibacterial drug clofoctol was identified as a novel inhibitor of prostate cancer cell proliferation. Morphologically, cells treated with clofoctol were found to undergo massive vacuolization, reminiscent of endoplasmic reticulum stress. Indeed, all three unfolded protein response pathways including inositol requiring enzyme 1, double-stranded RNA-activated PK-like ER kinase and activating transcription factor 6 were found to be activated by clofoctol. Activation of unfolded protein response pathways by clofoctol led to the inhibition of protein translation in cells and the induction of G1 cell cycle arrest in prostate cancer cells. Clofoctol also inhibited prostate cancer xenograft growth in vivo without apparent toxicity.
Conclusion and Implications
Our findings revealed clofoctol as a novel activator of the unfolded protein response pathways and a promising inhibitor of prostate cancer. As clofoctol has been used in the clinic for years, it is ready for clinical evaluation as a novel antiprostate cancer drug candidate.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| eIF2α | Thapsigargin |
| IRE1 | SP600125 |
| PERK | Cycloheximide |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Drug discovery and development is costly and a time-consuming process. Discovering new pharmacological activity among known drugs, or drug repositioning, allows for dramatic acceleration of the discovery and development of new drugs (Ashburn and Thor, 2004). Existing drugs have favourable pharmacokinetic and pharmacodynamic properties with tolerable side effects. Thus, old drugs can quickly enter human clinical testing for newly identified indications using the existing drug dosage regimen. Over 10 years ago, we began to assemble a library of clinical drugs, dubbed the Johns Hopkins Drug Library (JHDL), which contains approximately 3000 drugs that have been approved by either the US Food and Drug Administration or its foreign equivalents, or are under the late phases of clinical trials (Chong et al., 2006b). We have screened the JHDL in a variety of biological assays and disease models, including cancers, diabetes and infectious diseases, and identified a number of promising hits (Chong et al., 2006a,b; 2007; McMahon et al., 2007; Ren et al., 2008; Zhang et al., 2008; 2009; Lee et al., 2009; Kim et al., 2010; Shim et al., 2010; 2012; Lin et al., 2011; Platz et al., 2011; Rovira et al., 2011; Liu-Chittenden et al., 2012; Choi et al., 2013). Among the hits is the antifungal drug itraconazole that was identified as a potent inhibitor of angiogenesis and the hedgehog signalling pathway respectively (Chong et al., 2007; Kim et al., 2010). To date, itraconazole has entered multiple phase 2 clinical studies and shown promise in treating both non-small cell lung cancer and prostate cancer (Antonarakis et al., 2011; Rudin et al., 2013). One of the most potent hits from our screen for inhibitors of prostate cancer is digoxin, a cardiac glycoside, which was also identified as a potent inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α) (Zhang et al., 2008; Platz et al., 2011). These findings prompted us to conduct a large-scale cohort study with male patients having heart diseases who have taken digoxin for 5–10 years. Digoxin was found to significantly reduce prostate cancer incidence among patients who have used it compared with the control group. These findings suggest that digoxin is a promising drug candidate for the prevention and treatment of prostate cancer. Emerging from the same screen for inhibitors of prostate cancer is the antibacterial drug clofoctol that is likely to work through the activation of the unfolded protein response (UPR) pathway in the endoplasmic reticulum (ER).
The ER is a major cellular organelle in which folding and maturation of secretory proteins take place (Rutkowski and Kaufman, 2004; Ron and Walter, 2007). It plays an important role in the quality control of proteins, determining whether proteins are properly folded, which allows proteins to reach their final destination. When proteins are misfolded, they are retained in the ER and delivered into cytosol for proteosomal degradation in a process known as ER-associated degradation (Bernasconi and Molinari, 2011). If misfolded proteins accumulate over the limit of the capacity of ER-associated degradation, a process called UPR is activated (Sidrauski et al., 1998). UPR is a cellular protective mechanism to cope with an excess of misfolded proteins in the ER. UPR is activated by one of three sensing proteins including inositol requiring enzyme 1 (IRE1), double-stranded RNA-activated PK-like ER kinase (PERK) and activating transcription factor 6 (ATF6) (Walter and Ron, 2011). These three UPR pathways activate the transcription of UPR target genes which increase the protein folding capacity in the ER. In addition to transcriptional activation, they also activate signalling pathways which down-regulate protein translation and enhance the degradation of ER-bound mRNAs to eventually reduce the ER folding load (Harding et al., 2000).
The inhibition of protein translation under UPR conditions is governed by PERK that phosphorylates eukaryotic translation initiation factor 2α (eIF2α) (Harding et al., 2000). The phosphorylation of eIF2α inhibits the formation of the 43S translation initiation complex and, thereby blocking global translation. Inhibition of protein translation largely affects short-lived proteins such as cyclin D1, a rate-limiting regulator of cell cycle progression during the G1 phase. Activation of UPR decreases cyclin D1 translation, thereby inhibiting cyclin D-dependent kinase activity. This also leads to a concomitant inhibition of cyclin E- and A-dependent kinases, which culminate in G1 phase cell cycle arrest (Brewer et al., 1999). Upon prolonged activation of UPR, cells cannot alleviate ER stress and undergo cell death. If cells fail to establish homeostasis under ER stress, UPR activates apoptotic and/or autophagic cell death to remove these cells from the body to protect the entire organism (Woehlbier and Hetz, 2011). Highly proliferative cancer cells have a higher demand for ER to synthesize more proteins to keep up with the increased metabolic demand compared with normal cells (Suh et al., 2012). The more proteins the ER processes, the larger the amount of misfolded proteins that accumulate in the ER. Several lines of evidence suggest that many types of cancer cells have higher levels of ER stress and as a consequence, the UPR pathways are already active to maintain cellular homeostasis (Luo and Lee, 2013). Therefore, further increasing the ER stress, to overload the machinery of stress responses in cancer cells that can in turn induce cell cycle arrest or cell death, is recognized as an alternative approach to selectively inhibit cancer cells (Leleu et al., 2009; Suh et al., 2012). Several anticancer agents under development, including heat shock protein 90 (HSP90) inhibitors and proteasome inhibitors, have been shown to induce ER stress in cancer cells and have entered phase I/II clinical trials for cancer therapy (Obeng et al., 2006; Davenport et al., 2007).
Clofoctol is an antibiotic that has been used for the treatment of upper respiratory tract infections in France and Italy among other countries for decades (Danesi and Del Tacca, 1985). The proposed mechanism for its antibacterial activity is the alteration of bacterial membrane permeability owing to its hydrophobic nature (Yablonsky, 1983). Recently, it was reported that clofoctol inhibits protein translation in mammalian cells and as such, could serve as a potential anticancer drug (Pelletier et al., 2007). However, the mechanism by which clofoctol inhibits translation and whether this inhibition of translation is responsible for its anticancer activity is not known. In the present study, we demonstrated that clofoctol inhibits the growth of prostate cancer cells at clinically achievable concentrations. It was also found to induce ER stress and activate all three UPR pathways in prostate cancer cells, which led to indirect inhibition of protein translation. Inhibition of protein translation by clofoctol caused a decrease in the levels of cyclins that are critical for G1/S transition, leading to G1 cell cycle arrest. We further demonstrated that clofoctol was effective in an animal xenograft model of prostate cancer. Taken together, these results suggest that clofoctol is a novel small molecule activator of the UPR pathway that has potential as a new therapeutic agent for cancer.
Methods
Cell culture
LNCaP, C42B, PC-3 and CWR22Rv1 were grown in RPMI 1640 media plus 10% FBS (Life Technologies, Grand Island, NY, USA). DU145 were grown in DMEM with 10% FBS (Life Technologies). LAPC4 cells were grown in Iscove’s media with 10% FBS (Life Technologies) and 1 nM R1881. All cell lines were maintained at 37°C in a humidified incubator adjusted with 5% CO2. Authentication of these cell lines was carried out by the Powerplex 2.1 STR genotyping assay (Promega, Madison, WI, USA) as described previously (Platz et al., 2011).
Cell viability assay
Approximately 2 × 103 cells per well were seeded in 96-well plates with a total media volume of 100 μL per well and allowed to adhere overnight. The cells were treated with various concentrations of clofoctol for 72 h before incubation with 10 μL per well of alamarBlue dye (Life Technologies) for 2 h. The alamarBlue fluorescence from cells was measured at 560EX nm/590EM nm using a BMG FLUOStar OPTIMA plate reader (BMG Labtech, Cary, NC, USA). The IC50 values were calculated using the GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA, USA).
Cell cycle analysis
PC3 cells were plated in a 6-well plate or in 100-mm dishes and allowed to adhere overnight. After treatment with drugs for 24 h, the cells were harvested by trypsinization. The cell were then fixed with ice-cold 70% ethanol with gentle vortexing and stained with 20 μg·mL−1 propidium iodide in the presence of 0.2 μg·mL−1 RNase A for 30 min. The cells were passed through a mesh filter and subjected to cell cycle analysis using BD FACScan System (BD Biosciences, San Jose, CA, USA).
In vitro translation assay
In vitro translation of luciferase mRNA was conducted using Flexi® Rabbit Reticulocyte Lysate System (Promega) according to the manufacturer’s instructions. Briefly, rabbit reticulocyte lysate was incubated with 20 μM amino acid mixture, 70 mM KCl, 2 mM DTT, 0.4 U RNase inhibitor and 20 μg·mL−1 luciferase mRNA. Drugs (clofoctol or cycloheximide) were added to the reaction mixture and the reaction was continued at 30°C for 30 min before lysis buffer was added to stop the reaction. An aliquot of the reaction mixture was mixed with the luciferase substrate solution and the luciferase activity was measured using the MicroBeta luminescence plate reader (PerkinElmer, Waltham, MA, USA).
In vivo translational assay
PC3 cells (6 × 104 cells per well) were plated in 12-well plates and allowed to adhere overnight. The cells were then treated with clofoctol at the indicated concentrations. The cells were metabolically labelled with [35S]-methionine by adding the mixture of 1 μCi [35S]-methionine/Cys (PerkinElmer), per well, for 15 min. The cells were lysed with RIPA buffer containing 50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1× protease inhibitor cocktail, 1 mM Na3VO4 and 1 mM NaF. Equal amounts of total proteins were resolved by SDS-PAGE and were stained with Coomassie brilliant blue. The gels were destained and dried and subjected to the autoradiography.
X-box binding protein-1 (XBP-1) splicing assay
PC3 cells (1.5 × 105 cells per well) were plated in 6-well plates and allowed to adhere overnight. The cells were treated with various concentrations of clofoctol or 1 μM thapsigargin for 24 h. Total RNA were harvested using RNeasy mini kit (Qiagen, Valencia, CA, USA) and subjected to a reverse transcriptase-PCR analysis using specific primer pairs for XBP-1 (forward: 5′-AAACAGAGTAGCAGCTCAGACTGC-3′ and reverse: 5′-TCCTTCTGGGTAGACCTCTGGGAG-3′). The PCR products were resolved on 1.5% agarose gel and observed under the Kodak Image Station 440CF (Kodak, Rochester, NY, USA). To further distinguish the unspliced XBP-1 mRNA from the spliced form, the PCR products of XBP-1 mRNA were digested with PstI before gel electrophoresis.
Western blot analysis
Cells were lysed by adding 1 volume of 2× Laemmli buffer followed by boiling in water for 10 min. The samples were separated by SDS-PAGE and transferred onto nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). Proteins were detected using primary antibodies for eIF2α, transcription factor C/EBP homologous protein (CHOP), BiP, phospho-c-Jun, microtubule-associated protein light chain 3B (LC3B), PARP (Cell Signaling Technology, Danvers, MA, USA), phospho-eIF2α (Enzo Life Sciences, Farmingdale, NY, USA) and α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA); followed by incubation with HRP–conjugated antimouse or antirabbit antibodies (GE Healthcare, Pittsburgh, PA, USA) and enhanced chemiluminescence (GE Healthcare).
Confocal microscopy
Human pEGFP-LC3 (Addgene plasmid 24920) was obtained from Addgene (Cambridge, MA, USA) (Lee et al., 2008). PC3 cells (1 × 105) were seeded on a MatTek glass bottom culture dish (Fisher Scientific, Pittsburgh, PA, USA) and allowed to adhere for 24 h. The cells were transiently transfected with pEGFP-LC3 using the SuperFect transfection reagent (Qiagen) for 24 h. The cells were then treated with either DMSO or drugs for 6 h and the cellular EGFP fluorescence was observed under the Zeiss 510 Meta multiphoton confocal microscope (Carl Zeiss, Thornwood, NY, USA).
In vivo prostate cancer xenograft assay
Male athymic nude mice (BALB/c, nu/nu-NCr) aged 4–6 weeks were purchased from the National Cancer Institute (Frederick, MD, USA) and treated in accordance with Johns Hopkins Animal Care and Use Committee procedures. The study protocol was approved by the Johns Hopkins University Animal Care and Use Committee (MO10M221). All studies involving animals were conducted and reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Animals were housed in temperature- and humidity-controlled rooms on a 12 h light-dark cycle and had free access to food and water. A total of 13 animals were used in the experiments described herein. PC3 cells (1 million cells per injection) were implanted s.c. into the mice. After the tumours had become palpable, mice were randomly assigned to two groups. One group of mice (n = 6) were treated with vehicle (peanut oil with 5% DMSO) and the other group (n = 7) were treated with clofoctol (175 mg·kg−1, injected i.p.) every day. The tumour volume was measured periodically using a vernier caliper and calculated according to the modified ellipsoid formula: tumour volume (mm3) = (short axis)2 × (long axis) × π/6. After 37 days of treatment, mice were killed by an i.p. injection of 0.5 mL of 250 mg·kg−1 tribromoethanol (Sigma-Aldrich, St. Louis, MO, USA), followed by treatment with 100% CO2 when fully anaesthetized. Death was verified by an observed lack of breathing, below normal body temperature and absence of a palpable heartbeat. The tumours were harvested for wet weight measurement.
Statistical analysis
The statistical significance of differences between control and experimental groups was determined by Student’s t-test. All statistical tests were two-sided and P values less than 0.05 were considered statistically significant.
Results
Clofoctol inhibited prostate cancer cell growth and cell cycle progression
We have previously screened JHDL using two prostate cancer cell lines, LNCaP and PC3, to identify potential antiprostate cancer drugs (Platz et al., 2011). Clofoctol, an antibacterial drug, was among the hits that showed inhibitory activity against prostate cancer cell proliferation (Supporting Information Figure S1A). We thus tested clofoctol on six different prostate cancer cell lines including LNCaP, DU145, PC3, LAPC4, CWR22Rv1 and C42B. Clofoctol was active against all six prostate cancer cell lines with IC50 values ranging from 10 to 15 μM (Figure 1A and B). We further determined the effect of clofoctol on cell cycle progression of the PC3 line and found that it induced a G1 arrest (Figure 1C). Although the potency of clofoctol for inhibition of prostate cancer cell proliferation appeared relatively low in comparison with other hits (Platz et al., 2011), the average plasma concentrations of clofoctol in humans are significantly higher than its IC50 values for inhibition of prostate cancer cell growth (Del Tacca et al., 1987; Danesi et al., 1988), giving rise to a sufficiently wide therapeutic window and prompting us to characterize the antiproliferative activity on prostate cancer further.
Figure 1.
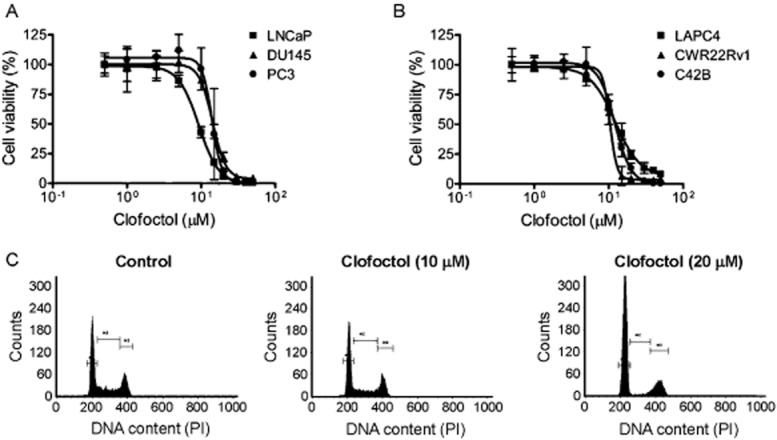
Clofoctol inhibited prostate cancer cell growth and cell cycle. (A and B) Six prostate cancer cell lines were grown in 96-well plates and treated with various concentrations of clofoctol for 72 h. An AlamarBlue assay was conducted to assess cell viability. Data represent mean + 95% confidence interval from three independent experiments performed in triplicate. (C) PC3 cells were grown in a 6-well plate and treated with clofoctol at indicated concentrations for 24 h. The cells were then harvested and subjected to cell cycle analysis. Representative cell cycle histograms from three independent experiments with similar results are shown.
Clofoctol induced ER stress and activated UPR pathways
To investigate the cellular and molecular mechanisms underlying the anticancer activity of clofoctol, we began by assessing the morphological changes of PC3 cells upon treatment with clofoctol. Clofoctol induced massive vacuolization in PC3 cells upon treatment for 24 h (Supporting Information Figure S1B). The induction of extensive cellular vacuolization and the G1 cell cycle arrest (Figure 1C) caused by clofoctol are both hallmarks of ER stress. We thus determined the effect of clofoctol on the activity of UPR pathways using ER stress markers in PC3 cells. We first determined the effect of clofoctol on the processing of XBP-1, which is a transcription factor that regulates expression of genes important for ER stress response (Calfon et al., 2002). When UPR is activated, IRE1 oligomerizes and activates its ribonuclease domain which catalyzes splicing of ubiquitously expressed form of XBP-1 mRNA (XBP-1u). This process will generate a spliced isoform of XBP-1 mRNA (XBP-1s), which serves as a marker of IRE1 activation. Using reverse transcriptase-PCR, we observed that clofoctol increased the splicing of XBP-1 mRNA in PC3 cells in a dose-dependent manner (Figure 2A). Unspliced form of XBP-1 mRNA contains a PstI digestion site that is removed upon splicing. We observed that the PstI digestion products from XBP-1 mRNA were dose-dependently decreased by clofoctol (Figure 2B). These results demonstrate that clofoctol activates IRE1 pathway in PC3 cells.
Figure 2.
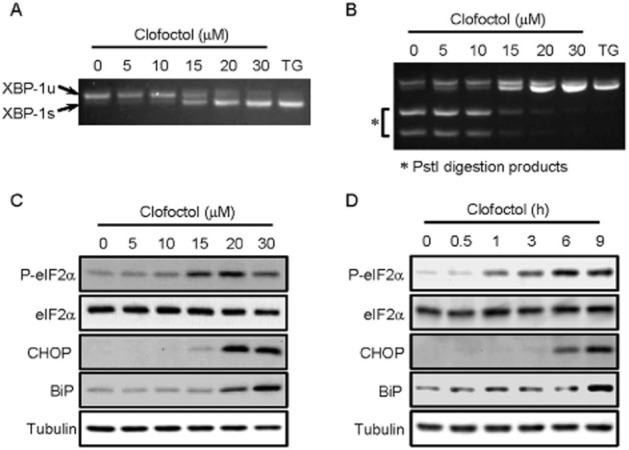
Clofoctol activates three UPR pathways in PC3 cells. (A and B) PC3 cells were treated with the indicated concentrations of clofoctol or thapsigargin (TG, 1 μM) for 24 h. RT-PCR analysis of XBP-1 mRNA was performed to observe the spliced form (XBP-1s) and unspliced form (XBP-1u) of XBP-1 in PC3 cells (A). Thapsigargin was used as a positive control compound for XBP-1 splicing. The RT-PCR products of XBP-1 mRNA were digested with PstI to further distinguish the XBP-1s from XBP-1u. XBP-1s does not contain a PstI digestion site, thus showing no digestion products (B). (C and D) PC3 cells were treated with various concentrations of clofoctol for 24 h (C). PC3 cells were treated with 20 μM clofoctol for indicated time points (D). The levels of phosphorylated and total proteins were assessed by Western blot analysis.
The second branch of UPR is mediated by PERK. PERK undergoes oligomerization and phosphorylates eIF2α, thereby inhibiting protein translation. In PC3 cells, clofoctol dose- and time-dependently increased the level of phosphorylated eIF2α (Figure 2C and D). It is known that when eIF2α is inhibited, certain mRNA, such as ATF4, that contains short open reading frames in their 5′-untranslated region are preferentially translated. ATF4 is a transcription factor that drives the expression of CHOP (Walter and Ron, 2011). In parallel with the induction of eIF2α phosphorylation, clofoctol up-regulated CHOP expression in a dose- and time-dependent manner (Figure 2C and D) consistent with the activation of the PERK-eIF2α-ATF4-CHOP signalling pathway.
The third branch of UPR involves the activation of ATF6 by two Golgi proteases, S1P and S2P that sequentially remove the luminal domain and the transmembrane anchor of ATF6 (Ye et al., 2000). The activated N-terminal fragment of ATF6 translocates into the nucleus and activates its target genes such as BiP. As shown in Figure 2C and D, clofoctol increased the expression of BiP in dose- and time-dependent manner. Activation of ATF6 pathway by clofoctol was further demonstrated by the nuclear translocation of the ATF6 fragment in PC3 cells (Supporting Information Figure S2). Together, these results indicate that clofoctol induces ER stress and activates all three UPR pathways in PC3 cells.
Clofoctol did not induce apoptosis or autophagy
Activation of IRE1 is known to activate JNK pathway, which eventually leads to an induction of apoptosis or autophagy during ER stress. We thus determined if clofoctol activated JNK pathway and autophagy in PC3 cells. Treatment of PC3 cells with clofoctol dose-dependently increased the level of phosphorylated c-Jun (Figure 3A). Conversion of LC3B-I to LC3B-II, a marker of autophagy induction, was also observed by increasing concentrations of clofoctol. In contrast, cleavage of PARP, a marker of caspase-dependent apoptosis, was not observed upon clofoctol treatment. To further determine whether clofoctol induced autophagy, we detected the autophagosome formation using EGFP-LC3-overexpressing PC3 cells. An increase in fluorescently labelled LC3 puncta together with formation of massive vacuoles was observed in the presence of clofoctol (Figure 3B). We next examined whether the induction of autophagy is mediated by JNK activation. PC3 cells were treated with a specific JNK inhibitor, SP600125 and the levels of phosphorylated c-Jun and LC3B were measured. The level of phosphorylated c-Jun was significantly increased by clofoctol and this increase was completely blocked by addition of SP600125 (Figure 3C). The conversion of LC3B by clofoctol, however, was not blocked by SP600125 (Figure 3C). These results suggested that JNK was not involved in the clofoctol-induced LC3B conversion. As inhibition of JNK did not affect growth inhibitory activity of clofoctol (Supporting Information Figure S3C), we wondered whether clofoctol-induced LC3B conversion was the indication of bona fide autophagy induction.
Figure 3.

Clofoctol inhibits autophagic flux in PC3 cells. (A) PC3 cells were treated with the indicated concentrations of clofoctol for 24 h. The protein levels of phosphorylated c-Jun, LC3B-I/II, PARP and tubulin were assessed by western blot analysis. (B) PC3 cells were transiently transfected with pEGFP-LC3 for 24 h and were treated with DMSO or clofoctol (20 μM) for 6 h. The EGFP-LC3 fluorescence was observed under the confocal microscope. Intense green dots are indicative of LC3 puncta. (C) PC3 cells were treated with indicated concentrations of clofoctol (CFT) and a JNK inhibitor, SP600125 (SP) for 24 h. The protein levels of PARP, phosphorylated c-Jun, LC3B-I/II and tubulin were assessed by Western blot analysis. (D) PC3 cells were treated with either DMSO (Ctrl), 20 μg·mL−1 tunicamycin (TM), 3 μM thapsigargin (TG) or 25 μM clofoctol (CFT) for 6 h and then were harvested for Western blot analysis of LC3B-I/II. Bafilomycin A1 (BAF-A1, 100 nM) was added 2 h before harvesting the cells. (E) PC3 cells were transiently transfected with pEGFP-LC3 for 24 h and were treated with either DMSO (Ctrl), 20 μg·mL−1 tunicamycin (TM), 3 μM thapsigargin (TG) or 25 μM clofoctol (CFT) for 6 h. Either DMSO (–BAF-A1) or bafilomycin A1 (+BAF-A1) was added 2 h before processing for the confocal microscope analysis of EGFP-LC3.
The autophagosome formation is a dynamic process as it is balanced by turnover through fusion with lysosomes and subsequent lysosome-mediated degradation. Inhibition of either fusion with lysosomes or lysosome-mediated degradation should prevent LC3 from degradation, which also causes increase in LC3 staining. Under these conditions, basal autophagic flux is inhibited but LC3 is accumulated, which often leads to misinterpretation of the autophagy-inducing activity of compounds. Thapsigargin, for example, had been known to induce autophagy as it is capable of increasing LC3 punta (Ogata et al., 2006). Subsequently, it was found to inhibit fusion between autophagosomes and lysosomes, thereby blocking basal autophagy (Ganley et al., 2011). An inhibitor of vacuolar type H+-ATPase (V-ATPase), bafilomycin A1, is known to raise lysosomal pH and inhibit lysosome-mediated degradation (Klionsky et al., 2008). Bafilomycin A1 also blocks basal autophagy while significantly increasing LC3 punta. We thus investigated whether clofoctol is a genuine inducer of autophagy. PC3 cells treated with bafilomycin increased LC3B conversion, indicating blockade of LC3 degradation (Figure 3D). Tunicamycin, an inhibitor of N-linked glycosylation and a genuine autophagy inducer, slightly increased the conversion of LC3B. The LC3B conversion induced by tunicamycin was further enhanced by bafilomycin, indicating that tunicamycin induced autophagic flux with active LC3B turnover. In contrast, thapsigargin and clofoctol strongly increased LC3B conversion with no further enhancement by bafilomycin. Similar results were observed from the LC3 staining. Cells treated with tunicamycin increased the number of LC3 puncta which were further enhanced by bafilomycin (Figure 3E). However, there was no significant enhancement in the number of LC3 puncta by bafilomycin in the cells treated with either thapsigargin or clofoctol. These results suggested that the increase in LC3B conversion by clofoctol was due to a blockade of basal autophagic flux rather than an induction of autophagy.
Clofoctol inhibited prostate cancer cell growth through induction of ER stress
To further investigate the underlying mechanism of anticancer activity of clofoctol, we applied several pathway inhibitors together with clofoctol and determined if they could rescue growth inhibitory activity of clofoctol. Thus, neither a specific autophagy inhibitor, 3-methyladenine nor apoptosis inhibitor, zVAD-fmk, affected the growth inhibition by clofoctol (Supporting Information Figure S3A and B), indicating that neither autophagy nor caspase-dependent apoptosis was involved in the anticancer activity of clofoctol. As mentioned previously, a JNK inhibitor SP600125 failed to rescue the growth inhibition by clofoctol (Supporting Information Figure S3C), suggesting that JNK pathway activation was not critical for anticancer activity of clofoctol. Similarly, both necroptosis inhibitor (necrostatin-1) and ferroptosis inhibitor (ferrostatin-1) failed to rescue the growth inhibition by clofoctol (Supporting Information Figure S3D and E), indicating that none of the pertinent cell death mechanisms are involved in the clofoctol activity. Lastly, we determined the effect of an ER stress inhibitor, phenylbutyrate, on the activity of clofoctol. Phenylbutyrate is the most popular chemical chaperone that can reduce cellular ER stress by correcting protein misfolding (Yam et al., 2007). We observed that 2 mM of phenylbutyrate partially, but significantly, reversed the growth inhibition of PC3 cells by clofoctol (Figure 4A). We further confirmed the reversal of growth inhibition by phenylbutyrate in the cells treated with a lower concentration of clofoctol (Supporting Information Figure S4). Reduction of ER stress by phenylbutyrate, that is decrease in the levels of phosphorylated eIF2α and CHOP, was observed in the cells treated with clofoctol (Figure 4B). These results suggested that anticancer activity of clofoctol is, at least in part, mediated through the induction of ER stress.
Figure 4.
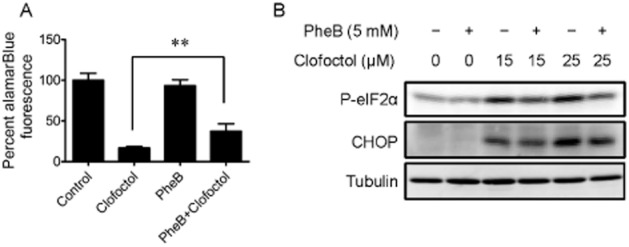
Clofoctol inhibits prostate cancer growth through induction of ER stress. (A) PC3 cells were grown in 96-well plates and treated with 20 μM clofoctol and 2 mM phenylbutyrate (PheB) for 72 h. An AlamarBlue assay was conducted to assess the cell viability. Data represent mean ± 95% confidence interval from three independent experiments (**P < 0.001). (B) PC3 cells were treated with the indicated concentrations of phenylbutyrate (PheB) and clofoctol for 24 h. Cells were then harvested for Western blot analysis of phosphorylated eIF2α and CHOP.
Clofoctol inhibited translation
Clofoctol has been reported to inhibit translation, which is consistent with the induction of phosphorylation of eIF2α we observed (Pelletier et al., 2007). To determine the effect of clofoctol on translation in prostate cancer cells, total proteins in PC3 cells were metabolically labelled with [35S]-methionine in the presence or absence of clofoctol. Clofoctol inhibited global protein synthesis in a dose- and time-dependent manner, as judged by the amounts of [35S]-methionine incorporated into newly synthesized proteins (Figure 5A and B). In contrast, no obvious difference in total protein levels as visualized by Coomassie brilliant blue staining between control and clofoctol-treated cells was observed within the time frame of the experiments, suggesting that newly synthesized protein levels were selectively reduced by clofoctol (Supporting Information Figure S5). To determine if the inhibition of translation is a direct effect by clofoctol, in vitro translation assay was conducted using luciferase mRNA and rabbit reticulocyte lysates. In this assay system, clofoctol showed no inhibition of the translation of luciferase mRNA at concentrations up to 100 μM. In contrast, cycloheximide, a known inhibitor of elongation, completely inhibited the translation of luciferase (Figure 5C). These results suggest that clofoctol inhibited in vivo translation indirectly through induction of the phosphorylation of eIF2α following ER stress induction in PC3 cells.
Figure 5.
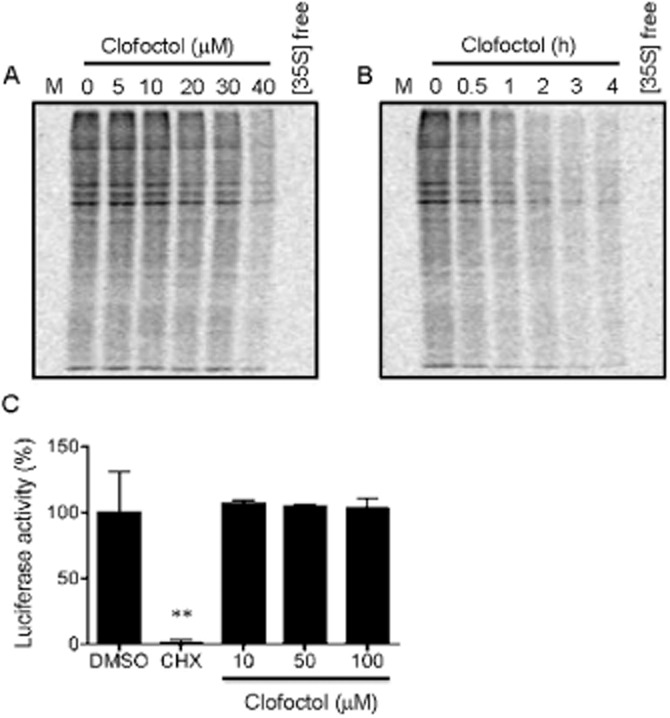
Clofoctol inhibits in vivo translation. (A and B) Total proteins in PC3 cells were metabolically labelled with [35S]-Met. The cells were then treated with various concentrations of clofoctol for 24 h (A) or treated with 30 μM clofoctol for indicated time points (B). The total proteins were resolved by the SDS-PAGE. 35S-labelled proteins were visualized by autoradiography. Cells without treatment with [35S]-Met ([35S] free) were used as a negative control. (C) The luciferase mRNA was translated in vitro using a rabbit reticulocyte lysate system. The reaction mixture was treated with either clofoctol or cycloheximide (CHX, 10 μM) and the luciferase activity assay was conducted to assess the in vitro translation of luciferase mRNA. Cycloheximide was used as a positive control compound to inhibit in vitro translation. Data represent mean + 95% confidence interval from three independent experiments performed in triplicate (**P < 0.001 vs. DMSO).
Clofoctol induced G1 phase cell cycle arrest through activation of UPR
Pharmacological activation of UPR leads to a G1 cell cycle arrest through inhibition of translation of cyclins that are responsible for G1/S transition (Brewer et al., 1999). Indeed, we observed that clofoctol induced G1 cell cycle arrest in PC3 cells (Figure 1C). Next, we determined whether clofoctol affected the levels of G1/S cyclins in PC3 cells. Treatment of PC3 cells with clofoctol for 24 h led to a dose-dependent decrease in the levels of cyclin A and cyclin D1 (Figure 6A). A subtle decrease in the level of cyclin E was also observed. Concurrently, the levels of phosphorylated eIF2α and CHOP were strongly increased. These results demonstrated that activation of UPR by clofoctol accompanied down-regulation of G1/S cyclins. As shown previously, an ER stress inhibitor phenylbutyrate could partially reverse inhibition of PC3 cell growth by clofoctol (Figure 4A; Supporting Information Figure S4). We thus examined whether phenylbutyrate could also reverse the clofoctol-induced G1 arrest in PC3 cells. As shown in Figure 6B, clofoctol increased the cell population in G1 phase, while phenylbutyrate partially increased the G2/M population. When applied together, phenylbutyrate reversed G1 cell population to the control level compared with that in clofoctol-treated cells (Figure 6B). These results suggested that the G1 cell cycle arrest caused by clofoctol is mediated through activation of UPR.
Figure 6.
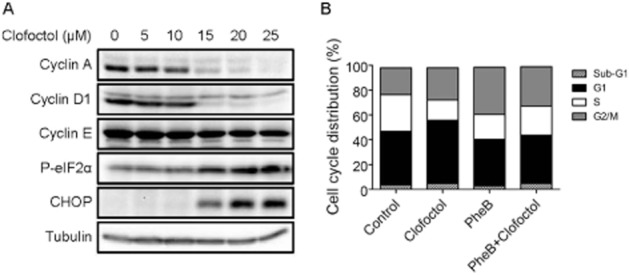
Clofoctol decreases G1/S cyclin levels. (A) PC3 cells were treated with indicated concentrations of clofoctol for 24 h. Cells were then harvested for Western blot analysis of cyclin A, cyclin D1, cyclin E, phosphorylated eIF2α and CHOP. (B) PC3 cells were treated with clofoctol and/or phenylbutyrate (PheB) for 24 h. The cells were then harvested and subjected to a cell cycle analysis. The % of cell population in each cell cycle phase is shown as a bar graph.
Clofoctol inhibited tumour growth in a mouse xenograft model
A mouse model of human prostate cancer xenograft was employed to determine the in vivo anticancer activity of clofoctol. PC3 cells were implanted s.c. into athymic nude mice and the mice were treated with either vehicle or clofoctol for 37 days. Tumour volume was measured during the course of the treatment. As shown in Figure 7A, PC3 tumour growth was significantly inhibited by clofoctol. Tumour weight was also reduced by 60% by clofoctol (Figure 7B). There was no significant change in mice body weight during the course of the drug treatment, suggesting that clofoctol did not have gross toxicity at the dose applied (Figure 7C). These results demonstrate that clofoctol is capable of blocking tumour growth in vivo without apparent toxicity.
Figure 7.
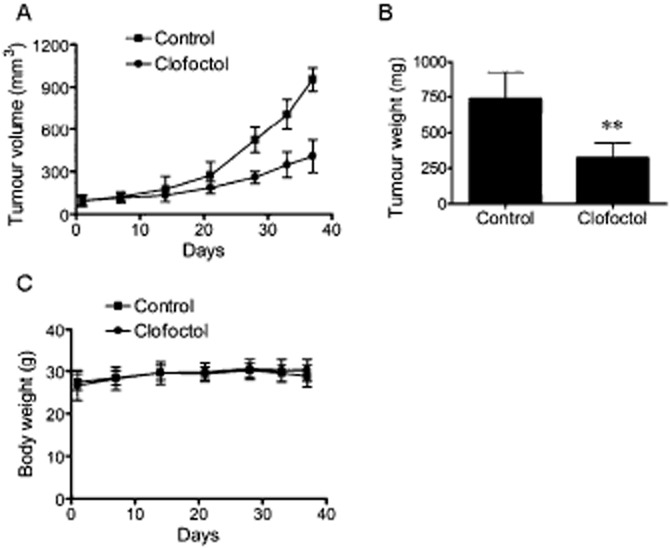
Clofoctol inhibits the growth of PC3 tumour xenografts in mice. (A–C) PC3 cells were implanted s.c. into nude mice. The mice were given vehicle (n = 6) or clofoctol (175 mg·kg−1) (n = 7) once daily via i.p. injection for 37 days. The tumour size was measured periodically and the tumour volume was calculated using a modified ellipsoid formula (A). After mice were killed, tumours were isolated from mice and the tumour wet weight was measured (B). Mice body weight was measured periodically to assess drug toxicity (C). In A–C, data represent mean + 95% confidence interval (**P < 0.001 vs. control).
Discussion
In this study, we identified clofoctol as an inhibitor of prostate cancer growth from JHDL with great potential as a translatable lead for developing antiprostate cancer drugs. Clofoctol is an antibacterial drug that has been used to treat upper respiratory tract infections in European countries for years. It is administered either orally or rectally. It showed relatively high peak plasma (Cmax: 38.1 μg·mL−1 or 104 μM) and lung (Cmax: 93.1 mg·kg−1) levels (Del Tacca et al., 1987; Danesi et al., 1988). In the present work, we found that clofoctol inhibited prostate cancer cell growth with IC50 values ranging from 10 to 15 μM, which are far below the clinically achievable plasma concentrations in vivo. It also inhibited the growth of a prostate cancer xenograft in mice with no apparent toxicity. These results suggest that clofoctol should have anticancer activity under its current antibacterial drug regimen.
Clofoctol is quite unique in its mechanism of action on cancer cells – it inhibits prostate cancer cell growth through activation of the ER stress and all three branches of the UPR pathway including IRE1, PERK and ATF6. As the three upstream UPR sensor protein receptors have distinct specificity, it is unlikely that clofoctol acts at the level of those UPR sensors. It is likely that clofoctol acts on a target that lies upstream of the UPR sensors which leads to the generation of unfolded proteins in the ER lumen. Prolonged induction of ER stress and activation of UPR result in cell growth arrest. It is well established that UPR uses PERK to coordinate inhibition of global protein translation by phosphorylating eIF2α (Harding et al., 2000). Inhibition of global translation causes a rapid reduction of short-lived cell cycle regulatory proteins, such as cyclin D1, which eventually leads to inhibition of G1/S transition (Brewer et al., 1999). Clofoctol indeed inhibited global protein translation and reduced the levels of cyclins A and D1 that are critical for G1/S transition. Furthermore, clofoctol induction of G1 arrest was partially reversed by an ER stress inhibitor, phenylbutyrate. Phenylbutyrate also partially reversed the inhibition of cell growth by clofoctol. These results suggested that anticancer mechanism of clofoctol can be explained by induction of ER stress, followed by inhibition of translation and G1 cell cycle arrest. We also tested inhibitors of the known cell death pathways including apoptosis, autophagy, necroptosis and ferroptosis in the cells treated with clofoctol. None of those inhibitors could reverse clofoctol effect on the viability of PC3 cells, suggesting that inhibition of prostate cancer cell growth by clofoctol does not involve conventional cell death mechanisms, but is likely due to the inhibition of cell cycle progression.
Tunicamycin and thapsigargin are both well-known inducers of ER stress, although their mechanisms of induction of ER stress are quite distinct. They are both known to activate JNK through activation of IRE1 and to increase LC3 puncta following ER stress (Ogata et al., 2006). Tunicamycin induces autophagy through IRE1-JNK pathway. In contrast, thapsigargin inhibits autophagy even though it activates JNK and increases LC3 puncta (Ganley et al., 2011). Clofoctol acted very similarly to thapsigargin in prostate cancer cells. It induced ER stress and activated the JNK pathway, followed by an increase in LC3 puncta. However, neither the JNK inhibitor nor autophagy inhibitor rescued the growth inhibitory effect of clofoctol on prostate cancer cells. In addition, bafilomycin treatment did not further enhance LC3 puncta formation by clofoctol, suggesting that clofoctol inhibited autophagic flux. These observations imply that clofoctol, similar to thapsigargin, might inhibit either fusion between autophagosome and lysosome or lysosome-dependent degradation pathway. Further studies including identification of the molecular target of clofoctol in eukaryotic cells would be necessary to elucidate its underlying mechanism of induction of ER stress and inhibition of autophagy.
Clofoctol is a structurally unique drug, with a largely hydrophobic core containing a single polar phenol moiety. In a preliminary structure-activity relationship study, we synthesized a number of analogues and found that the phenolic oxygen can tolerate most chemical modifications without significant loss in antiproliferative activity (Supporting Information Table S1). Interestingly, conversion of the methylene bridge between the two phenyl rings into a polar carbonyl group (CFT-CO) led to inactivation of the compound, underscoring a critical role of the large hydrophobic core in the activity of clofoctol.
Recently, ER stress and the UPR pathway have been proposed as a promising target for developing drugs for several human diseases. A number of small molecules have long been known to induce ER stress and activate the UPR pathway, including oxidizing agents, thapsigargin, tunicamycin and brefeldin A among others (Ma and Hendershot, 2004; Kim et al., 2008). The majority of those inducers, however, are too toxic to be developed into therapeutic agents. As clofoctol has been used as a drug in humans for a long time with tolerable side effects, it is ready to be evaluated in human clinical studies for the treatment of prostate cancer. Its unique accumulation in the lung suggests that it could also be effective for treating lung cancer. Moreover, activation of UPR pathway has been implicated in the clearance of protein aggregates in a number of neurodegenerative diseases (Kim et al., 2008), our findings also suggest that clofoctol may have therapeutic effects on those diseases in addition to prostate and other cancers.
Acknowledgments
This work was supported by grants from the NIH/NCI (CA122814), FAMRI and Prostate Cancer Foundation.
Glossary
- ATF6
activating transcription factor 6
- CHOP
transcription factor C/EBP homologous protein
- eIF2α
eukaryotic translation initiation factor 2α
- IRE1
inositol requiring enzyme 1
- JHDL
Johns Hopkins Drug Library
- LC3B
microtubule-associated protein light chain 3B
- PERK
double-stranded RNA-activated PK-like ER kinase
- UPR
unfolded protein response
- XBP-1
X-box binding protein-1
Author contributions
J. O. L., M. W., J. S. S. and R-J. L. designed the experiments; M. W., J. S. S., R-J. L., Y. D., Q. H. and M. D. performed the experiments; J. O. L., M. W., J. S. S., R-J. L. and M. D. analysed the results; J. O. L., J. S. S., M. W. and R-J. L. wrote the manuscript.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site: http://dx.doi.org/10.1111/bph.12800
Figure S1 (A) Chemical structure of clofoctol. (B) Clofoctol induces massive vacuolization in PC3. PC3 cells were treated with 20 μM clofoctol for 24 h and the cell morphology was observed under a phase contrast microscope.
Figure S2 Clofoctol induces ATF6 nuclear translocation in PC3 cells. PC3 cells were treated with or without 10 μM clofoctol for 24 h. Endogenous ATF6 was labelled with an antibody specific for ATF6. Confocal microscopy demonstrated that clofoctol treatment induced the nuclear translocation of ATF6 in PC3 cells.
Figure S3 (A–E) PC3 cells were grown in 96-well plates and treated with 20 μM clofoctol and various inhibitors including 3-methyladenine (3-MA; 5 mM), zVAD-fmk (zVAD; 20 μM), SP600125 (SP; 10 μM), necrostatin-1 (Nec-1; 2 μM) and ferrostatin-1 (Fer-1; 2 μM) for 72 h. AlamarBlue assay was conducted to assess the cell viability. Data represent mean ± 95% confidence interval from three independent experiments. NS denotes not significant (P > 0.01).
Figure S4 Reversal effect of phenylbutyrate (2 mM) on inhibition of PC3 cell growth by clofoctol (15 μM). Data represent mean ± 95% confidence interval from three independent experiments (*P < 0.01).
Figure S5 (A and B) Coomassie brilliant blue (CBB) staining of total proteins used in Figure 5A and B is shown.
Table S1 IC50 Values for clofoctol and its analogues for inhibition of PC3.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1862. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Heath EI, Smith DC, Rathkopf D, Blackford AL, Danila DC, et al. A non-comparative randomized phase 2 study of two dose levels of itraconazole in men with metastatic castration-resistant prostate cancer (mCRPC): a DOD/PCCTC trial. J Clin Oncol. 2011;29:Abstract 4532. [Google Scholar]
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- Bernasconi R, Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol. 2011;23:176–183. doi: 10.1016/j.ceb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JW, Hendershot LM, Sherr CJ, Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc Natl Acad Sci U S A. 1999;96:8505–8510. doi: 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Choi SM, Kim Y, Shim JS, Park JT, Wang RH, Leach SD, et al. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology. 2013;57:2458–2468. doi: 10.1002/hep.26237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ., Jr A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006a;2:415–416. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- Chong CR, Qian DZ, Pan F, Wei Y, Pili R, Sullivan DJ, Jr, et al. Identification of type 1 inosine monophosphate dehydrogenase as an antiangiogenic drug target. J Med Chem. 2006b;49:2677–2680. doi: 10.1021/jm051225t. [DOI] [PubMed] [Google Scholar]
- Chong CR, Xu J, Lu J, Bhat S, Sullivan DJ, Jr, Liu JO. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem Biol. 2007;2:263–270. doi: 10.1021/cb600362d. [DOI] [PubMed] [Google Scholar]
- Danesi R, Del Tacca M. Clinical study on the efficacy of clofoctol in the treatment of infectious respiratory diseases. Int J Clin Pharmacol Res. 1985;5:175–179. [PubMed] [Google Scholar]
- Danesi R, Gasperini M, Senesi S, Freer G, Angeletti CA, Del Tacca M. A pharmacokinetic study of clofoctol in human plasma and lung tissue by using a microbiological assay. Drugs Exp Clin Res. 1988;14:39–43. [PubMed] [Google Scholar]
- Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, et al. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–2649. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
- Del Tacca M, Danesi R, Senesi S, Gasperini M, Mussi A, Angeletti CA. Penetration of clofoctol into human lung. J Antimicrob Chemother. 1987;19:679–683. doi: 10.1093/jac/19.5.679. [DOI] [PubMed] [Google Scholar]
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42:731–743. doi: 10.1016/j.molcel.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388–399. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4:849–850. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci U S A. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Leleu X, Xu L, Jia XY, Sacco A, Farag M, Hunter ZR, et al. Endoplasmic reticulum stress is a target for therapy in Waldenstrom macroglobulinemia. Blood. 2009;113:626–634. doi: 10.1182/blood-2007-10-116848. [DOI] [PubMed] [Google Scholar]
- Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J, et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate. 2011;71:333–343. doi: 10.1002/pros.21247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004;4:966–977. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon MA, Jilek BL, Brennan TP, Shen L, Zhou Y, Wind-Rotolo M, et al. The HBV drug entecavir – effects on HIV-1 replication and resistance. N Engl J Med. 2007;356:2614–2621. doi: 10.1056/NEJMoa067710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Bordeleau M, Lindqvist L, Francis R, Sukarieh R, Tanaka J. 2007. Chemotherapeutic agents for inhibition of protein translation Vol. WO2007/009264 A1, A61K 31/58 edn.
- Platz EA, Yegnasubramanian S, Liu JO, Chong CR, Shim JS, Kenfield SA, et al. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov. 2011;1:68–77. doi: 10.1158/2159-8274.CD-10-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren YR, Pan F, Parvez S, Fleig A, Chong CR, Xu J, et al. Clofazimine inhibits human Kv1.3 potassium channel by perturbing calcium oscillation in T lymphocytes. PLoS ONE. 2008;3:e4009. doi: 10.1371/journal.pone.0004009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rovira M, Huang W, Yusuff S, Shim JS, Ferrante AA, Liu JO, et al. Chemical screen identifies FDA-approved drugs and target pathways that induce precocious pancreatic endocrine differentiation. Proc Natl Acad Sci U S A. 2011;108:19264–19269. doi: 10.1073/pnas.1113081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Brahmer JR, Juergens RA, Hann CL, Ettinger DS, Sebree R, et al. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J Thorac Oncol. 2013;8:619–623. doi: 10.1097/JTO.0b013e31828c3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Shim JS, Matsui Y, Bhat S, Nacev BA, Xu J, Bhang HE, et al. Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J Natl Cancer Inst. 2010;102:1855–1873. doi: 10.1093/jnci/djq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JS, Rao R, Beebe K, Neckers L, Han I, Nahta R, et al. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J Natl Cancer Inst. 2012;104:1576–1590. doi: 10.1093/jnci/djs396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Chapman R, Walter P. The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol. 1998;8:245–249. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann N Y Acad Sci. 2012;1271:20–32. doi: 10.1111/j.1749-6632.2012.06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36:329–337. doi: 10.1016/j.tibs.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Yablonsky F. Alteration of membrane permeability in Bacillus subtilis by clofoctol. J Gen Microbiol. 1983;129:1089–1095. doi: 10.1099/00221287-129-4-1089. [DOI] [PubMed] [Google Scholar]
- Yam GH, Gaplovska-Kysela K, Zuber C, Roth J. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Invest Ophthalmol Vis Sci. 2007;48:1683–1690. doi: 10.1167/iovs.06-0943. [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Byun Y, Ren YR, Liu JO, Laterra J, Pomper MG. Identification of inhibitors of ABCG2 by a bioluminescence imaging-based high-throughput assay. Cancer Res. 2009;69:5867–5875. doi: 10.1158/0008-5472.CAN-08-4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) Chemical structure of clofoctol. (B) Clofoctol induces massive vacuolization in PC3. PC3 cells were treated with 20 μM clofoctol for 24 h and the cell morphology was observed under a phase contrast microscope.
Figure S2 Clofoctol induces ATF6 nuclear translocation in PC3 cells. PC3 cells were treated with or without 10 μM clofoctol for 24 h. Endogenous ATF6 was labelled with an antibody specific for ATF6. Confocal microscopy demonstrated that clofoctol treatment induced the nuclear translocation of ATF6 in PC3 cells.
Figure S3 (A–E) PC3 cells were grown in 96-well plates and treated with 20 μM clofoctol and various inhibitors including 3-methyladenine (3-MA; 5 mM), zVAD-fmk (zVAD; 20 μM), SP600125 (SP; 10 μM), necrostatin-1 (Nec-1; 2 μM) and ferrostatin-1 (Fer-1; 2 μM) for 72 h. AlamarBlue assay was conducted to assess the cell viability. Data represent mean ± 95% confidence interval from three independent experiments. NS denotes not significant (P > 0.01).
Figure S4 Reversal effect of phenylbutyrate (2 mM) on inhibition of PC3 cell growth by clofoctol (15 μM). Data represent mean ± 95% confidence interval from three independent experiments (*P < 0.01).
Figure S5 (A and B) Coomassie brilliant blue (CBB) staining of total proteins used in Figure 5A and B is shown.
Table S1 IC50 Values for clofoctol and its analogues for inhibition of PC3.