

Abstract
Background and Purpose
Dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel causes the genetic disease cystic fibrosis (CF). Towards the development of transformational drug therapies for CF, we investigated the channel function and action of CFTR potentiators on A561E, a CF mutation found frequently in Portugal. Like the most common CF mutation F508del, A561E causes a temperature-sensitive folding defect that prevents CFTR delivery to the cell membrane and is associated with severe disease.
Experimental Approach
Using baby hamster kidney cells expressing recombinant CFTR, we investigated CFTR expression by cell surface biotinylation, and function and pharmacology with the iodide efflux and patch-clamp techniques.
Key Results
Low temperature incubation delivered a small proportion of A561E-CFTR protein to the cell surface. Like F508del-CFTR, low temperature-rescued A561E-CFTR exhibited a severe gating defect characterized by brief channel openings separated by prolonged channel closures. A561E-CFTR also exhibited thermoinstability, losing function more quickly than F508del-CFTR in cell-free membrane patches and intact cells. Using the iodide efflux assay, CFTR potentiators, including genistein and the clinically approved small-molecule ivacaftor, partially restored function to A561E-CFTR. Interestingly, ivacaftor restored wild-type levels of channel activity (as measured by open probability) to single A561E- and F508del-CFTR Cl− channels. However, it accentuated the thermoinstability of both mutants in cell-free membrane patches.
Conclusions and Implications
Like F508del-CFTR, A561E-CFTR perturbs protein processing, thermostability and channel gating. CFTR potentiators partially restore channel function to low temperature-rescued A561E-CFTR. Transformational drug therapy for A561E-CFTR is likely to require CFTR correctors, CFTR potentiators and special attention to thermostability.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| Cystic fibrosis transmembrane conductance regulator, CFTR | Forskolin |
| Genistein | |
| Ivacaftor, VX-770 | |
| Lumacaftor, VX-809 | |
| UCCF-853 |
This Table lists the protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The genetic disease cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), an epithelial Cl− channel with complex regulation (Riordan et al., 1989; Welsh et al., 2001; Gadsby et al., 2006) (channel nomenclature follows Alexander et al., 2013). To date, almost 2000 disease-causing mutations have been identified in the CFTR gene (http://www.genet.sickkids.on.ca/cftr/). The most common and best understood CF mutation is F508del, deletion of the phenylalanine residue at position 508 of the CFTR protein sequence; F508del accounts for about 70% of CF mutations worldwide and is associated with a severe disease phenotype (Welsh et al., 2001; Rowe et al., 2005). The molecular basis of the F508del defect is protein misfolding, leading to defective processing and intracellular transport of CFTR (see Lukacs and Verkman, 2012; Farinha et al., 2013b). However, any F508del-CFTR that reaches the cell membrane exhibits two further defects – protein instability (Lukacs et al., 1993) and defective channel gating (Dalemans et al., 1991).
Restoration of channel function to CF mutants requires two types of small molecules. First, CFTR correctors that overcome protein folding defects allowing the mutant protein to traffic to the apical membrane of epithelia (Lukacs and Verkman, 2012; Hanrahan et al., 2013). Second, CFTR potentiators that repair gating defects to enhance greatly channel activity (Verkman and Galietta, 2009; Cai et al., 2011). In 2012, the CFTR potentiator ivacaftor (Kalydeco™; VX-770; Vertex Pharmaceuticals Incorporated, Boston, MA, USA) was approved for use in CF patients aged 6 years and older with the gating mutation G551D-CFTR (Van Goor et al., 2009; Ramsey et al., 2011) and initial results of clinical trials of ivacaftor and the CFTR corrector VX-809 (lumacaftor; Vertex Pharmaceuticals Incorporated) in CF patients with F508del-CFTR were announced (Van Goor et al., 2011; Boyle et al., 2012). To ensure that transformational drug therapies are available to all CF patients, it is necessary to elucidate how individual CF mutations disrupt CFTR function and determine their responses to small molecule CFTR modulators.
The second most common CF mutation in Portugal is A561E (Mendes et al., 2003). CF patients homozygous for A561E have a clinical phenotype similar to that of F508del homozygotes and rectal biopsies lack residual Cl− channel function (Mendes et al., 2003; Hirtz et al., 2004). Like F508del, the mutation affects a residue located within the first nucleotide-binding domain (NBD1) of CFTR (Lewis et al., 2004). More interestingly, both mutants cause temperature-sensitive folding defects that prevent CFTR delivery to the apical membrane (Denning et al., 1992; Mendes et al., 2003). Because low temperature incubation restores some function to A561E-CFTR (Mendes et al., 2003) and because F508del-CFTR perturbs channel gating (Dalemans et al., 1991), whereas some other CF mutations in the NBDs are without effect (e.g. A455E; Sheppard et al., 1995), we were interested to assess the effects of the A561E mutation on CFTR channel function and the mutant’s response to CFTR potentiators, including ivacaftor. To address these aims, we studied low temperature-rescued A561E-CFTR using the iodide efflux and patch-clamp techniques. We discovered that A561E-CFTR is an unstable channel with a severe gating defect. However, some function could be restored to A561E-CFTR using CFTR potentiators. At the single-channel level, ivacaftor conferred wild-type levels of channel activity (as measured by open probability; Po) on A561E-CFTR, but accelerated channel deactivation. We conclude that transformational drug therapy for A561E-CFTR is likely to require CFTR correctors, CFTR potentiators and special attention to protein stability.
Methods
Cells and cell culture
We used untransfected baby hamster kidney (BHK) cells and BHK cells stably expressing wild-type human CFTR and the CF mutants F508del- and A561E-CFTR (Farinha et al., 2002; Roxo-Rosa et al., 2006), generously provided by MD Amaral (University of Lisboa, Lisboa, Portugal). Cells were cultured and used as described previously (Schmidt et al., 2008) with the exception that BHK cells expressing F508del- and A561E-CFTR were routinely incubated at 27°C for 24 h prior to use to promote the cell surface expression of these mutants (Denning et al., 1992; Mendes et al., 2003).
Cell-surface biotinylation
Cell-surface biotinylation was performed on untransfected and CFTR-expressing BHK cells as described previously (Toye et al., 2008; 2004). Cells were washed with pre-cooled borate buffer (10 mM boric acid, 154 mM NaCl, 7.2 mM KCl and 1.8 mM CaCl2, pH 9) and then treated with 3 mL of borate buffer containing 1 mg·mL−1 non-penetrating biotinylation reagent EZ-link™ Sulfo-NHS-biotin (Pierce, Rockford, IL, USA) at 4°C for 30 min. The reaction was quenched with several washes of pre-cooled 0.192 M glycine/25 mM Tris, pH 8.3 buffer. Cells were then lysed with immunoprecipitation buffer (20 mM Tris-HCl, pH 8.0, 137 mM NaCl, 10 mM EDTA, 100 mM NaF, 1% v·v−1 Nonidet P-40, 10% v·v−1 glycerol, 2 mM Na3VO4 containing protease inhibitors) and pre-cleared using 50 μL of empty protein G beads. CFTR was immunoprecipitated using the rabbit anti-CFTR polyclonal antibody Mr. Pink, which recognizes NBD1 of human CFTR [(Hoelen et al., 2010); a generous gift of B Kleizen and I Braakman (Utrecht University, Utrecht, The Netherlands)] (10 μg per 30 μL protein G beads). The immunoprecipitated CFTR was eluted twice from the protein G beads by boiling in elution buffer (10 mM Tris/HCl, pH 7.4; 1 mM EDTA containing 5% SDS and protease inhibitors) and resuspended in 50 vol of immunoprecipitation buffer. The biotinylated CFTR was isolated from the combined eluate using 50 μL streptavidin beads in sample buffer (50 mM Tris/HCl, pH 8; 2 mM EDTA, 12% glycerol and 10% SDS). Approximately 10–15% of the CFTR immunoprecipitate was reserved to provide an indication of the input for the streptavidin isolation. The remainder of the eluted proteins were separated by SDS-PAGE and then blotted onto PVDF membrane (Millipore Ltd., Watford, UK). CFTR was detected using the mouse anti-CFTR monoclonal antibody (596), which recognizes NBD2 of human CFTR [(Cui et al., 2007); a generous gift of T Jensen and JR Riordan (University of North Carolina) and Cystic Fibrosis Foundation Therapeutics (CFFT) (Bethesda, MD, USA)] and a rabbit antimouse HRP conjugated secondary (Dako UK Ltd., Ely, UK).
Iodide efflux experiments
We measured CFTR-mediated iodide efflux at 23°C as described previously (Lansdell et al., 1998b) using the cAMP agonist forskolin (10 μM) and CFTR potentiators (genistein, 50 μM; test potentiators, 10 μM). BHK cells were first incubated for 1 h in loading buffer containing (mM): 136 NaI, 3 KNO3, 2 Ca(NO3)2, 20 HEPES and 11 glucose, adjusted to pH 7.4 with 1 M NaOH. Next, BHK cells were washed thoroughly with efflux buffer (136 mM NaNO3 replacing 136 mM NaI in the loading buffer) before treatment with agonists. Finally, the amount of iodide in each sample of efflux buffer was determined using an iodide-selective electrode (Model: Orion 94–53, Thermo Scientific, Nijkerk, The Netherlands) and presented as cumulative iodide efflux (Chappe et al., 2003).
Patch-clamp experiments
CFTR Cl− channels were recorded in excised inside-out membrane patches using an Axopatch 200A patch-clamp amplifier and pCLAMP software (both from Molecular Devices, Union City, CA, USA) (Sheppard and Robinson, 1997). The pipette (extracellular) solution contained (mM): 140 N-methyl-D-glucamine (NMDG), 140 aspartic acid, 5 CaCl2, 2 MgSO4 and 10 N-tris[hydroxymethyl]methyl-2-aminoethanesulphonic acid (TES), adjusted to pH 7.3 with Tris ([Cl−], 10 mM). The bath (intracellular) solution contained (mM): 140 NMDG, 3 MgCl2, 1 CsEGTA and 10 TES, adjusted to pH 7.3 with HCl ([Cl−], 147 mM; free [Ca2+], <10−8 M) and was maintained at 23, 27 or 37°C using a temperature-controlled microscope stage (Brook Industries, Lake Villa, IL, USA).
After excision of inside-out membrane patches, we added the catalytic subunit of PKA (75 nM) and ATP (1 mM) to the intracellular solution within 5 min of patch excision to activate CFTR Cl− channels. To minimize channel rundown, we added PKA to all intracellular solutions, maintained the ATP concentration at 1 mM and clamped voltage at −50 mV. The effects of CFTR potentiators on mutant Cl− channels were tested by addition to the intracellular solution in the continuous presence of ATP (1 mM) and PKA (75 nM). Because of the rundown of F508del- and A561E-CFTR Cl− channels in excised membrane patches at 37°C and the difficulty of removing ivacaftor from the recording chamber, the effects of potentiators were tested at 27°C and not bracketed by control periods. Instead, specific interventions with test potentiators were compared with the pre-intervention control period made with the same concentration of ATP and PKA, but without the test potentiator.
To investigate the thermostability of F508del- and A561E-CFTR, membrane patches were excised at 27°C and channels activated by the addition of PKA and ATP to the intracellular solution. Once channels were fully activated, the temperature of the intracellular solution was increased to 37°C, which took 2–3 min. To evaluate mutant channel thermostability at 37°C, we calculated Po values in 30 s intervals over a 9 min period. As controls, we studied F508del- and A561E-CFTR Cl− channels at 23°C and wild-type CFTR at 37°C commencing Po measurements only after channels were fully activated.
In this study, we used membrane patches containing ≤5 active channels [wild-type CFTR, number of active channels (N) ≤ 4; F508del-CFTR, N ≤ 5; A561E-CFTR, N ≤ 5]. To determine channel number, we used the maximum number of simultaneous channel openings observed during the course of an experiment (Cai et al., 2006). To minimize errors, we used experimental conditions that robustly potentiate channel activity and verified that recordings were of sufficient length to ascertain the correct number of channels (Venglarik et al., 1994; Lansdell et al., 1998a). Despite our precautions, we cannot exclude the possibility of unobserved mutant Cl− channels in excised membrane patches. Therefore, Po values for F508del- and A561E-CFTR might possibly be overestimated.
We recorded, filtered and digitized data as described previously (Sheppard and Robinson, 1997). To measure single-channel current amplitude (i), Gaussian distributions were fit to current amplitude histograms. For Po and burst analyses, lists of open- and closed-times were created using a half-amplitude crossing criterion for event detection and dwell-time histograms constructed as described previously (Sheppard and Robinson, 1997). For burst analysis, we used a tc (the time that separates interburst closures from intraburst closures) determined from closed-time histograms [wild-type CFTR, tc = 14.6 ± 0.2 ms (n = 6); F508del-CFTR, tc = 23 ± 3 ms (n = 5); A561E-CFTR, tc = 19 ± 1 ms (n = 5)] (Cai et al., 2006). The mean interburst interval (TIBI) was calculated using the equation (Cai et al., 2006):
| (Eq. 1) |
where, Tb = (mean burst duration) × (open probability within a burst). Mean burst duration (TMBD) and open probability within a burst (Po(burst)) were determined directly from experimental data using pCLAMP software. For wild-type CFTR, only membrane patches that contained a single active channel were used for burst analyses, whereas for F508del- and A561E-CFTR, membrane patches contained no more than three active channels. We analysed only bursts of single-channel openings with no superimposed openings that were separated from one another by a time interval ≥tc.
Data analysis
Results are expressed as means ± SEM of n observations. To test for differences between groups of data, we used Student’s t-test. Differences were considered statistically significant when P < 0.05. All tests were performed using SigmaStat™ (Systat Software Inc., Richmond, CA, USA).
Materials
The CFTR potentiators PG-01 [CFFT CFTR Compound Program reference no. P2; Pedemonte et al. (2005)], SF-03 (P3; Pedemonte et al., 2005), UCCF-853 (P4; Caci et al., 2003), ΔF508act-02 (P5; Yang et al., 2003) and UCCF-180 (P9; Sammelson et al., 2003) were generous gifts of RJ Bridges (Rosalind Franklin University of Medicine and Science, Chicago, IL, USA) and CFFT while ivacaftor was purchased from Selleck Chemicals (Stratech Scientific Ltd., Newmarket, UK). Forskolin was purchased from the Sigma-Aldrich Company Ltd. (Gillingham, UK), genistein from LC Laboratories (Woburn, MA, USA) and PKA purified from bovine heart from Calbiochem (Merck Chemicals Ltd., Nottingham, UK). All other chemicals were of reagent grade and supplied by the Sigma-Aldrich Company Ltd.
ATP was dissolved in intracellular solution, forskolin in methanol and all other reagents in DMSO. Stock solutions were stored at −20°C with the exception of those of ATP, which were prepared directly before each experiment. Immediately before use, stock solutions were diluted to final concentrations. Precautions against light-sensitive reactions were observed when using CFTR potentiators. DMSO was without effect on CFTR activity (Sheppard and Robinson, 1997; Schmidt et al., 2008). On completion of experiments, the recording chamber was thoroughly cleaned before reuse [ivacaftor: soaked overnight in DMSO (40% v·v−1) before thorough washing with double distilled water].
Results
Low-temperature incubation restores channel function to A561E-CFTR
In this study, we investigated the impact of the CF mutation A561E on CFTR channel function and explored the action of CFTR potentiators on this mutant. To study A561E-CFTR, we used BHK cells stably expressing A561E-CFTR, hereafter termed BHK-A561E-CFTR. As controls, we employed BHK cells stably expressing wild-type and F508del-CFTR, termed BHK-WT-CFTR and BHK-F508del-CFTR respectively. Using the iodide efflux assay, we examined the behaviour of large numbers of CFTR channels in intact cells and with the patch-clamp technique, we explored the properties of individual CFTR channels in cell-free membrane patches.
The cAMP agonist forskolin (10 μM) and the CFTR potentiator genistein (50 μM) stimulated a robust efflux of iodide from BHK-WT-CFTR cells grown at 37°C, but were without effect on BHK-F508del-CFTR and BHK-A561E-CFTR cells cultured at this temperature (Figure 1A and C, Supporting Information Figure S1A); untransfected BHK cells are unresponsive to forskolin (10 μM) and genistein (50 μM) (Schmidt et al., 2008). After incubating BHK-F508del-CFTR and BHK-A561E-CFTR cells at 27°C for 24 h to allow low-temperature rescue of misfolded CFTR proteins (Denning et al., 1992; Mendes et al., 2003), these cells generated robust CFTR-mediated iodide efflux with a similar time course, but reduced magnitude compared to that of BHK-WT-CFTR cells (Figure 1B and C and Supporting Information Figure S1B).
Figure 1.
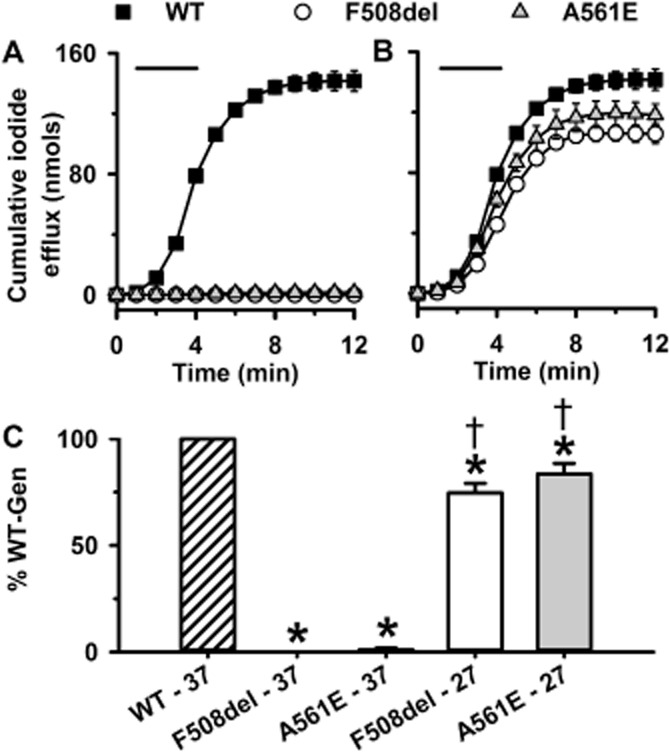
Genistein potentiates iodide efflux by F508del- and A561E-CFTR after their rescue by low-temperature incubation. (A and B) Time courses of cumulative iodide efflux from BHK cells expressing wild-type, F508del- and A561E-CFTR. BHK-WT-CFTR cells were grown at 37°C, whereas BHK-F508del-CFTR and BHK-A561E-CFTR cells were grown at 37°C (A) or 27°C for 24 h before the experiment (B). During the periods indicated by the bars, cells were treated with forskolin (10 μM) and genistein (50 μM). (C) Effects of low temperature incubation on iodide efflux by F508del- and A561E-CFTR. Cumulative iodide efflux at 12 min is expressed as a percentage of that of wild-type CFTR. Data are means ± SEM (n = 4); *P < 0.05 versus wild-type CFTR; †P < 0.05 versus same construct cultured at 37°C.
Figure 1 suggests that low-temperature incubation restores substantial amounts of function to F508del- and A561E-CFTR. However, for both mutants, the amount of mature protein rescued by low-temperature incubation is only a small fraction of that of wild-type CFTR (Denning et al., 1992; Mendes et al., 2003). To determine the amount of CFTR protein at the cell surface in the BHK cells used in this study, we performed cell surface biotinylation. Figure 2 shows that when BHK-F508del-CFTR and BHK-A561E-CFTR were grown at 37°C, only low levels of immunoprecipitated F508del- and A561E-CFTR protein were observed and no biotinylated protein was detected in marked contrast to BHK-WT-CFTR cells; no CFTR protein was detected in untransfected cells (Figure 2). When BHK-F508del-CFTR and BHK-A561E-CFTR cells were incubated at 27°C, the amount of immunoprecipitated CFTR protein increased for both F508del- and A561E-CFTR and a small, but variable amount was biotinylated, confirming rescue of a proportion of the mutant protein to the cell surface (Figure 2). Consistent with previous results (Gee et al., 2011), some biotinylated mutant protein appeared to be the immature form of CFTR (band B) (Figure 2). It was not possible to accurately quantify the amounts of F508del- and A561E-CFTR protein delivered to the cell surface due to the variable low levels of detectable protein present relative to the oversaturated total protein lanes, but we estimate it to be approximately 1%. These data suggest that iodide efflux from BHK-WT-CFTR cells might saturate when stimulated with forskolin and genistein (for discussion, see Xu et al., 2014).
Figure 2.
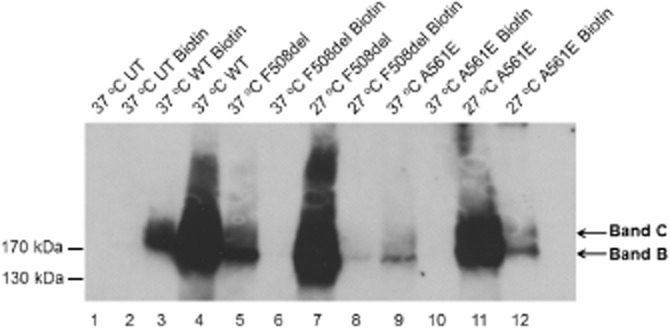
Cell-surface biotinylation of CFTR constructs in stably transfected BHK cells. BHK-WT-CFTR and control (untransfected; UT) cells were grown at 37°C, whereas BHK-F508del-CFTR and BHK-A561E-CFTR cells were grown at 37 or 27°C. For each CFTR construct, the protein detected at the cell surface is compared with a proportion of total CFTR expression (approximately 10%). One confluent T75 flask of cells for each condition were lysed and CFTR immunoprecipitated, the proteins eluted and the biotinylated fraction isolated using streptavidin beads and separated by SDS-PAGE (as described in the Methods section). The positions of bands B and C are indicated by arrows. Data shown are representative of multiple experiments (n = 3–4) performed in duplicate.
The A561E mutation disrupts CFTR gating and thermostability
When delivered to the cell surface by low-temperature incubation, F508del-CFTR forms a channel with a pronounced gating defect (Dalemans et al., 1991). To learn whether the A561E mutation perturbs CFTR function, we investigated the single-channel activity of A561E-CFTR at 37°C following its rescue by low-temperature incubation. Figure 3A shows representative single-channel recordings of wild-type F508del- and A561E-CFTR in excised inside-out membrane patches from BHK cells. Like F508del-CFTR (Cai et al., 2011), A561E-CFTR was without effect on i, but perturbed severely channel gating (Figure 3). The gating pattern of wild-type CFTR is characterized by bursts of channel openings interrupted by brief flickery closures and separated by longer closures between bursts. Interestingly, the Po of A561E-CFTR was reduced markedly compared with that of wild-type CFTR, but was equivalent to that of F508del-CFTR (Figure 3C). For both F508del- and A561E-CFTR, the interburst interval (IBI) was prolonged markedly, while mean burst duration (MBD) was little changed compared to wild-type CFTR (Figure 3D and E). Thus, the mutations F508del and A561E have very similar impact on CFTR channel gating.
Figure 3.
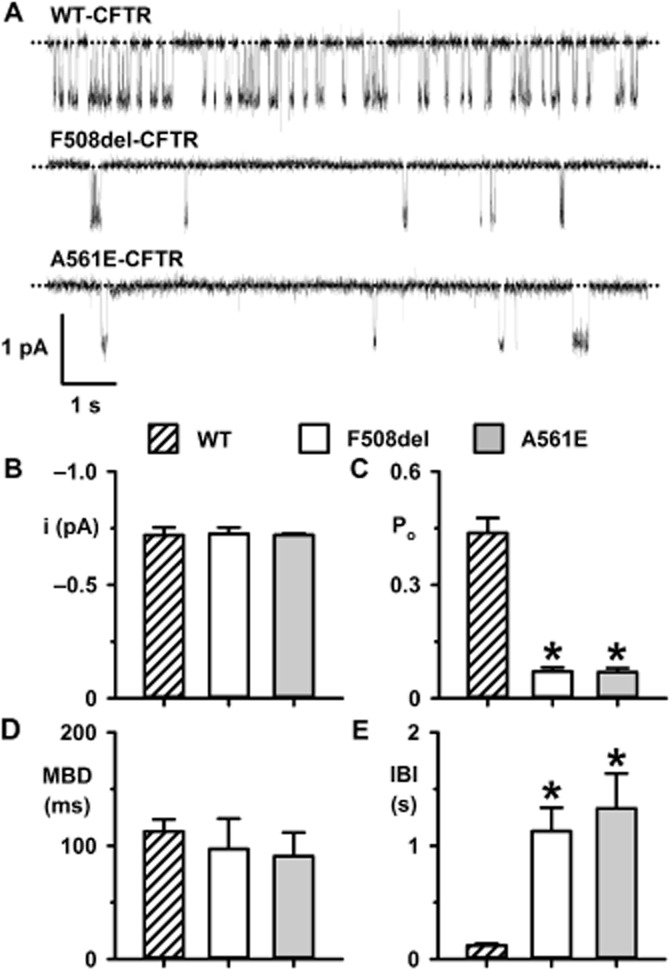
The mutations F508del and A561E have similar impact on CFTR channel gating. (A) Representative single-channel recordings of wild-type CFTR and low temperature-rescued F508del- and A561E-CFTR in excised inside-out membrane patches from BHK cells. ATP (1 mM) and PKA (75 nM) were continuously present in the intracellular solution. Unless otherwise indicated, in this and subsequent figures, voltage was −50 mV, there was a large Cl− concentration gradient across the membrane patch ([Cl−]int, 147 mM; [Cl−]ext, 10 mM) and temperature was 37°C. The dotted lines indicate closed channels and downward deflections of the traces denote channel openings. (B–E) i, Po, MBD and IBI of wild-type, F508del- and A561E-CFTR. Data are means + SEM quantified as described in the Methods section from 3980 ± 487 transitions (wild-type, number of active channels (N) = 1, n = 6); 1586 ± 394 transitions (F508del, N ≤ 2, n = 5) and 1012 ± 198 transitions (A561E, N ≤ 2, n = 5); *P < 0.05 versus wild-type CFTR.
The F508del mutation attenuates CFTR stability at the cell surface (Lukacs et al., 1993), which is evident in single-channel recordings as accelerated channel rundown at 37°C (e.g. Schultz et al., 1999). To investigate the impact of A561E-CFTR on CFTR thermostability, we made Po measurements to monitor the duration of channel activity in excised inside-out membrane patches at 23 and 37°C. As controls, we studied the single-channel activity of F508del-CFTR at 23 and 37°C and wild-type CFTR at 37°C, commencing Po measurements only after channels were fully activated by PKA-dependent phosphorylation. Figures 4 and 5 show the single-channel activity of wild-type, F508del- and A561E-CFTR, measured as Po, over the first 9 min at 23 and 37°C once channels were fully activated. Wild-type CFTR demonstrated robust channel activity at 37°C that was stable and sustained over the 9 min period (Figures 4A and 5A). For both F508del- and A561E-CFTR, channel activity was sustained at 23°C, albeit at a very low level compared with that of wild-type CFTR at 37°C (Figure 4A and B, and Figure 5A and B). At 37°C, Po values for F508del- and A561E-CFTR were initially greater than those at 23°C, but they declined rapidly to zero (Figure 4B and C, and Figure 5B and C). Comparison of normalized Po values reveals that the rundown of F508del- and A561E-CFTR was completed within 7 and 6 min, respectively (Figures 4C and 5C). Addition of fresh PKA and ATP to the intracellular solution was ineffective, suggesting that channel rundown was irreversible (F508del, n = 3; A561E, n = 2; Y. Wang et al., data not shown).
Figure 4.
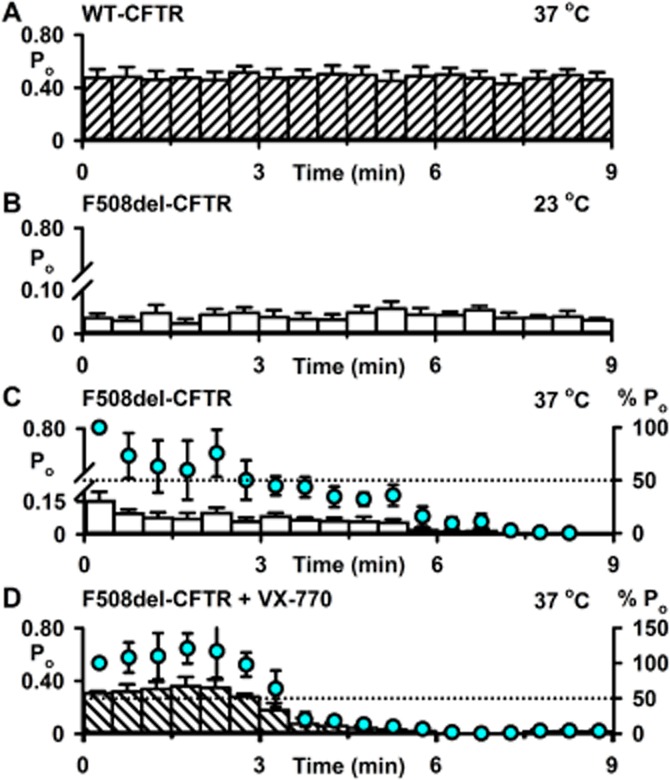
Thermal instability of F508del-CFTR channel activity in excised inside-out membrane patches. (A–D) Time courses of Po for wild-type CFTR and low temperature-rescued F508del-CFTR in excised inside-out membrane patches once channel activation was complete. Low temperature-rescued F508del-CFTR Cl− channels were studied at 23°C (B), at 37°C (C) and at 37°C in the presence of ivacaftor (VX-770; 10 μM) (D). Po values were calculated for each 30 s interval. In C and D, the left and right ordinates show Po (bars) and normalized Po (circles), respectively; Po values were normalized to that measured immediately temperature reached 37°C (t = 0–30 s). For wild-type CFTR, the membrane patch was excised, channels activated and studied all at 37°C. For F508del-CFTR, the membrane patch was excised and channels activated at either 23°C (B) or 27°C (C and D). In B, temperature was maintained at 23°C, whereas in C and D, it was increased to 37°C before Po values were measured. Data are means ± SEM quantified as described in the Methods section from 12 883 ± 228 transitions (wild-type, N = 1, n = 4); 9574 ± 3909 transitions (F508del: 23°C, N ≤ 5, n = 5); 3173 ± 1107 transitions (F508del: 37°C, N ≤ 4, n = 5) and 30 279 ± 16 750 transitions (F508del: 37°C + VX-770, N ≤ 5, n = 4).
Figure 5.
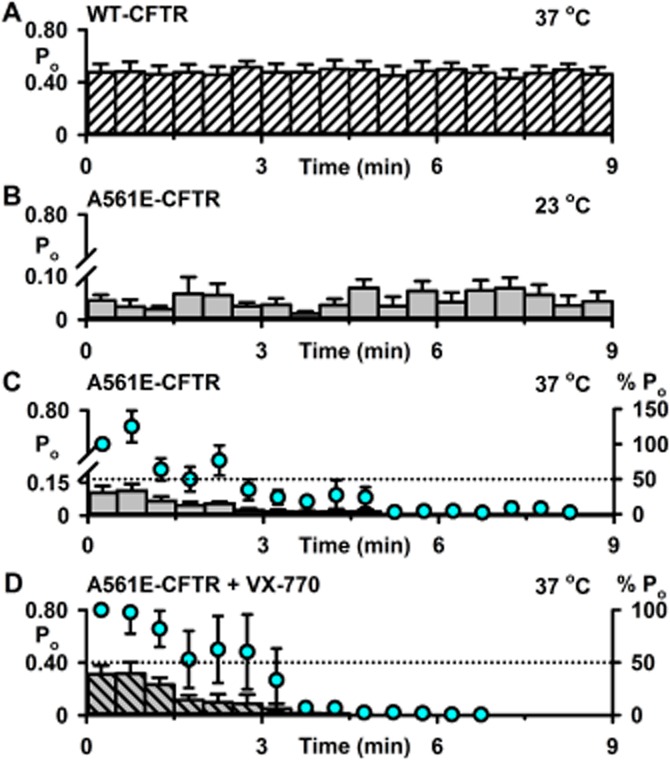
Loss of A561E-CFTR channel activity in excised inside-out membrane patches. (A–D) Time courses of Po for wild-type CFTR and low temperature-rescued A561E-CFTR in excised inside-out membrane patches once channel activation was complete. Low temperature-rescued A561E-CFTR Cl− channels were studied at 23°C (B), at 37°C (C) and at 37°C in the presence of ivacaftor (VX-770; 10 μM) (D). Po values were calculated for each 30 s interval. In C and D, the left and right ordinates show Po (bars) and normalized Po (circles), respectively; Po values were normalized to that measured immediately temperature reached 37°C (t = 0–30 s). The wild-type CFTR data are the same as Figure 3A. For A561E-CFTR, the membrane patch was excised and channels activated at either 23°C (B) or 27°C (C and D). In B, temperature was maintained at 23°C, whereas in C and D, it was increased to 37°C before Po values were measured. Data are means ± SEM quantified from 4165 ± 1626 transitions (A561E: 23°C, N ≤ 3, n = 4); 6076 ± 2300 transitions (A561E: 37°C, N ≤ 5, n = 6) and 5164 ± 1639 transitions (A561E: 37°C + VX-770, N ≤ 2, n = 4).
To explore further the thermostability of A561E-CFTR, we used the iodide efflux assay to study large numbers of mutant Cl− channels in intact cells. Following incubation at 27°C for 24 h to deliver F508del- and A561E-CFTR to the cell surface, BHK-F508del-CFTR and BHK-A561E-CFTR cells were returned to 37°C and CFTR-mediated iodide efflux was monitored over an 8 h period. Figure 6 shows the magnitude of CFTR-mediated iodide efflux normalized to that measured at time t = 0 h when cells expressing temperature-rescued mutants were returned to 37°C. CFTR-mediated iodide efflux generated by BHK-WT-CFTR cells was stable over time, whereas that produced by BHK-F508del-CFTR and BHK-A561E-CFTR cells decreased progressively (Figure 6). Interestingly, loss of CFTR-mediated iodide efflux was quicker for A561E-CFTR (F508del; t½ CFTR-mediated iodide efflux ∼7 h; A561E; t½ CFTR-mediated iodide efflux ∼4 h; Figure 6), suggesting that A561E-CFTR is less stable than F508del-CFTR. We conclude that the A561E mutation perturbs severely CFTR stability at the cell surface.
Figure 6.
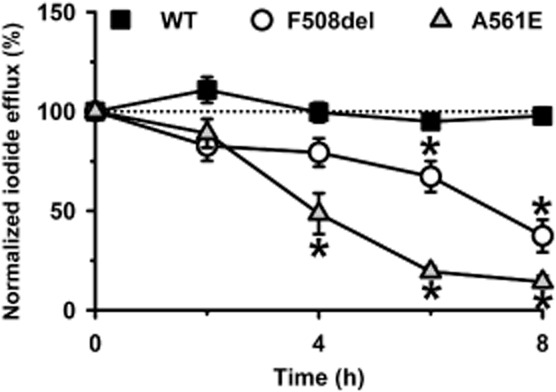
F508del- and A561E-CFTR exhibit a time-dependent loss of channel activity in intact cells studied at 37°C. The magnitude of CFTR-mediated iodide efflux elicited by BHK-WT-CFTR, BHK-F508del-CFTR and BHK-A561E-CFTR cells is shown for an 8 h period at 37°C. Prior to study, BHK-WT-CFTR cells were grown at 37°C, whereas BHK-F508del-CFTR and BHK-A561E-CFTR cells were grown at 27°C for 24 h. Values represent cumulative iodide efflux stimulated by forskolin (10 μM) and potentiated by genistein (50 μM) normalized to that measured at time t = 0 h, when cells expressing low temperature-rescued mutants were returned to 37°C. Data are means ± SEM (wild-type, n = 4–5; F508del-CFTR, n = 4–6; A561E-CFTR, n = 4–9); *P < 0.05 versus response of same cell line at t = 0 h.
CFTR potentiators augment A561E-CFTR channel function
High-throughput screening has identified many chemically diverse small molecules that rescue the gating defect of F508del-CFTR (Verkman and Galietta, 2009; Cai et al., 2011). To investigate whether CFTR potentiators might restore function to A561E-CFTR, we used the iodide efflux assay to test the effects of ivacaftor, the first CFTR potentiator to be approved for patient use (Van Goor et al., 2009; Ramsey et al., 2011) and five CFTR potentiators from the CFFT CFTR Compound Program (P2, P3, P4, P5 and P9) that robustly restore channel function to F508del-CFTR (see the heading ‘Materials’ and Supporting Information Figure S2). As a control, we employed genistein, the best-studied CFTR potentiator (Hwang and Sheppard, 1999; Lansdell et al., 2000; Ai et al., 2004). Based on published data (Caci et al., 2003; Sammelson et al., 2003; Yang et al., 2003; Pedemonte et al., 2005; Van Goor et al., 2009), we studied test CFTR potentiators at 10 μM and genistein at 50 μM, a concentration that generates close to maximum CFTR-mediated iodide efflux (Schmidt et al., 2008).
Figure 7 and Supporting Information Figures S3 and S4 show iodide efflux from BHK-WT-CFTR cells and low temperature-rescued BHK-F508del-CFTR and BHK-A561E-CFTR cells treated with different CFTR potentiators. For BHK-WT-CFTR cells, P2, P5 and P9 generated cumulative iodide efflux with comparable profiles to that of genistein, whereas those elicited by P3, P4 and ivacaftor were reduced in magnitude (Figure 7A). For BHK-F508del-CFTR cells, with the exception of ivacaftor, all test potentiators generated cumulative iodide efflux that was attenuated greatly in magnitude compared with genistein (Figure 7B). For BHK-A561E-CFTR cells, P3 and P5 generated cumulative iodide efflux similar to that observed with BHK-F508del-CFTR cells treated with test potentiators (Figure 7B and C). By contrast, P2, P4, P9 and ivacaftor all generated noticeable cumulative iodide efflux from BHK-A561E-CFTR cells, albeit reduced in magnitude compared with that of genistein (Figure 7C).
Figure 7.
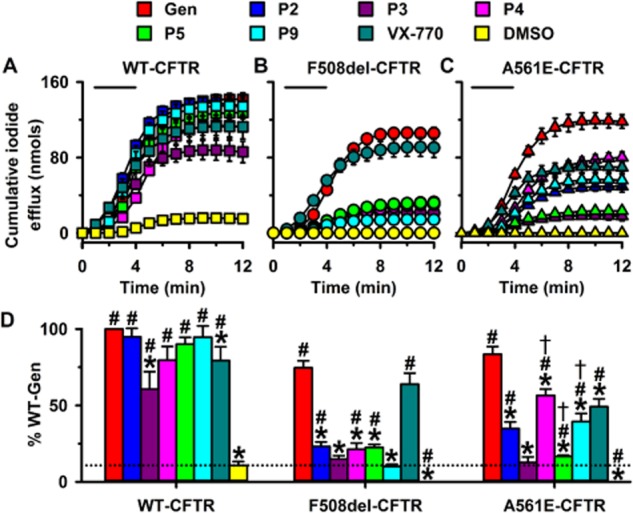
Rescue of F508del- and A561E-CFTR by CFTR potentiators. (A–C) Time courses of cumulative iodide efflux from BHK-WT-CFTR cells and low temperature-rescued BHK-F508del-CFTR and BHK-A561E-CFTR cells treated with forskolin (10 μM) and CFTR potentiators (genistein (Gen), 50 μM; test potentiators, 10 μM) or the vehicle DMSO (0.1% v·v−1) during the periods indicated by the bars. (D) Magnitude of iodide efflux elicited by different CFTR potentiators from BHK-WT-CFTR, BHK-F508del-CFTR and BHK-A561E-CFTR cells. Cumulative iodide efflux at 12 min is expressed as a percentage of that generated by wild-type CFTR treated with forskolin (10 μM) and genistein (50 μM). The dashed line indicates the DMSO response of wild-type CFTR; the DMSO responses of F508del- and A561E-CFTR are too small to be observed. Data are means ± SEM [n = 4, except ivacaftor (VX-770) where n = 6]; *P < 0.05 versus genistein data for same construct; #P < 0.05 versus wild-type CFTR DMSO response; †P < 0.05 versus F508del-CFTR data for same potentiator. Other details as in Figure 1.
To compare further the effects of CFTR potentiators, we expressed the magnitude of cumulative iodide efflux measured at the end of experiments as a percentage of that achieved by wild-type CFTR potentiated with genistein (50 μM) (Figure 7D). For wild-type CFTR, the rank order of potentiation was genistein ≥ P2 ≥ P9 ≥ P5 ≥ P4 ≥ ivacaftor > P3; for F508del-CFTR, it was genistein ≥ ivacaftor > > P2 ≥ P5 ≥ P4 ≥ P3 ≥ P9 and for A561E-CFTR, it was genistein > P4 ≥ ivacaftor ≥ P9 ≥ P2 > P5 ≥ P3 (Figure 7D). Two conclusions can be drawn from these data. First, most CFTR potentiators restored some function to F508del- and A561E-CFTR, but none achieved wild-type levels. Second, several CFTR potentiators restored more function to A561E-CFTR than F508del-CFTR: genistein, P2, P4 and P9, whereas P5 worked best on F508del-CFTR.
For ivacaftor, we determined the relationship between drug concentration and CFTR-mediated iodide efflux. Figure 8A and B and Supporting Information Figure S4 show CFTR-mediated iodide efflux from low temperature-rescued BHK-F508del-CFTR and BHK-A561E-CFTR cells treated with different concentrations of ivacaftor and Figure 8C summary data. At all concentrations of ivacaftor tested, cumulative iodide efflux from BHK-F508del-CFTR cells was greater than that from BHK-A561E-CFTR (F508del-CFTR, EC50 = 29 ± 3 nM; A561E-CFTR, EC50 = 136 ± 4 nM; n = 5 for both) (Figure 8C). These data suggest that ivacaftor potentiates F508del-CFTR with almost fivefold greater affinity than A561E-CFTR.
Figure 8.
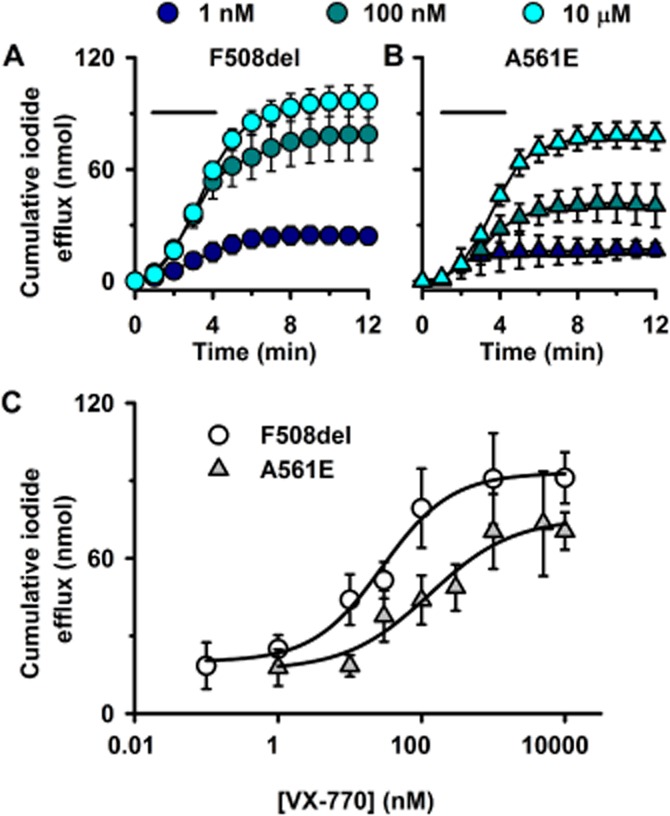
Ivacaftor potentiation of CFTR-mediated iodide efflux by F508del- and A561E-CFTR is concentration-dependent. (A and B) Time courses of cumulative iodide efflux from low temperature-rescued BHK-F508del-CFTR and BHK-A561E-CFTR cells treated with forskolin (10 μM) and the indicated concentrations of ivacaftor during the periods indicated by the bars. (C) Effects of ivacaftor concentration on cumulative CFTR-mediated iodide efflux from BHK-F508del and BHK-A561E-CFTR. Data are means ± SEM (n = 4–8). The continuous lines are the fit of the dose-response equation [log(agonist) vs. response, three parameters] using GraphPad Prism 6.03.
Among the test potentiators studied, P4 and ivacaftor restored greatest levels of function to A561E-CFTR. Therefore, we investigated their effects on the single-channel activity of low temperature-rescued F508del- and A561E-CFTR. To maximize channel activity and minimize channel rundown, we studied F508del- and A561E-CFTR channels at 27°C (Y. Wang, Z. Cai and D. N. Sheppard, unpubl. obs.). Figures 9A and 10A demonstrate that both P4 (10 μM) and ivacaftor (10 μM) enhanced F508del- and A561E-CFTR channel activity by altering channel gating without modifying current flow through open channels. Visual inspection of single-channel recordings suggests that P4 (10 μM) enhanced the frequency of channel openings, whereas ivacaftor (10 μM) augmented markedly both the frequency and duration of channel openings (Figures 9A and 10A). P4 (10 μM) increased Po fivefold for F508del-CFTR and twofold for A561E-CFTR without restoring channel activity to wild-type levels (Figure 9). By contrast, ivacaftor (10 μM) increased Po sevenfold for F508del-CFTR and fourfold for A561E-CFTR to restore wild-type levels of channel activity (but not gating pattern) to both mutants (Figure 10).
Figure 9.
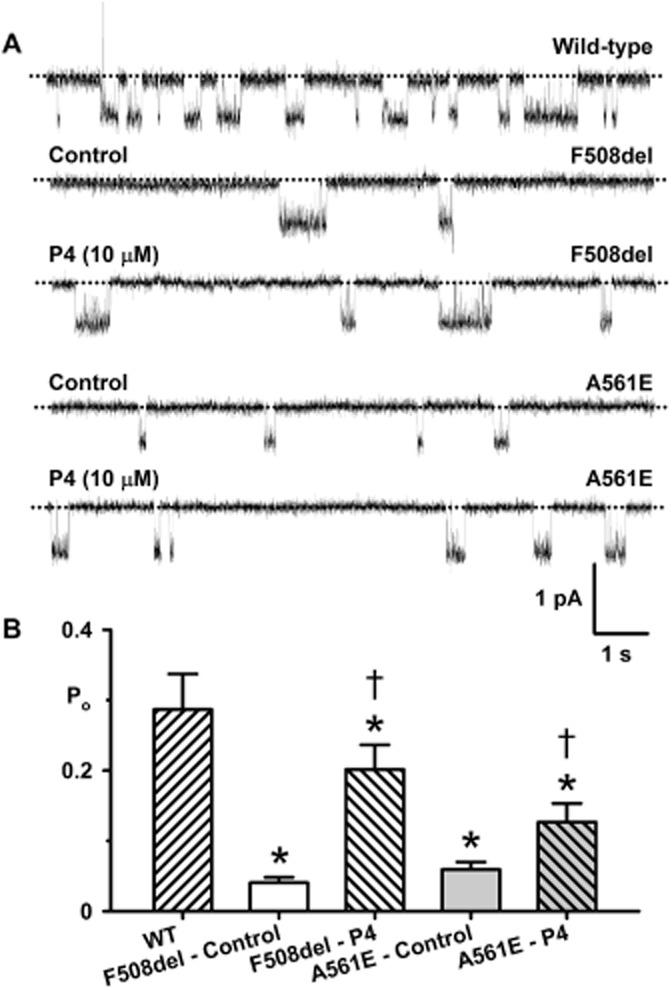
Potentiator P4 enhances F508del- and A561E-CFTR channel gating. (A) Representative single-channel recordings of wild-type CFTR and low temperature-rescued F508del- and A561E-CFTR in the absence and presence of P4 (10 μM). ATP (1 mM) and PKA (75 nM) were continuously present in the intracellular solution; temperature was 27°C. The dotted lines indicate closed channels and downward deflections of the traces denote channel openings. (B) Effects of P4 (10 μM) on the Po of wild-type CFTR and low temperature-rescued F508del- and A561E-CFTR studied at 27°C. Data are means + SEM quantified from 10 087 ± 2449 transitions (wild-type, N ≤ 3, n = 6); 1475 ± 176 transitions (F508del: control, N ≤ 5, n = 4); 3430 ± 1070 transitions (F508del: P4, N ≤ 5, n = 4); 1749 ± 619 transitions (A561E: control, N ≤ 5, n = 8); 10 470 ± 4123 transitions (A561E: P4, N ≤ 5, n = 8); *P < 0.05 versus wild-type CFTR; †P < 0.05 versus control without P4. Other details as in Figure 3.
Figure 10.
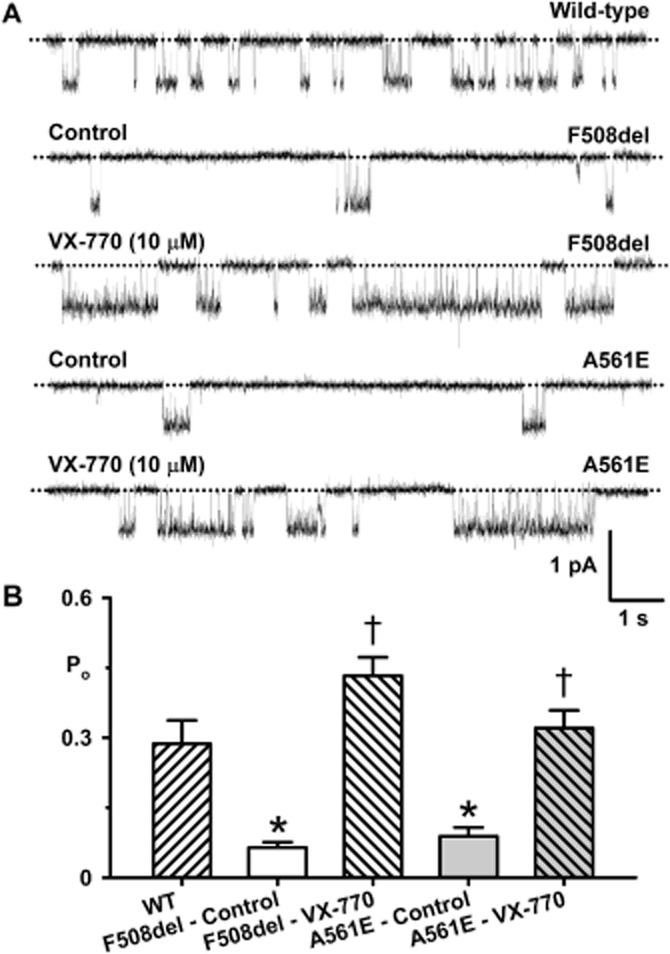
Ivacaftor restores wild-type levels of channel activity to F508del- and A561E-CFTR. (A) Representative single-channel recordings of wild-type CFTR and low temperature-rescued F508del- and A561E-CFTR in the absence and presence of ivacaftor (VX-770; 10 μM). ATP (1 mM) and PKA (75 nM) were continuously present in the intracellular solution; temperature was 27°C. The dotted lines indicate closed channels and downward deflections of the traces denote channel openings. (B) Effects of ivacaftor (10 μM) on the Po of wild-type CFTR and low temperature-rescued F508del- and A561E-CFTR studied at 27°C. The wild-type CFTR data are the same as Figure 9B. Data are means + SEM quantified from 2072 ± 679 transitions (F508del: control, N ≤ 2, n = 4); 30993 ± 8525 transitions (F508del: VX-770, N ≤ 2, n = 4); 1831 ± 537 transitions (A561E: control, N ≤ 2, n = 4); 8903 ± 1576 transitions (A561E: VX-770, N ≤ 2, n = 4); *P < 0.05 versus wild-type CFTR; †P < 0.05 versus control without VX-770. Other details as in Figure 3.
Because ivacaftor restored Po values to wild-type levels, we investigated its impact on the thermostability of mutant Cl− channels. Figures 4D and 5D show the single-channel activity of F508del- and A561E-CFTR measured as Po, over the first 9 min at 37°C once channels were fully potentiated by ivacaftor (10 μM). When compared with recordings made in the absence of the drug, Po values in the presence of ivacaftor (10 μM) were initially markedly greater, but they declined to zero noticeably quicker (Figures 4C and D and Figure 5C and D). Analysis of normalized Po values indicated that rundown of F508del-CFTR in the presence of ivacaftor (10 μM) occurred ∼90 s earlier, while for A561E-CFTR rundown occurred ∼150 s earlier (Figures 4C and D and Figure 5C and D). We conclude that ivacaftor potentiation of mutant Cl− channels accelerates channel rundown in excised inside-out membrane patches.
Discussion and conclusions
This study investigated the impact of the CF mutation A561E on the single-channel behaviour of CFTR and its rescue by CFTR potentiators. We demonstrated that A561E disrupts channel gating as severely as F508del, but its effects on thermostability are noticeably worse. With some differences in efficacy compared to F508del-CFTR, CFTR potentiators partially restore function to A561E-CFTR.
The mechanism of dysfunction of A561E-CFTR shows striking similarities to F508del-CFTR, but also some important differences. First, both mutations affect residues located within the ATP-binding cassette α-subdomain of NBD1 (Lewis et al., 2004). F508 is located on the surface of NBD1 in a loop between α-helices H3 and H4 (Lewis et al., 2004; 2005) where it interacts with the coupling helix of intracellular loop 4 (Mornon et al., 2008; Serohijos et al., 2008) (Supporting Information Figure S5). By contrast, A561 located within the H5 α-helix, which links the LSGGQ and Walker B motifs of NBD1, is partially buried in the tertiary structure of NBD1 (Lewis et al., 2004; Roxo-Rosa et al., 2006) (Supporting Information Figure S5). At first sight, the structural impact of F508del is widespread, whereas that of A561E might be localized to NBD1. However, the proximity of A561E to the NBD1: NBD2 dimer interface (Lewis et al., 2004; Roxo-Rosa et al., 2006) argues that it might also disrupt domain–domain interactions in CFTR.
Second, both F508del- and A561E-CFTR cause temperature-sensitive folding defects that prevent the acquisition of the correct tertiary conformation required to leave the endoplasmic reticulum and traffic to the Golgi apparatus, where CFTR is matured prior to delivery to the plasma membrane (Denning et al., 1992; Mendes et al., 2003). Using cell surface biotinylation, we found that after low temperature incubation the expression of F508del- and A561E-CFTR at the cell surface in recombinant BHK cells was equivalent and very low compared with that of wild-type CFTR. Interestingly, Roxo-Rosa et al. (2006) demonstrated that the revertant mutations G550E and 4RK (the simultaneous mutation of four arginine-framed tripeptides: R29K, R516K, R555K and R766K) were less effective at rescuing the expression and function of A561E- than F508del-CFTR. These data argue that the A561E-CFTR folding defect is less amenable than that of F508del-CFTR to rescue by alteration of NBD1 structure or arginine-framed tripeptide-mediated escape from endoplasmic reticulum retention/retrieval (Roxo-Rosa et al., 2006). This suggests that the processing defect of A561E-CFTR might be distinct from that of F508del-CFTR.
Third, F508del and A561E had very similar, if not identical, effects on CFTR channel gating. Both mutants prolonged IBI markedly, but did not change MBD with the result that Po was decreased sixfold compared to that of wild-type CFTR. The prolonged channel closures observed with both F508del- and A561E-CFTR suggest that both mutants disrupt the formation of the NBD1: NBD2 dimer in the ATP-driven NBD dimerization model of CFTR channel gating (Vergani et al., 2003; 2005). In this model, the LSGGQ motif of NBD1, which is located in close proximity to A561, contributes to the canonical ATP-binding site (site 2), where ATP binding initiates channel opening and ATP hydrolysis leads promptly to channel closure (Gadsby et al., 2006; Hwang and Sheppard, 2009). This suggests that A561E might distort the structure of ATP-binding site 2 to adversely affect channel gating. Some previous work argues that F508del might destabilize NBD1, hampering ATP binding to sites 1 and 2 (Jih et al., 2011). However, the location of F508 suggests that the mutation interferes with CFTR gating by disrupting coupling between the NBD1: NBD2 dimer and CFTR pore (Mornon et al., 2008; Serohijos et al., 2008).
Fourth, in marked contrast to wild-type CFTR, F508del- and A561E-CFTR activity was labile in both excised inside-out membrane patches and intact cells with channel deactivation noticeably quicker for A561E-CFTR. These data demonstrate that A561E-CFTR exhibits thermal instability similar to F508del-CFTR. They also suggest that this defect is worse for A561E-CFTR than for F508del-CFTR. The observation that thermal instability occurs in intact cells and cell-free membrane patches argues that it is not simply the result of severing ties with CFTR-interacting proteins (e.g. Valentine et al., 2012), but also a consequence of a change in CFTR structure. Consistent with this idea, Wang et al. (2011) and Liu et al. (2012) demonstrated that revertant mutations, which have direct effects on CFTR structure (e.g. G550E; Roxo-Rosa et al., 2006; Hoelen et al., 2010), restored some thermal stability to F508del-CFTR.
Understanding the mechanisms of CFTR dysfunction in CF is crucial for rational therapy development. Using the molecular classification of CF mutations (Welsh and Smith, 1993; Zielenski and Tsui, 1995; Rowe et al., 2005), both F508del and A561E are class II mutations (defective protein processing), which also exhibit characteristics of classes III (defective channel regulation) and VI (reduced protein stability). Thus, transformational drug therapies for both CF mutants will necessitate the use of (i) CFTR correctors to traffic the mutant protein to the cell surface; (ii) small molecules to stabilize the mutant protein’s conformation at the cell surface and (iii) CFTR potentiators to augment greatly mutant channel gating.
Although the mutations F508del and A561E have similar impacts on CFTR expression and function, their distinct locations in NBD1 raise the possibility that different small molecules will be required to traffic each mutant to the cell surface. For F508del-CFTR, the CFTR corrector lumacaftor restored to CF bronchial epithelia levels of transepithelial ion transport equivalent to 14% those of human bronchial epithelia expressing endogenous levels of wild-type CFTR (Van Goor et al., 2011). However, by itself, lumacaftor did not improve lung function in CF patients (Clancy et al., 2012). A potential explanation for the lack of clinical efficacy of lumacaftor is the impact of F508del on NBD1 folding and the interaction of NBD1 with intracellular loop 4 (Mendoza et al., 2012; Rabeh et al., 2012). These and other data (Farinha et al., 2013a; Okiyoneda et al., 2013) argue that combinations of CFTR correctors are likely to be required to restore fully the surface expression of F508del-CFTR. We speculate that the same is likely to be the case for A561E-CFTR.
With some differences in efficacy, most likely as a result of differences in experimental protocols (Pedemonte et al., 2010; present results), CFTR potentiators partially restored function to F508del- and A561E-CFTR. Of note, ivacaftor robustly potentiated both F508del- and A561E-CFTR. The EC50 value for ivacaftor potentiation of F508del-CFTR observed in the present study agrees well with that reported by Van Goor et al. (2009), while that for ivacaftor potentiation of A561E-CFTR was comparable with EC50 values for ivacaftor potentiation of various CF mutants including gating mutations (e.g. G551D-CFTR) and mutations with multiple mechanisms of CFTR dysfunction (e.g. R117H-CFTR) (Van Goor et al., 2009; 2014). In excised membrane patches, ivacaftor restored the activity of individual channels (measured by Po) to wild-type levels by increasing the frequency and duration of channel openings. These data suggest that ivacaftor accelerates the formation of the NBD1: NBD2 dimer and stabilizes its conformation in the ATP-driven NBD dimerization model of CFTR channel gating (Vergani et al., 2003; 2005). However, work by Eckford et al. (2012) and Jih and Hwang (2013) demonstrated that ivacaftor enhances ATP-independent channel gating by G551D-CFTR. Using a new model of CFTR channel gating, Jih and Hwang (2013) argue that ivacaftor acts on an ATP-independent pathway to favour occupancy of an open-state. Consistent with this idea, ivacaftor increases the open-time of wild-type CFTR (Jih and Hwang, 2013). Disappointingly, the robust potentiation of F508del- and A561E-CFTR by ivacaftor accelerated channel rundown. These and other data (Liu et al., 2012) suggest that channel activity is negatively correlated with thermal stability. These results have important implications for the development of CF therapy. They raise the possibility that high levels of mutant CFTR activity might be detrimental to protein stability. Future studies should explore this possibility further.
In conclusion, the CF mutation A561E causes CFTR dysfunction by several mechanisms. First, it induces a temperature-sensitive folding defect that prevents CFTR processing and intracellular transport (Mendes et al., 2003). Second, it severely destabilizes CFTR at the plasma membrane. Third, it perturbs strongly channel gating. Thus, the consequences for CFTR expression and function of A561E are very similar to those of F508del, even though F508del and A561E have distinct effects on CFTR structure (Lewis et al., 2004; Roxo-Rosa et al., 2006; Mornon et al., 2008; Serohijos et al., 2008). Importantly, the present results demonstrate that some channel function can be restored to A561E-CFTR with small molecules that potentiate F508del- and G551D-CFTR Cl− channels. We conclude that transformational drug therapy for A561E-CFTR will require a combination of small molecules designed to traffic the mutant protein to the plasma membrane, confer it with thermostability and rescue its gating defect.
Acknowledgments
We thank GS Banting, B Kleizen, I Braakman, I Callebaut, P Lehn and R Tarrant for valuable discussions and assistance. We are very grateful to MD Amaral (University of Lisboa) for generous gifts of cells; RJ Bridges (Rosalind Franklin University of Medicine and Science) and CFFT (Bethesda, MD, USA) for generous gifts of small molecules, and B Kleizen and I Braakman (Utrecht University), JR Riordan and T Jensen (University of North Carolina) and CFFT for generous gifts of anti-CFTR antibodies. This work was supported by the Cystic Fibrosis Trust and the University of Bristol; Y. W. was supported by a scholarship from Beijing Sun-Hope Intellectual Property Ltd.; J. L. was supported by scholarships from the University of Bristol and the Overseas Research Student Awards Scheme of Universities UK; A. L. was supported by an INSPIRE vacation studentship award; A. M. T. and B. R. H. were supported by NHS Blood and Transplant R&D and H. L. in part by the Engineering and Physical Sciences Research Council (grant no. EP/F03623X/1).
Glossary
- BHK
cells, baby hamster kidney cells
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- i
single-channel current amplitude
- IBI
interburst interval
- MBD
mean burst duration
- NBD
nucleotide-binding domain
- Po
open probability
Author contributions
H. L., A. M. T. and D. N. S. conceived and designed the experiments; Y. W., J. L., A. L., L. A. B., R. W., B. R. H., A. M. T. and H. L. performed the research; Y. W., J. L., A. L., L. A. B., R. W., B. R. H., Z. C., A. M. T., D. N. S. and H. L. analysed and interpreted the data; D. N. S., Z. C., A. M. T. and H. L. drafted the manuscript and revised it critically for important intellectual content. All authors approved the final version of the manuscript.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site: http://dx.doi.org/10.1111/bph.12791
Figure S1 Low-temperature incubation restores iodide efflux to F508del- and A561E-CFTR. (A and B) Time courses of iodide efflux from BHK cells expressing wild-type, F508del- and A561E-CFTR. BHK-F508del-CFTR and BHK-A561E-CFTR cells were grown at 37°C (A) or 27°C for 24 h before the experiment (B). In (A) and (B), BHK-WT-CFTR cells were grown at 37°C. During the periods indicated by the bars, cells were treated with forskolin (10 μM) and genistein (50 μM). Data are means ± SEM (n = 4); error bars smaller than symbol size are not shown.
Figure S2 Chemical structures of small molecule CFTR potentiators. The structures of genistein and the test CFTR potentiators used in this study are shown. Abbreviations: PG-01, 2-[(2-1H-indol-3-yl-acetyl)-methyl-amino]-N-(4-isopropyl-phenyl)-2-phenyl-acetamide; SF-03, 6-(Ethyl-phenyl-sulfonyl)-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid 2-methoxy-benzylamide; UCCF-853, 1-(3-chlorophenyl)-5-trifluoromethyl-3-hydrobenzimidazol-2-one; ΔF508act-02, 2-(2-chloro-benzoyl-amino)-4,5,6,7-tetrahydro-benzo[b]thiophene-3-carboxylic acid amide; UCCF-180, 3-but-3-ynyl-5-methoxy-1-phenyl-1H-pyrazole-4-carbaldehyde; ivacaftor (VX-770), N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide.
Figure S3 Small-molecule potentiation of iodide efflux from BHK cells expressing wild-type, F508del and A561E-CFTR. Time courses of iodide efflux from BHK cells expressing wild-type CFTR (A and D) and low temperature-rescued F508del- (B and E) and A561E-CFTR (C and F). (A–C) Effects of the test potentiators P2, P3 and P5, genistein and the vehicle DMSO (0.1% v·v−1). (D–F) Effects of the test potentiators P4 and P9, genistein and DMSO (0.1% v·v−1). During the periods indicated by the bars, cells were treated with forskolin (10 μM) and CFTR potentiators (genistein, 50 μM; test potentiators, 10 μM). Data are means ± SEM (n = 4); error bars smaller than symbol size are not shown.
Figure S4 Ivacaftor potentiates iodide efflux from BHK cells expressing F508del- and A561E-CFTR. Time courses of iodide efflux from BHK cells expressing low temperature-rescued F508del- (A) and A561E-CFTR (B). During the periods indicated by the bars, cells were treated with forskolin (10 μM) and the indicated concentrations of ivacaftor (VX-770). Data are means ± SEM (n = 4–8); error bars smaller than symbol size are not shown.
Figure S5 Molecular model of human CFTR showing the location of F508 and A561. (A and C) show two orthogonal views of CFTR using a ribbon representation to depict the overall architecture of the channel. In C, the model has been rotated ∼45° relative to that shown in A. (B and D) show enlargements of the boxed sections in A and C respectively. Different domains of CFTR are identified using different colours: dark blue, membrane-spanning domain 1 (MSD1); red, MSD2; light blue, nucleotide-binding domain 1 (NBD1); orange, NBD2; grey, regulatory domain (RD). F508 in NBD1 is coloured green and A561 pink. The positions of nucleotides bound to the NBD dimer are shown (carbon, white; nitrogen, blue; oxygen, red; phosphorus, orange). These models of CFTR are based on the crystal structure of Sav1866 (Dawson and Locher, 2006). They represent CFTR in a nucleotide-bound outward-facing (i.e. open-channel) configuration. Modified, with permission, from Mornon et al. (2008).
References
- Ai T, Bompadre SG, Wang X, Hu S, Li M, Hwang T-C. Capsaicin potentiates wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride-channel currents. Mol Pharmacol. 2004;65:1415–1426. doi: 10.1124/mol.65.6.1415. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Bell S, Konstan M, McColley SA, Kang L, Patel N, et al. The investigational CFTR corrector, VX-809 (lumacaftor) co-administered with the oral potentiator ivacaftor improved CFTR and lung function in F508del homozygous patients: phase II study results. Pediatr Pulmonol Suppl. 2012;35:315. [Google Scholar]
- Caci E, Folli C, Zegarra-Moran O, Ma T, Springsteel MF, Sammelson RE, et al. CFTR activation in human bronchial epithelial cells by novel benzoflavone and benzimidazolone compounds. Am J Physiol Lung Cell Mol Physiol. 2003;285:L180–L188. doi: 10.1152/ajplung.00351.2002. [DOI] [PubMed] [Google Scholar]
- Cai Z, Taddei A, Sheppard DN. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J Biol Chem. 2006;281:1970–1977. doi: 10.1074/jbc.M510576200. [DOI] [PubMed] [Google Scholar]
- Cai Z-W, Liu J, Li H-Y, Sheppard DN. Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis. Acta Pharmacol Sin. 2011;32:693–701. doi: 10.1038/aps.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappe V, Hinkson DA, Zhu T, Chang X-B, Riordan JR, Hanrahan JW. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J Physiol. 2003;548:39–52. doi: 10.1113/jphysiol.2002.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Aleksandrov L, Chang X-B, Hou Y-X, He L, Hegedus T, et al. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol. 2007;365:981–994. doi: 10.1016/j.jmb.2006.10.086. [DOI] [PubMed] [Google Scholar]
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, et al. Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature. 1991;354:526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Eckford PDW, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem. 2012;287:36639–36649. doi: 10.1074/jbc.M112.393637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, Nogueira P, Mendes F, Penque D, Amaral MD. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J. 2002;366:797–806. doi: 10.1042/BJ20011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, King-Underwood J, Sousa M, Correia AR, Henriques BJ, Roxo-Rosa M, et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem Biol. 2013a;20:943–955. doi: 10.1016/j.chembiol.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Matos P, Amaral MD. Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: not just from the endoplasmic reticulum to the Golgi. FEBS J. 2013b;280:4396–4406. doi: 10.1111/febs.12392. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146:746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Hanrahan JW, Sampson HM, Thomas DY. Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol Sci. 2013;34:119–125. doi: 10.1016/j.tips.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Hirtz S, Gonska T, Seydewitz HH, Thomas J, Greiner P, Kuehr J, et al. CFTR Cl− channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology. 2004;127:1085–1095. doi: 10.1053/j.gastro.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Hoelen H, Kleizen B, Schmidt A, Richardson J, Charitou P, Thomas PJ, et al. The primary folding defect and rescue of ΔF508 CFTR emerge during translation of the mutant domain. PLoS ONE. 2010;5:e15458. doi: 10.1371/journal.pone.0015458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T-C, Sheppard DN. Molecular pharmacology of the CFTR Cl− channel. Trends Pharmacol Sci. 1999;20:448–453. doi: 10.1016/s0165-6147(99)01386-3. [DOI] [PubMed] [Google Scholar]
- Hwang T-C, Sheppard DN. Gating of the CFTR Cl− channel by ATP-driven nucleotide-binding domain dimerisation. J Physiol. 2009;587:2151–2161. doi: 10.1113/jphysiol.2009.171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih K-Y, Hwang T-C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci U S A. 2013;110:4404–4409. doi: 10.1073/pnas.1215982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih K-Y, Li M, Hwang T-C, Bompadre SG. The most common cystic fibrosis-associated mutation destabilizes the dimeric state of the nucleotide-binding domains of CFTR. J Physiol. 2011;589:2719–2731. doi: 10.1113/jphysiol.2010.202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell KA, Delaney SJ, Lunn DP, Thomson SA, Sheppard DN, Wainwright BJ. Comparison of the gating behaviour of human and murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in mammalian cells. J Physiol. 1998a;508:379–392. doi: 10.1111/j.1469-7793.1998.379bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell KA, Kidd JF, Delaney SJ, Wainwright BJ, Sheppard DN. Regulation of murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in Chinese hamster ovary cells. J Physiol. 1998b;512:751–764. doi: 10.1111/j.1469-7793.1998.751bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell KA, Cai Z, Kidd JF, Sheppard DN. Two mechanisms of genistein inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in murine cell line. J Physiol. 2000;524:317–330. doi: 10.1111/j.1469-7793.2000.t01-1-00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004;23:282–293. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, et al. Impact of the ΔF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem. 2005;280:1346–1353. doi: 10.1074/jbc.M410968200. [DOI] [PubMed] [Google Scholar]
- Liu X, O’Donnell N, Landstrom A, Skach WR, Dawson DC. Thermal instability of ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) channel function: protection by single suppressor mutations and inhibiting channel activity. Biochemistry. 2012;51:5113–5124. doi: 10.1021/bi300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Chang X-B, Bear C, Kartner N, Mohamed A, Riordan JR, et al. The ΔF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane: determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–21598. [PubMed] [Google Scholar]
- Mendes F, Roxo-Rosa M, Dragomir A, Farinha CM, Roomans GM, Amaral MD, et al. Unusually common cystic fibrosis mutation in Portugal encodes a misprocessed protein. Biochem Biophys Res Commun. 2003;311:665–671. doi: 10.1016/j.bbrc.2003.10.048. [DOI] [PubMed] [Google Scholar]
- Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, et al. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148:164–174. doi: 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornon J-P, Lehn P, Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci. 2008;65:2594–2612. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol. 2013;9:444–454. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Research. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, et al. Phenylglycine and sulfonamide correctors of defective ΔF508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol. 2005;67:1797–1807. doi: 10.1124/mol.105.010959. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Tomati V, Sondo E, Galietta LJV. Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol. 2010;298:C866–C874. doi: 10.1152/ajpcell.00404.2009. [DOI] [PubMed] [Google Scholar]
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148:150–163. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1671. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- Roxo-Rosa M, Xu Z, Schmidt A, Neto M, Cai Z, Soares CM, et al. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc Natl Acad Sci U S A. 2006;103:17891–17896. doi: 10.1073/pnas.0608312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammelson RE, Ma T, Galietta LJV, Verkman AS, Kurth MJ. 3-(2-benzyloxyphenyl)isoxazoles and isoxazolines: synthesis and evaluation as CFTR activators. Bioorg Med Chem Lett. 2003;13:2509–2512. doi: 10.1016/s0960-894x(03)00482-7. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hughes LK, Cai Z, Mendes F, Li H, Sheppard DN, et al. Prolonged treatment of cells with genistein modulates the expression and function of the cystic fibrosis transmembrane conductance regulator. Br J Pharmacol. 2008;153:1311–1323. doi: 10.1038/sj.bjp.0707663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz BD, Frizzell RA, Bridges RJ. Rescue of dysfunctional ΔF508-CFTR chloride channel activity by IBMX. J Membr Biol. 1999;170:51–66. doi: 10.1007/s002329900537. [DOI] [PubMed] [Google Scholar]
- Serohijos AWR, Hegedús T, Aleksandrov AA, He L, Cui L, Dokholyan NV, et al. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J Physiol. 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Ostedgaard LS, Winter MC, Welsh MJ. Mechanism of dysfunction of two nucleotide binding domain mutations in cystic fibrosis transmembrane conductance regulator that are associated with pancreatic sufficiency. EMBO J. 1995;14:876–883. doi: 10.1002/j.1460-2075.1995.tb07069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toye AM, Banting G, Tanner MJA. Regions of human kidney anion exchanger 1 (kAE1) required for basolateral targeting of kAE1 in polarised kidney cells: mis-targeting explains dominant renal tubular acidosis (dRTA) J Cell Sci. 2004;117:1399–1410. doi: 10.1242/jcs.00974. [DOI] [PubMed] [Google Scholar]
- Toye AM, Williamson RC, Khanfar M, Bader-Meunier B, Cynober T, Thibault M, et al. Band 3 Courcouronnes (Ser667Phe): a trafficking mutant differentially rescued by wild-type band 3 and glycophorin A. Blood. 2008;111:5380–5389. doi: 10.1182/blood-2007-07-099473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine CD, Lukacs GL, Verkman AS, Haggie PM. Reduced PDZ interactions of rescued ΔF508CFTR increases its cell surface mobility. J Biol Chem. 2012;287:43630–43638. doi: 10.1074/jbc.M112.421172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros. 2014;13:29–36. doi: 10.1016/j.jcf.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Venglarik CJ, Schultz BD, Frizzell RA, Bridges RJ. ATP alters current fluctuations of cystic fibrosis transmembrane conductance regulator: evidence for a three-state activation mechanism. J Gen Physiol. 1994;104:123–146. doi: 10.1085/jgp.104.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Nairn AC, Gadsby DC. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J Gen Physiol. 2003;121:17–36. doi: 10.1085/jgp.20028673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433:876–880. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS, Galietta LJV. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Okeyo GO, Tao B, Hong JS, Kirk KL. Thermally unstable gating of the most common cystic fibrosis mutant channel (ΔF508): ‘rescue’ by suppressor mutations in nucleotide binding domain 1 and by constitutive mutations in the cytosolic loops. J Biol Chem. 2011;286:41937–41948. doi: 10.1074/jbc.M111.296061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. Eighth edn. New York: McGraw-Hill; 2001. pp. 5121–5188. [Google Scholar]
- Xu Z, Pissarra LS, Farinha CM, Liu J, Cai Z, Thibodeau PH, et al. Revertant mutants modify, but do not rescue, the gating defect of the cystic fibrosis mutant G551D-CFTR. J Physiol. 2014;592:1931–1947. doi: 10.1113/jphysiol.2014.271817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, et al. Nanomolar affinity small molecule correctors of defective ΔF508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- Zielenski J, Tsui L-C. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Low-temperature incubation restores iodide efflux to F508del- and A561E-CFTR. (A and B) Time courses of iodide efflux from BHK cells expressing wild-type, F508del- and A561E-CFTR. BHK-F508del-CFTR and BHK-A561E-CFTR cells were grown at 37°C (A) or 27°C for 24 h before the experiment (B). In (A) and (B), BHK-WT-CFTR cells were grown at 37°C. During the periods indicated by the bars, cells were treated with forskolin (10 μM) and genistein (50 μM). Data are means ± SEM (n = 4); error bars smaller than symbol size are not shown.
Figure S2 Chemical structures of small molecule CFTR potentiators. The structures of genistein and the test CFTR potentiators used in this study are shown. Abbreviations: PG-01, 2-[(2-1H-indol-3-yl-acetyl)-methyl-amino]-N-(4-isopropyl-phenyl)-2-phenyl-acetamide; SF-03, 6-(Ethyl-phenyl-sulfonyl)-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid 2-methoxy-benzylamide; UCCF-853, 1-(3-chlorophenyl)-5-trifluoromethyl-3-hydrobenzimidazol-2-one; ΔF508act-02, 2-(2-chloro-benzoyl-amino)-4,5,6,7-tetrahydro-benzo[b]thiophene-3-carboxylic acid amide; UCCF-180, 3-but-3-ynyl-5-methoxy-1-phenyl-1H-pyrazole-4-carbaldehyde; ivacaftor (VX-770), N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide.
Figure S3 Small-molecule potentiation of iodide efflux from BHK cells expressing wild-type, F508del and A561E-CFTR. Time courses of iodide efflux from BHK cells expressing wild-type CFTR (A and D) and low temperature-rescued F508del- (B and E) and A561E-CFTR (C and F). (A–C) Effects of the test potentiators P2, P3 and P5, genistein and the vehicle DMSO (0.1% v·v−1). (D–F) Effects of the test potentiators P4 and P9, genistein and DMSO (0.1% v·v−1). During the periods indicated by the bars, cells were treated with forskolin (10 μM) and CFTR potentiators (genistein, 50 μM; test potentiators, 10 μM). Data are means ± SEM (n = 4); error bars smaller than symbol size are not shown.
Figure S4 Ivacaftor potentiates iodide efflux from BHK cells expressing F508del- and A561E-CFTR. Time courses of iodide efflux from BHK cells expressing low temperature-rescued F508del- (A) and A561E-CFTR (B). During the periods indicated by the bars, cells were treated with forskolin (10 μM) and the indicated concentrations of ivacaftor (VX-770). Data are means ± SEM (n = 4–8); error bars smaller than symbol size are not shown.
Figure S5 Molecular model of human CFTR showing the location of F508 and A561. (A and C) show two orthogonal views of CFTR using a ribbon representation to depict the overall architecture of the channel. In C, the model has been rotated ∼45° relative to that shown in A. (B and D) show enlargements of the boxed sections in A and C respectively. Different domains of CFTR are identified using different colours: dark blue, membrane-spanning domain 1 (MSD1); red, MSD2; light blue, nucleotide-binding domain 1 (NBD1); orange, NBD2; grey, regulatory domain (RD). F508 in NBD1 is coloured green and A561 pink. The positions of nucleotides bound to the NBD dimer are shown (carbon, white; nitrogen, blue; oxygen, red; phosphorus, orange). These models of CFTR are based on the crystal structure of Sav1866 (Dawson and Locher, 2006). They represent CFTR in a nucleotide-bound outward-facing (i.e. open-channel) configuration. Modified, with permission, from Mornon et al. (2008).