

Abstract
Background and Purpose
Different protease-activated receptors (PARs) activated by thrombin are involved in cardiovascular disease, via up-regulation of inflammatory proteins including COX-2. However, the mechanisms underlying thrombin-regulated COX-2 expression in human cardiomyocytes remain unclear.
Experimental Approach
Human cardiomyocytes were used in the study. Thrombin-induced COX-2 protein and mRNA expression, and signalling pathways were determined by Western blot, real-time PCR and COX-2 promoter luciferase reporter assays, and pharmacological inhibitors or siRNAs. PGE2 generation and cell proliferation were also determined.
Key Results
Thrombin-induced COX-2 protein and mRNA expression, promoter activity and PGE2 release was attenuated by the PAR1 antagonist (SCH79797) or the inhibitors of proteinase activity (PPACK), MEK1/2 (U0126), p38 MAPK (SB202190) or JNK1/2 (SP600125), and transfection with small interfering RNA (siRNA) of PAR1, p38, p42 or JNK2. These results suggested that PAR1-dependent MAPKs participate in thrombin-induced COX-2 expression in human cardiomyocytes. Moreover, thrombin stimulated phosphorylation of MAPKs, which was attenuated by PPACK and SCH79797. Furthermore, thrombin-induced COX-2 expression was blocked by the inhibitors of AP-1 (tanshinone IIA) and NF-κB (helenalin). Moreover, thrombin-stimulated phosphorylation of c-Jun/AP-1 and p65/NF-κB was attenuated by tanshinone IIA and helenalin, respectively, suggesting that thrombin induces COX-2 expression via PAR1/MAPKs/AP-1 or the NF-κB pathway. Functionally, thrombin increased human cardiomyocyte proliferation through the COX-2/PGE2 system linking to EP2 receptors, as determined by proliferating cell nuclear antigen and cyclin D1 expression.
Conclusions and Implications
These findings demonstrate that MAPKs-mediated activation of AP-1/NF-κB pathways is, at least in part, required for COX-2/PGE2/EP2-triggered cell proliferation in human cardiomyocytes.
Table of Links
| TARGETS | LIGANDS | |
|---|---|---|
| COX-2 | AH6809 | Thrombin |
| EP2 prostanoid receptor | Celecoxib | U0126 |
| Erk1/2 | PGE | |
| JNK1/2 | SB202190 | |
| p38 MAPK | SP600125 | |
| PAR1 | TFLLR-NH2 |
This Table lists the protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a, Alexander et al., 2013b).
Introduction
Heart failure, one of the cardiovascular conditions with high morbidity and mortality, describes a situation where the heart is incapable of supplying sufficient blood for circulation (Heineke and Molkentin, 2006). The characteristic response of heart failure occurs in the ventricular chambers. In response to cytokines, neurohormones, growth factors and cardiac injury, ventricular cardiomyocytes increase in size and thickness of walls and reorganize the sarcomeres but reduce the internal dimensions of the ventricular chamber in an attempt to provide sufficient blood for peripheral tissues and organs (Ritter and Neyses, 2003).
The major action of thrombin is to prevent blood loss at the sites of injury through converting fibrinogen to fibrin by forming rigid blood clots (Ariens, 2013) and most studies concerning thrombin have focused on vascular endothelium, platelets and other cardiovascular components, but little is known about its role in the heart. Thrombin exerts its physiological and pathological processes via cellular surface receptors, known as protease-activated receptors (PARs), a class of the GPCR family (Coughlin, 2000). The PARs are divided into four subtypes, PAR1, PAR-2, PAR-3 and PAR-4, in cardiovascular systems. PAR1 is widespread in cells and tissues and its activation regarding platelet activation and vasodilatation (Coughlin, 1999). PAR1, PAR-2 and PAR-4 are expressed in myocardium where activation of PAR1 activation leads to a broad range of signalling events in cardiomyocytes (Sabri et al., 2003). PAR1 is functionally linked to G-protein, PLC, MAPKs and Akt (Barnes et al., 2004). Moreover, PAR1 and PAR-2 may share the common signalling pathways and contribute to dilated hypertrophy in neonatal rat cardiomyocytes (Sabri et al., 2000; Moshal et al., 2005a). Activation of PAR1 by thrombin increases atrial natriuretic peptide mRNA levels in neonatal rat ventricular cardiomyocyte (Glembotski et al., 1993). Furthermore, up-regulation of PAR1 has been demonstrated to contribute to cardiac hypertrophy and remodelling, suggesting that PAR1 may be a novel therapeutic target in heart failure (Moshal et al., 2005b; Pawlinski et al., 2007).
Two COX isoforms have been identified as the enzymes responsible for PG synthesis in most tissues (Duvivier et al., 1975). The COXs convert arachidonic acid into its hydroperoxy-endoperoxide, PGG2, which is quickly reduced to PGH2 and thence to various PG end-products (Streicher and Wang, 2008). Among COXs, COX-1 is ubiquitously expressed in mammals mediating physiological homeostasis (Rolin et al., 2007). COX-2 can be rapidly induced in response to many proinflammatory mediators, suggesting that COX-2 may participate in inflammation (Smith et al., 2000). Moreover, up-regulation of COX-2 has been found in cardiomyocytes from heart failure patients whereas it was undetectable in healthy controls (Norman et al., 1998; Saito and Giaid, 1999). In addition to COX-2, high levels of PGs also have been found in patients with heart failure (Kotlyar et al., 2006). However, the relationship between thrombin and COX-2 in cardiomyocytes remains to be investigated.
In the present study, we sought to elucidate the mechanisms underlying thrombin-induced COX-2 expression in human cardiomyocytes. The results demonstrated that thrombin-induced COX-2 expression and PGE2 generation is mediated through a PAR1-dependent signalling pathway, including p38 MAPK, Erk1/2, JNK1/2, AP-1 and NF-κB. Moreover, we confirmed that thrombin could induce cell proliferation via PAR1-dependent COX-2/PGE2/EP2 recptor signalling pathway. These results provided new insight into therapeutic targets on the mechanisms of thrombin acts that could potentially ameliorate heart failure.
Methods
Primary human cardiomyocyte culture
Primary human neonatal cardiomyocytes were purchased from ScienCell Research Laboratories (San Diego, CA, USA). The origin of cells was isolated and cultured from one neonatal donor, and confirmed by immunostaining for sarcomeric α-actinin myosin staining. The cell population consisted of 90∼95% cardiomyocytes. The cells were cultured in a commercial growth medium supplemented with 5% FBS, growth supplements (10 μg·mL−1 BSA, 10 μg·mL−1 apo-transferrin, 5 μg·mL−1 insulin, 2 ng·mL−1 EGF, 2 ng·mL−1 FGF-2, 2 ng·mL−1 IGF-1, 1 μg·mL−1 hydrocortisone and 100 nM retinoic acid), and antibiotics (100 U·mL−1 penicillin G, 100 μg·mL−1 streptomycin and 250 ng·mL−1 fungizone) at 37°C in a humidified 5% CO2 atmosphere. When the cultures reached confluence (4 days), cells were treated with 0.05% trypsin/1 mM EDTA for 3 min at 37°C. The cell suspension was diluted with commercial growth medium to a concentration of 2 × 105 cells·mL−1. The cell suspension was plated onto (2 mL per well) 6-well culture plates and (10 mL per dish) 10 cm culture dishes for the measurement of protein expression and mRNA accumulation respectively. Culture medium was changed after 48 h and then every 3 days.
Sample preparation and Western blot analysis
Human cardiomyocytes were plated onto 6-well culture plates and made quiescent at confluence by incubation in DMEM/F-12 with 0.05% BSA and 2 mM glutamine for 24 h. Growth-arrested cells were incubated with or without different concentrations of thrombin at 37°C for the indicated time intervals. When inhibitors were used, they were added 1 h prior to the application of thrombin. After incubation, the cells were then rapidly washed with ice-cold PBS, scraped and collected by centrifugation at 1000× g for 10 min. The collected whole cells were lysed with ice-cold lysis buffer containing: 25 mM Tris-HCl, pH 7.4, 25 mM NaCl, 25 mM NaF, 25 mM sodium pyrophosphate, 1 mM sodium vanadate, 2.5 mM EDTA, 2.5 mM EGTA, 0.05% Triton X-100, 0.5% SDS, 0.5% deoxycholate, 0.5% NP-40, 5 μg·mL−1 leupeptin, 5 μg·mL−1 aprotinin, and 1 mM phenylmethylsulfonyl fluoride. The lysates were centrifuged at 45 000× g for 1 h at 4°C to yield the whole cell extract. The protein concentration was determined by using BCA reagents according to the instructions of the manufacturer. Samples from these supernatant fractions (30 μg protein) were denatured and subjected to SDS-PAGE using a 10% running gel. Proteins were transferred to nitrocellulose membrane and incubated successively at room temperature with 5% BSA in Tween-Tris buffered saline (50 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20, pH 7.4) for 1 h. Membranes were incubated overnight at 4°C with their respective component antibody or anti-GAPDH antibody used at a dilution of 1:2000 in Tween-Tris buffered saline. Membranes were washed with Tween-Tris buffered saline four times for 5 min each, incubated with a 1:1500 dilution of anti-mouse horseradish peroxidase antibody for 1 h. Following each incubation, the membrane was washed extensively with Tween-Tris buffered saline. The immunoreactive bands were detected by ECL reagents and captured by a UVP BioSpectrum 500 Imaging System (Upland, CA, USA). The image densitometry analysis was quantified by an UN-SCAN-IT gel software (Orem, UT, USA).
Total RNA extraction and RT-PCR analysis
Total RNA was isolated from human cardiomyocytes (10 cm culture dishes) incubated with thrombin for the indicated time intervals, using TRIzol according to the protocol of the manufacturer. RNA concentration was spectrophotometrically determined at 260 nm. First strand cDNA synthesis was performed with 2 μg of total RNA using random hexamers as primers in a final volume of 20 μL (5 μg·μL−1 random hexamers, 1 mM dNTPs, 2 units·μL−1 RNasin and 10 units·μL−1 Moloney murine leukaemia virus reverse transcriptase). The reaction was carried out at 37°C for 60 min. cDNAs encoding COX-2, β-actin, PAR1-4 and EP1-4 were amplified from 3–5 μL of the cDNA reaction mixture using specific gene primers. The primers, as previously described (Kunisch et al., 2009; Seminario-Vidal et al., 2009), were used for amplification reaction.
Real-time RT-PCR analysis
Real-time PCR was performed with the TaqMan gene expression assay system, using primers and probe mixes for COX-2 and endogenous GAPDH control genes. PCRs were performed using SYBR Green PCR reagents (Applied Biosystems, Branchburg, NJ, USA). Relative gene expression was determined by the ΔΔCt method, where Ct meant threshold cycle. All experiments were performed in triplicate (n = 3).
Human COX-2, AP-1 and NF-κB promoter cloning, transient transfection and promoter activity assays
The upstream region (−1280 to +19) of the human COX-2 promoter was cloned into the pGL3-basic vector containing the luciferase reporter system. Briefly, a 1.3 kb segment at the 5′-flanking region of the human COX-2 gene was amplified by PCR using specific primers for the human COX-2 gene (accession no. U36476): 5′-ccccggtaccGAAGGCGAAATGCTTTGCCC (forward/Kpn1) and 5′-ccccctcgaGGGTGAGAACCGAAGCTTCTG (reverse/Xho1). The pGL3-Basic vector, containing a polyadenylation signal upstream from the luciferase gene, was used to construct the expression vectors by subcloning PCR-amplified DNA of the COX-2 promoter into the Kpn1/Xho1 site of this vector. The PCR products (pGL3-COX-2WT) were confirmed by their sizes, as determined by electrophoresis and by DNA sequencing. Additionally, the introduction of a mismatched primer mutation into the AP-1 and NF-κB to generate pGL3-COX-2ΔAP-1 and pGL3-COX-2ΔNF-κB was performed, using the following (forward) primer: ΔAP-1: 5′-ACACACACCCTGAGTTGGCG-3′. ΔNF-κB: 5′-GGTAGGCTTACTGGGCCCCCAC-3′ respectively. All plasmids were prepared by using QIAGEN plasmid DNA preparation kits. The COX-2 promoter reporter construct was transfected into HCM cells using the Lipofectamine reagent according to the instructions of manufacture. To assess promoter activity, cells were collected and disrupted by sonication in lysis buffer (25 mM Tris-phosphate, pH 7.8, 2 mM EDTA, 1% Triton X-100 and 10% glycerol). After centrifugation, aliquots of the supernatants were tested for luciferase activity using the luciferase assay system (Promega, Madison, WI, USA). The chemiluminescence was determined by using a Synergy H1 Hybrid reader (BioTek, Winooski, VT, USA) and processed with Gene 5 software (BioTek) (version 2.0). Firefly luciferase activities were standardized to those of β-galactosidase activity. The data presented are summarized from three independent assays.
Measurement of PGE2 release
To determine the level of PGE2, human cardiomyocytes were treated with thrombin for indicated time intervals. Culture supernatants were analysed for secreted PGE2, using a PGE2 Enzyme Immunoassay Kit (Cayman Chem., Ann Arbor, MI, USA) according to the manufacturer’s instructions.
Transient transfection with siRNAs
Human siRNAs of scrambled, PAR1, p38, Erk1, JNK2 c-Jun and p65 were from Sigma. Transient transfection of siRNAs (100 nM) was performed using a Lipofectamine 2000 reagent according to the manufacturer’s instructions.
Cell proliferation assay
Human cardiomyocytes (2 × 104 cells per well) were seeded in 24-well culture plates and incubated at 37°C in commercial growth medium. After reaching 70% confluence, human cardiomyocytes were serum-starved for 24 h and incubated with thrombin for the indicated time intervals. Proliferation of human cardiomyocytes was assessed by a cell counting kit (CCK-8), by measuring mitochondria-metabolized formazan generation, directly proportional to live cells. The absorbance of samples was measured with a wavelength of 450 nm on an elisa reader according to the manufacturer’s instructions. Cell counting assay was also performed to assess the cell proliferation. Trypsin-digested human cardiomyocytes were collected, centrifuged and resuspended in PBS. Subsequently, Trypan blue solution was mixed with cell suspension in a 1:1 ratio, and the cells were counted in a haemocytometer via microscope.
Data analysis
Data are expressed as mean ± SEM and analysed by one-way anova followed with Tukey’s post hoc test. Analysis was carried out with the GraphPad Prism program (Graph Pad, San Diego, CA, USA). A value of P < 0.05 was considered significant.
Materials
Anti-COX-2 antibody (#2169-1) was from Epitomics (Burlingame, CA, USA). Anti-phospho-c-Jun (#2361), anti-phospho-p42/p44 MAPK (#9101), anti-phospho-p38 MAPK (#9211) and anti-phospho-JNK1/2 (#4668) antibodies were from Cell Signaling (Danver, MA, USA). Anti-PAR1(sc-154), anti-Erk2 (sc-154), anti-JNK2 (sc-827), anti-p38 MAPK (sc-535), anti-p65 (sc-7151), anti-c-Jun (sc-822), anti-PCNA (sc-56) and anti-cyclin D1 (sc-8396) antibodies were from Santa Cruz (Santa Cruz, CA, USA). Anti-PAR1 antibody (#659102) was from BioLegend (San Diego, CA, USA). Anti-GAPDH (#MCA-1D4) was from Encor (Gainesville, FL, USA). PPACK, SCH79797, U0126, SP600125, SB202190, tanshinone IIA and helenalin were from Biomol (Plymouth Meeting, PA, USA). Thrombin (T4648), AH6809, Enzymes and Cell Counting Kit-8 (CCK-8) were from Sigma (St. Louis, MO, USA). TFLLR-NH2 was from R&D systems (Minneapolis, MN, USA). SDS-PAGE supplies were from MDBio Inc (Taipei, Taiwan).
Results
Thrombin up-regulates COX-2 expression and PGE2 generation
Thrombin-induced COX-2 expression has been reported in rat vascular smooth muscle cells (Hsieh et al., 2008). In our experiments, as shown in Figure 1A, thrombin induced COX-2 protein expression in a time- and concentration-dependent manner and a significant increase within 8 h and at 3–10 U·mL−1 of thrombin. Thrombin also time-dependently induced COX-2 mRNA expression, reaching a peak within 4 h and slightly declining after 6 h (Figure 1B) and increased COX-2 promoter activity within 2 h. The induction of COX-2 protein by thrombin was accompanied by increased PGE2 biosynthesis (Figure 1C). Thrombin time dependently induced PGE2 generation with a maximum within 16–24 h (the basal level of PGE2 ranged from 2 to −4 ng·mL−1). To further determine whether thrombin induced COX-2 expression via transcription and translation processes, an inhibitor of transcription, actinomycin D, or of translation, cycloheximide were used. Figure 1D showed that pretreatment with actinomycin D or CHI concentration-dependently attenuated thrombin-induced COX-2 expression. Moreover, pretreatment with actinomycin D, but not cycloheximide, attenuated thrombin-induced COX-2 mRNA expression (Figure 1E). These results indicated that thrombin-induced COX-2 expression is mediated through de novo mRNA and protein synthesis, resulting in PGE2 production.
Figure 1.
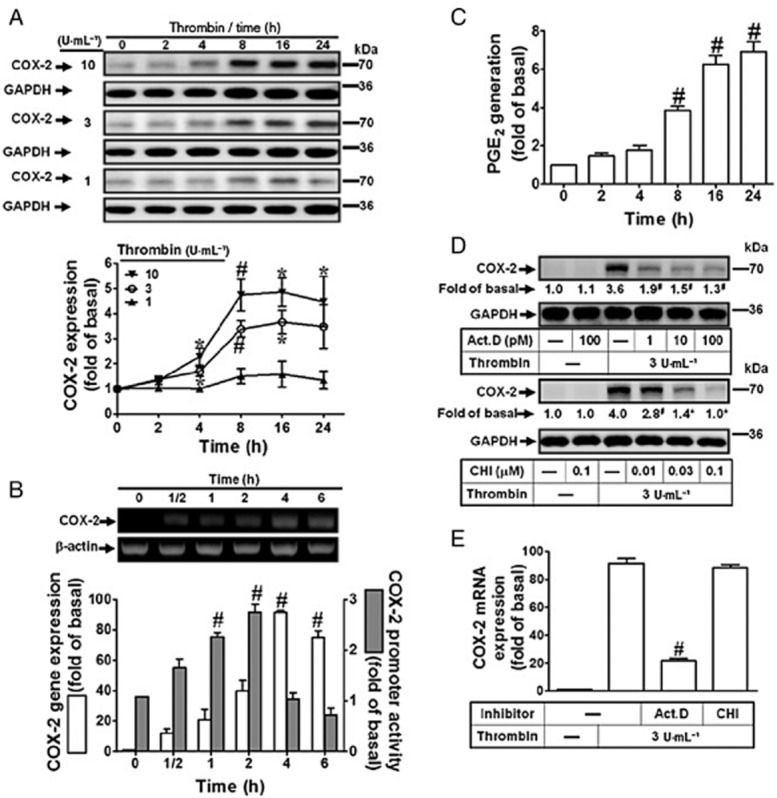
Thrombin induces COX-2 expression and PGE2 generation in human cardiomyocytes. (A) Human cardiomyocytes were incubated with various concentrations of thrombin for the indicated time intervals. The cell lysates were subjected to Western blot analysis. (B) The cells were incubated with thrombin (3 U·mL−1) for the indicated time intervals. The levels of COX-2 mRNA were analysed by RT-PCR and real-time PCR. Human cardiomyocytes were co-transfected with a COX-2 promoter luciferase reporter gene with a β-galactosidase plasmid and then incubated with thrombin (3 U·mL−1) for indicated time intervals (black bar). The COX-2 luciferase activity was detected and normalized to β-galactosidase activity. (C) The conditioned media were collected for PGE2 generation analysis by an EIA kit. The basal value of PGE2 is ranged from 2–4 ng·mL−1. (D–E) Human cardiomyocytes were pretreated with actinomycin D (Act. D) or cycloheximide CHI for 1 h and then incubated with thrombin (3 U·mL−1) for (D) 16 h or (E) 4 h. The COX-2 protein and mRNA levels was determined by Western blot (D) and real-time PCR (E). Data are expressed as mean ± SEM (n = 3). *P < 0.05; #P < 0.01, significantly different from cells incubated with vehicle alone (A–C) or thrombin alone (D–E).
PAR1 mediates thrombin-induced COX-2 expression
PARs mediate biological effects of thrombin via relaying downstream signalling cascades in hearts (Shah, 2009). Therefore, we identified which subtype of PARs is present in human cardiomyocytes. As shown in Figure 2A, PAR1, -2 and -3 were expressed in human cardiomyocytes. Pretreatment with a proteolytic activity inhibitor PPACK or a PAR1 antagonist SCH79797 both concentration dependently blocked thrombin-induced COX-2 expression (Figure 2B and 2C). Moreover, thrombin-induced COX-2 mRNA expression and promoter activity was also attenuated by pretreatment with PPACK or SCH79797 (Figure 2D). To further ensure the role of PAR1 in these responses, as shown in Figure 2E, transfection with PAR1 siRNA attenuated thrombin-induced COX-2 expression. Pretreatment with PPACK or SCH79797 also attenuated thrombin-induced PGE2 production (Figure 2F). To further confirm the role of PAR1 in these responses, a PAR1 activation peptide, TFLLR-NH2, was used. The results showed that TFLLR-NH2 also induced COX-2 expression, including protein, mRNA and promoter activity levels, which were inhibited by pretreatment with SCH79797 (Supporting Information Fig. S1A and S1B), suggesting that activation of PAR1 is critical for COX-2 expression in human cardiomyocytes. These results suggested that thrombin-induced COX-2 expression and PGE2 production is mediated through its proteolytic activity and PAR1 in human cardiomyocytes.
Figure 2.
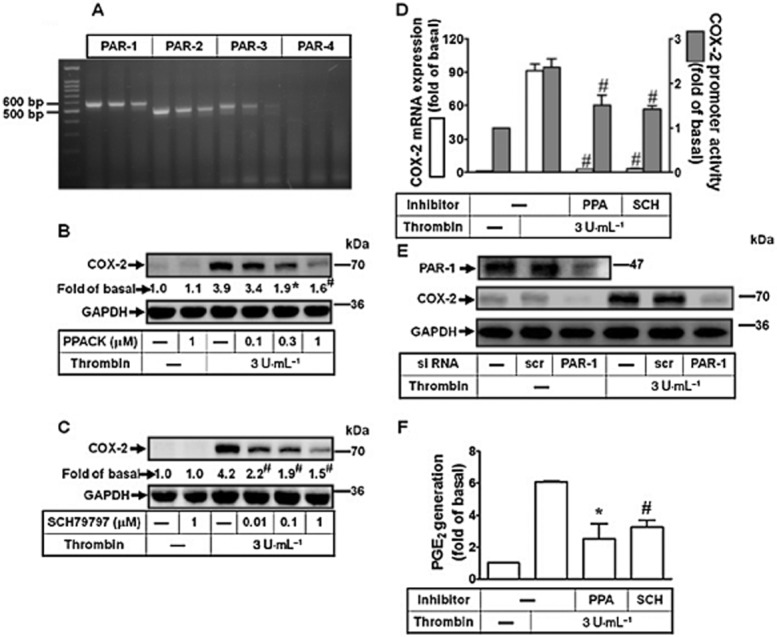
Thrombin induces COX-2 expression via a PAR1-dependent manner. (A) Expression of PAR subtypes in human cardiomyocytes was analysed by RT-PCR. (B–C) Cells were pretreated with PPACK (B) or SCH79797 (C) for 1 h and then incubated with thrombin for 16 h. The COX-2 protein was determined by Western blot. (D) Cells were pretreated with PPACK (PPA, 1 μM) or SCH79797 (SCH, 10 μM) for 1 h and then incubated with thrombin for 4 h. The COX-2 mRNA and promoter luciferase activity were detected by real-time PCR and promoter assay. (E) Cells were transfected with siRNA for scramble (scr) or PAR1 and then incubated with thrombin for 16 h. The COX-2 protein expression was determined by Western blot. (F) Cells were pretreated with PPACK (PPA, 1 μM) or SCH79797 (SCH, 1 μM) for 1 h and then incubated with thrombin for 16 h. The conditioned media were collected for PGE2 generation analysed by an EIA kit. Data are expressed as mean ± SEM (n = 3). *P < 0.05; #P < 0.01, significantly different from cells incubated with thrombin alone.
Involvement of MAPKs in thrombin-induced COX-2 expression
MAPKs are activated through PARs and contribute to cardiac hypertrophy (Wang, 2007). We explored the roles of p38 MAPK, Erk1/2 and JNK1/2 in thrombin-induced COX-2 expression. Human cardiomyocytes were pretreated with the corresponding MAPK inhibitors before thrombin challenge. As shown in Figure 3A and 3B, thrombin-induced COX-2 protein and mRNA expression, and COX-2 promoter activity was attenuated by pretreatment with the inhibitor of p38 MAPK (SB202190), MEK1/2 (U0126) or JNK1/2 (SP600125). Thrombin also time-dependently stimulated phosphorylation of p38 MAPK, Erk1/2 and JNK1/2, which were attenuated by their respective inhibitors (i.e. SB202190, U0126 and SP600125), during the period of observation (Figure 3C). The protein levels of MAPKs were not changed by incubation with thrombin or inhibitor. To further confirm the roles of these three MAPKs in thrombin-mediated responses, as shown in Figure 3D, transfection with p38 MAPK, p42 or JNK2 siRNA knocked down their respective proteins and subsequently attenuated thrombin-induced COX-2 expression. Moreover, pretreatment with SB202190, U0126 and SP600125 also inhibited thrombin-induced PGE2 production (Figure 3E). The data demonstrated that thrombin-induced COX-2 expression and PGE2 production is mediated through these MAPKs (i.e. p38 MAPK, Erk1/2, and JNK1/2) in human cardiomyocytes.
Figure 3.
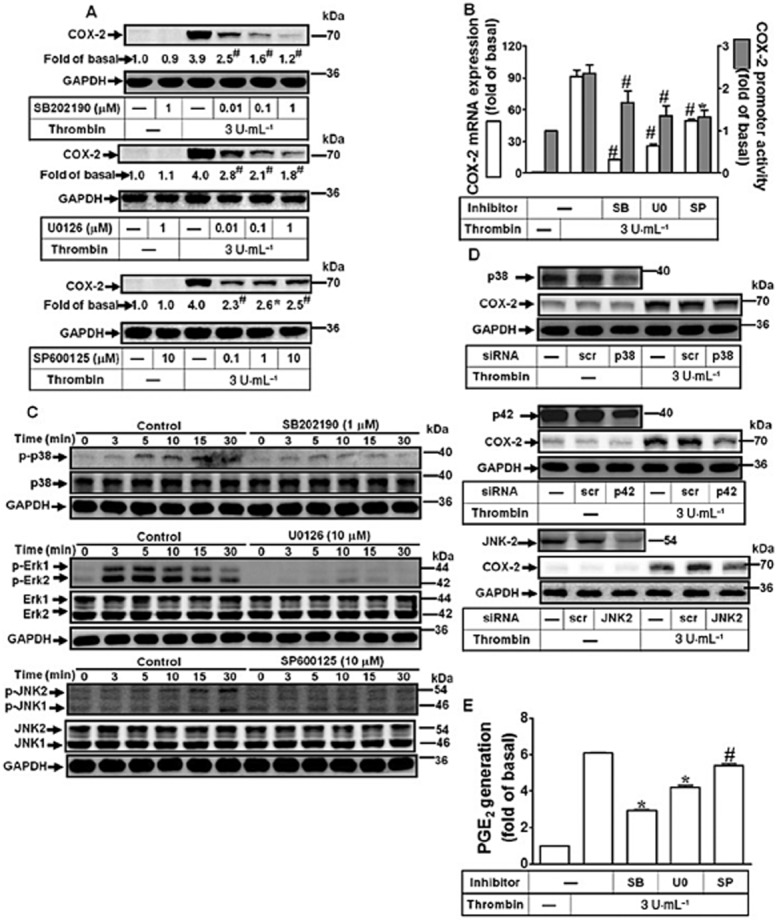
Effects of MAPKs in thrombin-induced COX-2 expression. (A) Cells were pretreated with SB202190, U0126, or SP600125 for 1 h and then incubated with thrombin for 16 h. The COX-2 protein expression was determined by Western blot. (B) Cells were pretreated with SB202190 (1 μM), U0126 (10 μM) or SP600125 (10 μM) for 1 h and then incubated with thrombin for 4 h. The COX-2 mRNA and promoter luciferase activity were detected. (C) Cells were pretreated with SB202190 (1 μM), U0126 (10 μM) or SP600125 (10 μM) for 1 h and then challenged with thrombin (3 U·mL−1) for the indicated time intervals. The cell lysates were analysed by Western blot using an anti-phospho-p38, anti-phospho-Erk1/2, anti-phospho-JNK1/2 or anti-GAPDH (as an internal control) antibody. (D) Cells were transfected with siRNA for scramble (scr), p38, p42 (Erk2) or JNK2 and then incubated with thrombin for 16 h. The COX-2 protein expression was determined by Western blot. (E) Cells were pretreated with SB202190 (1 μM), U0126 (1 μM) or SP600125 (10 μM) for 1 h and then incubated with thrombin for 16 h. The conditioned media were collected for PGE2 generation analysed by an EIA kit. Data were expressed as mean ± SEM (n = 3). *P < 0.05; #P < 0.01, significantly different from thrombin alone.
AP-1 and NF-κB are required for thrombin-induced COX-2 expression
AP-1 and NF-κB are two important transcription factors for regulating COX-2 gene expression in various cells upon diverse stimuli (Kang et al., 2007). Here, to investigate the roles of AP-1 and NF-κB in thrombin-induced COX-2 expression, the inhibitors of AP-1 (tanshinone IIA) and NF-κB (helenalin) were used. As shown in Figure 4A and 4B, pretreatment with either inhibitor attenuated thrombin-induced COX-2 protein, mRNA expression and promoter activity. Furthermore, the phosphorylation of c-Jun/AP-1 and p65 NF-κB stimulated by thrombin was blocked by tanshinone IIA or helenalin respectively (Figure 4C). To investigate whether thrombin can regulate AP-1 and NF-κB transcriptional activity, the AP-1 and NF-κB promoter plasmids were used. As shown in Figure 4D, thrombin enhanced the transcriptional activities of AP-1 and NF-κB, which were attenuated by PPACK, SCH79797, SB202190, U0126 and SP600125. We further confirmed that AP-1 and NF-κB are required for thrombin-induced COX-2 promoter luciferase activity by transfection of human cardiomyocytes with the wild-type (WT), mutated AP-1 (mtAP-1) or mutated NF-κB (mtNF-κB) of COX-2 promoter plasmid. As shown in Figure 4E, transfection with mt-AP-1 or mt-NF-κB COX-2 promoter plasmid significantly blocked thrombin-stimulated COX-2 promoter activity in human cardiomyocytes. To confirm the roles of c-Jun/AP-1 and p65 NF-κB in thrombin-induced COX-2 expression, as shown in Figure 4F, transfection with c-Jun or p65 siRNA knocked down c-Jun or p65 protein and attenuated thrombin-induced COX-2 expression. Finally, we also found that pretreatment with tanshinone IIA or helenalin significantly inhibited thrombin-induced PGE2 generation (Figure 4G). These results indicated that both AP-1 and NF-κB are essential for thrombin-induced COX-2 expression in human cardiomyocytes.
Figure 4.
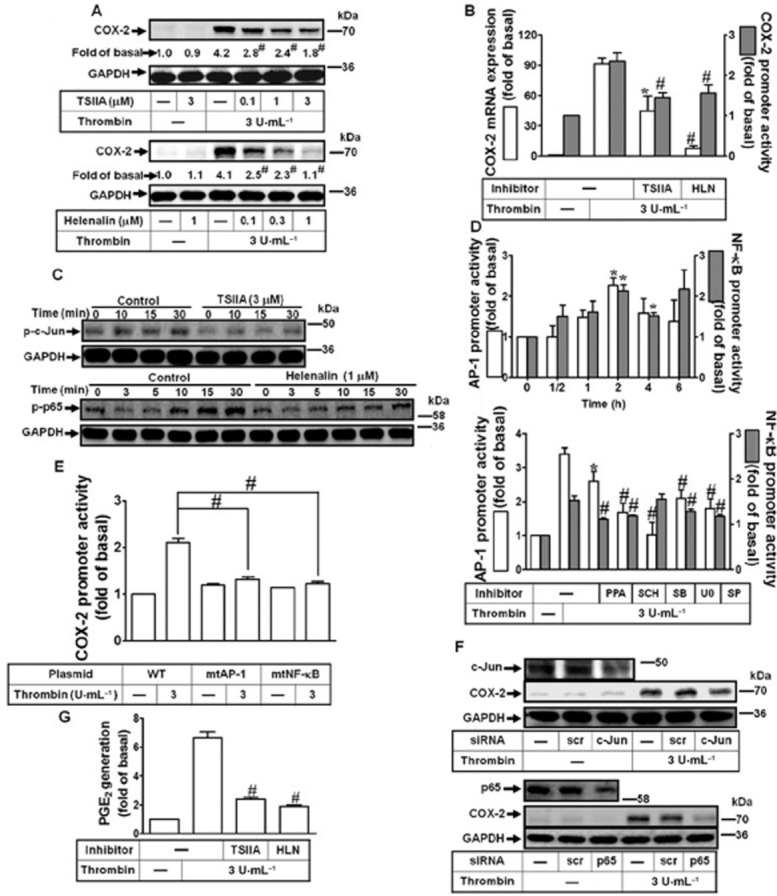
AP-1 and NF-κB are required for thrombin-induced COX-2 expression. (A) Cells were pretreated with tanshinone IIA (TSIIA) or helenalin (HLN) for 1 h and then incubated with thrombin for 16 h. The COX-2 protein expression was determined by Western blot. (B) Cells were pretreated with TSIIA (3 μM) or HLN (1 μM) for 1 h and then incubated with thrombin for 4 h. The COX-2 mRNA levels and promoter luciferase activity were detected. (C) Cells were pretreated with TSIIA (3 μM) or HLN (1 μM) for 1 h and then challenged with thrombin (3 U·mL−1) for the indicated time intervals. The cell lysates were analysed by Western blot using an anti-phospho-c-Jun, anti-phospho-p65 or anti-GAPDH (as an internal control) antibody. (D) Cells were transfected with AP-1 or NF-κB report gene and treated with thrombin for the indicated time intervals. The luciferase activity was detected and normalized to β-galactosidase activity. (E) Cells were transfected with wild-type (WT), mutated AP-1 (mtAP-1) or mutated NF-κB (mtNF-κB) COX-2 promoter gene and then challenged with thrombin (3 U·mL−1) for 2 h. (F) Cells were transfected with siRNA for scramble (scr), c-Jun or p65 and then incubated with thrombin for 16 h. The COX-2 protein expression was determined by Western blot. (G) Cells were pretreated with TSIIA (3 μM), or HLN (1 μM) for 1 h and then incubated with thrombin for 16 h. The conditioned media were collected for PGE2 generation analysed by an EIA kit. Data are expressed as mean ± SEM (n = 3). *P < 0.05; #P < 0.01, significantly different from cells incubated thrombin alone.
Involvement of PAR1-mediated activation of MAPKs in thrombin-stimulated AP-1 and NF-κB
We demonstrated that PAR1, p38 MAPK, Erk1/2, JNK1/2, AP-1 and NF-κB are required for thrombin-induced COX-2 expression in human cardiomyocytes. Next, to investigate the relationships among these signalling molecules, their respective inhibitors were used. As shown in Figure 5A, pretreatment with PPACK or SCH79797 reduced phosphorylation of p38 MAPK, Erk1/2 and JNK1/2, suggesting that thrombin stimulated MAPKs phosphorylation via a PAR1-dependent manner. To further investigate whether thrombin-stimulated phosphorylation of c-Jun/AP-1 and p65/NF-κB is mediated through PAR1-dependent MAPKs cascade, as shown in Figure 5B, thrombin-stimulated c-Jun phosphorylation was attenuated by PPACK, SCH79797, SB202190, U0126 or SP600125. Moreover, thrombin-stimulated p65 phosphorylation was attenuated by PPACK, SCH79797, U0126 or SP600125, but not SB202190. To ensure the phosphorylation of these MAPKs was due to activation of PAR1, we found that TFLLR-NH2 also stimulated p38 MAPK, Erk1/2, JNK1/2, p65 and c-Jun phosphorylation in a time-dependent manner, which was attenuated by pretreatment with SCH79797 (Supporting Information Fig. S1C). These results suggested that thrombin-stimulated AP-1 and NF-κB activation is mediated through PAR1-mediated MAPKs and leading to COX-2 expression in human cardiomyocytes.
Figure 5.
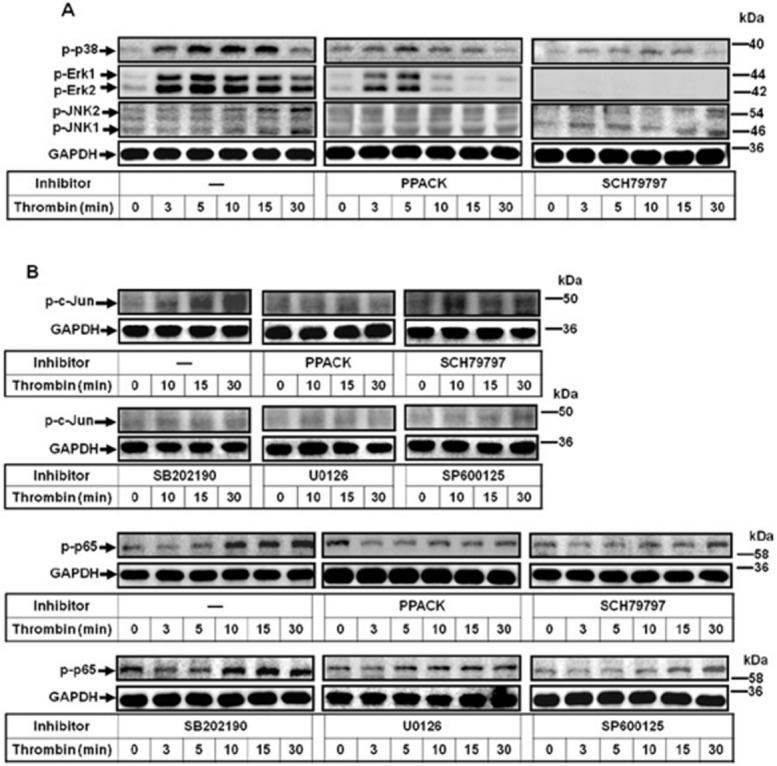
Thrombin stimulates MAPK-dependent activation of AP-1 and NF-κB via PAR1. (A) Cells were pretreated with PPACK (1 μM) or SCH79797 (10 μM) for 1 h and then challenged with thrombin (3 U·mL−1) for the indicated time intervals. The phosphorylation of p38 MAPK, Erk1/2, and JNK1/2 were determined by Western blot. (B) Cells were pretreated with PPACK (1 μM), SCH79797 (10 μM), SB202190 (1 μM), U0126 (10 μM) or SP600125 (10 μM) for 1 h and then challenged with thrombin (3 U·mL−1) for the indicated time intervals. The phosphorylation of c-Jun and p65 were determined by Western blot (n = 3).
Thrombin induces proliferation of human cardiomyocytes via PAR1-dependent MAPKs, AP-1 and NF-κB pathways
Thrombin has been shown to regulate several cellular functions such as proliferation in various cells (Isenovic et al., 2010). Hence, we investigated the effects of COX-2/PGE2 up-regulation by thrombin on human cardiomyocyte proliferation. The results showed that thrombin induced cell proliferation (Figure 6A), which was inhibited by pretreatment with COX-2 inhibitors, celecoxib and NS-398,as well as PPACK and SCH79797. Moreover, pretreatment with SB202190, U0126, SP600125, tanshinone IIA or helenalin also inhibited thrombin-induced cell proliferation, suggesting that COX-2/PGE2 may contribute to thrombin/PAR1-induced cardiomyocyte proliferation. Moreover, TFLLR-NH2 also induced cell proliferation (Supporting Information Fig. S1D). We further assayed the levels of proliferation marker molecules including a cell cycle regulator cyclin D1 and proliferating cell nuclear antigen (PCNA) in human cardiomyocytes challenged with thrombin. As shown in Figure 6B, thrombin time-dependently up-regulated cyclin D1 and PCNA expression, with a significant response within 36–48 h, and GAPDH expression was used as an indicator of internal protein control. Moreover, pretreatment with either of the COX-2 inhibitors attenuated thrombin-induced cyclin D1 and PCNA expression (Figure 6C). To further examine the effects of these signalling molecules of COX-2 induction by thrombin on cardiomyocyte proliferation (Figure 6D and 6E), pretreatment with PPACK, SCH79797, SB202190, U0126, SP600125, tanshinone IIA or helenalin attenuated thrombin-induced PCNA and cyclin D1 expression. To further confirm the proliferating response, the cell counting assay was also performed. The results showed that thrombin or TFLLR-NH2 increased cell numbers in a time-dependent manner (Supporting Information Fig. S2A). Collectively, these results indicated that thrombin induced COX-2/PGE2-mediated cell proliferation via PAR1-dependent MAPKs linking to AP-1 and NF-κB pathways in human cardiomyocytes.
Figure 6.
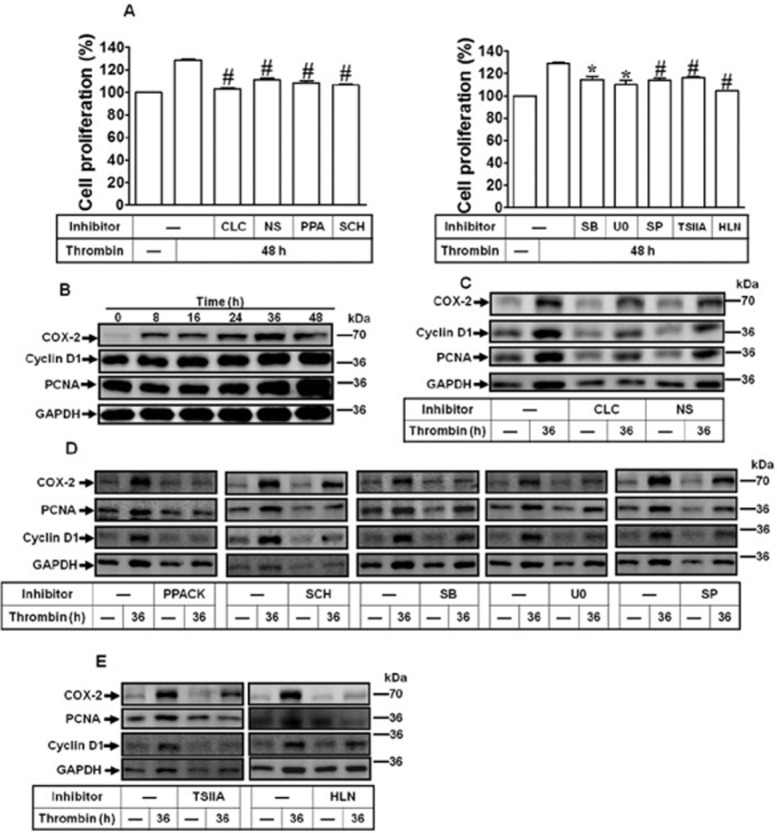
Thrombin-induced COX-2 expression contributes to cell proliferation. (A) The proliferating effects of thrombin on human cardiomyocytes were analysed by a CCK-8 kit. Cells were pretreated with CLC (10 μM), NS-398 (NS, 10 μM), PPACK (PPA, 1 μM), SCH79797 (SCH, 1 μM), SB202190 (SB, 1 μM), U0126 (U0, 1 μM), SP600125 (SP, 0.1 μM), tanshinone IIA (TSIIA; 3 μM) or helenalin (HLN; 1 μM) for 1 h and then incubated with thrombin for 48 h. (B) Cells were incubated with thrombin (3 U·mL−1) for the indicated time intervals. (C–D) Cells were pretreated with CLC or NS-398 (C), PPACK, SCH79797, SB202190, U0126, SP600125, TSIIA or HLN (D) for 1 h and then incubated with thrombin for 36 h. The COX-2, PCNA and cyclin D1 protein expression were determined by Western blot. Data are expressed as mean ± SEM (n = 3). *P < 0.05; #P < 0.01, significantly different from thrombin alone.
Thrombin-induced cell proliferation is mediated through the PG EP2 receptor
The prostanoid EP receptors mediate PGE2-activated biological events, including cell proliferation, in rat neonatal cardiomyocytes (Harding and Murray, 2011; Zhang et al., 2013). To examine whether EP receptors were involved in thrombin-triggered responses, we first used RT-PCR to analyse whether EP receptors are expressed in human cardiomyocytes. As shown in Figure 7A, the EP2 receptor was predominantly expressed in human cardiomyocytes. Next, pretreatment with an EP2 receptor antagonist AH6809 attenuated thrombin-induced cell proliferation at 48 h (Figure 7B). Moreover, when human cardiomyocytes were directly treated with PGE2 for the indicated time intervals. cell proliferation was induced in a time-dependent manner (Figure 7C), which was attenuated by pretreatment with AH6809 (Figure 7D). These results were also confirmed by a cell counting assay (Supporting Information Fig. S2). Pretreatment with AH6809 also attenuated thrombin-induced expression of proliferating markers such as PCNA and Cyclin D1 at 36 h (Figure 7E). Taken together, our reulst show that EP2 receptors played an important role in thrombin-induced human cardiomyocyte proliferation.
Figure 7.
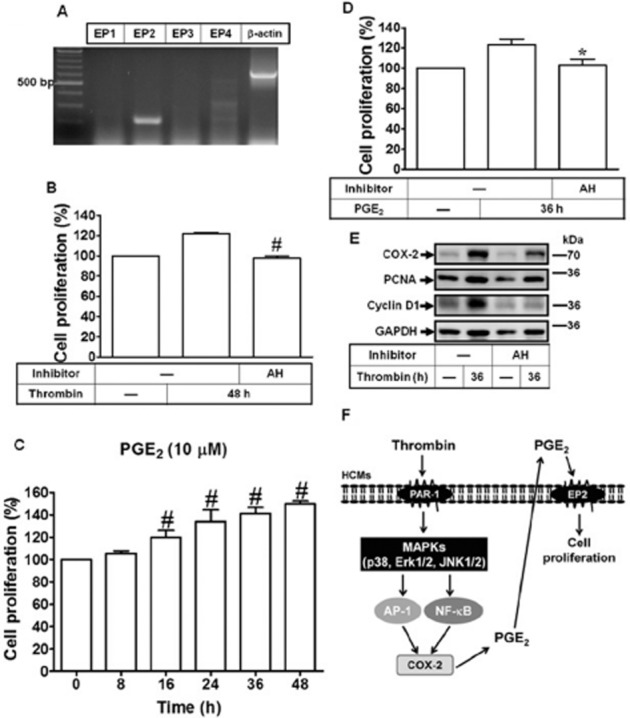
EP2 receptors mediate thrombin-induced cell proliferation. (A) Expression of EP receptor subtypes in human cardiomyocytes was analysed by RT-PCR. (B) The proliferating effects of thrombin on human cardiomyocytes were analysed by a CCK-8 kit. Cells were pretreated with AH6809 (AH, 10 μM) for 1 h prior to thrombin treatment for 48 h. (C) Cells were treated with PGE2 (10 μM) for the indicated times and analysed the proliferating effects by a XTT kit. (D) Cells were pretreated with AH6809 for 1 h and then incubated with thrombin for 36 h. (E) The COX-2, PCNA and cyclin D1 protein expression were determined by Western blot. Data are expressed as mean ± SEM (n = 3). *P < 0.05; #P < 0.01, significantly different from thrombin alone. (F) Each solid line and arrow represents a step in an activating pathway. Thrombin binds and activates its PAR1 receptor on the surface of primary human cardiomyocytes and relays intracellular signalling transduction cascades including MAPKs (i.e. p38, ERK1/2 and JNK1/2), subsequently initiated the transcription factors AP-1 and NF-κB pathways and ultimately leading to COX-2/PGE2-dependent EP2 receptor-activated cell proliferation in primary human cardiomyocytes.
Discussion and conclusions
Several factors have been implicated in heart failure, with an up-regulation of many proteases and inflammatory mediators (Yang et al., 2013). Among them, IL-1β, TNF-α and thrombin are elevated and highly related to heart failure progression (Li and Olshansky, 2011). COX-2 up-regulation is also detected in patients with chronic heart failure (Saito and Giaid, 1999). However, little is known about the detailed mechanisms of COX-2 up-regulation by thrombin in human cardiomyocytes. Thus, we investigated the molecular mechanisms underlying thrombin-induced COX-2 expression in human cardiomyocytes. Here, we demonstrated that in human cardiomyocytes, activation of PAR1-dependent MAPKs, c-Jun/AP-1 and NF-κB, were essential for thrombin-induced COX-2 expression and PGE2 production, and enhancement of cell proliferation. The results suggested that thrombin-induced COX-2 expression, PGE2 production and cell proliferation may contribute to heart inflammation and situations such as heart failure.
COX-2 is an inducible mediator of inflammation, which can be up-regulated via various signalling pathways and is involved in various pathological states and inflammatory diseases (Streicher et al., 2010). COX-2 is induced by many pro-inflammatory mediators in many different cell types (Kang et al., 2007). Several transcriptional factors have been identified to regulate human COX-2 gene expression, including PPAR, NF-κB, AP-1 and cyclic AMP-binding protein (CREB), leading to a complex signal coordination for COX-2 expression. In vascular smooth muscle cells and murine macrophages, thrombin triggers COX-2 induction via a PAR1-dependent pathway (Hsieh et al., 2008; Lo et al., 2009). Here, we first showed that, in human cardiomyocytes, thrombin induced the COX-2/PGE2 system, including expression of protein and mRNA, and PGE2 generation via a PAR1-dependent cascade. Moreover, the involvement of PAR1 in thrombin-induced responses via MAPKs was also confirmed by using a PAR1 activation peptide, which was attenuated by SCH79797 in human cardiomyocytes. However, in the A549 cell line, activation of PAR-2 also led to COX-2 expression via Erk1/2 and p38 MAPK and thus shared a mechanism, similar to those of PAR1 (Kawao et al., 2005; Wang et al., 2008). Further work will be necessary to determine whether PAR-2 can induce COX-2 expression. Moreover, PAR 4 was not expressed in human cardiomyocytes, consistent with a previous report using the same primers of PAR-4 (Kreda et al., 2010). Furthermore, pretreatment with a PAR-4 antagonist, Tcy-NH2, had no inhibitory effect on thrombin-induced COX-2 expression. Based on these data, we suggested that PAR1 may be critical for thrombin-induced COX-2 expression in human cardiomyocytes.
Examination of the mechanisms of increased potential risk factors contributing to patients combined stroke with heart failure showed that pro-coagulation factors, particularly thrombin, are up-regulated (Jug et al., 2009). Several proteases have been implicated in activation of PARs in vivo (Soh et al., 2010). These observations imply that thrombin may act as an important factor in the pathogenesis of heart failure. Thrombin activates PARs, which mediate diverse intracellular signals for cellular functions, including atrial natriuretic factor (ANF) generation and cardiac hypertrophy (Sabri et al., 2003). A recent study shows that PAR1 and thrombin are up-regulated in left atria and ventricle in patients with atrial fibrillation (Ito et al., 2013). Nevertheless, little is known for the expression and role of PARs in human cardiomyocytes. In this study, we confirmed that thrombin-induced COX-2 expression was attenuated by a proteolytic activity inhibitor PPACK, suggesting that the proteolytic activity of thrombin is involved in COX-2 expression. Furthermore, we confirmed that human cardiomyocytes expressed PAR1, PAR-2 and PAR-3. PAR1 is the predominant prototype of PAR-mediated thrombin responses. Moreover, we confirmed the role of PAR1 in thrombin-induced responses by transfection with a PAR1 siRNA. These results are consistent with previous reports indicating that PAR1 contributes to COX-2 induction in rat gastric epithelial cells and murine macrophages (Lo et al., 2009; Sekiguchi et al., 2011).
MAPKs have been shown to mediate various cellular functions, including proliferation, differentiation, motility, survival and apoptosis in response to various stimuli (Ravingerova et al., 2003). Activation of MAPKs has been well defined at different stages of heart diseases such as cardiac hypertrophy (Wang, 2007). Moreover, activation of PARs may increase MAPK activity and cause cardiac hypertrophy, although, the definite roles of each MAPK in thrombin-mediated responses in human cardiomyocytes remain to be investigated. Earlier work showed that thrombin could activate Erk1/2, p38 MAPK and JNK1/2 leading to cardiac hypertrophy and ANF expression (Jaffre et al., 2012). Here, we further confirmed that in human cardiomyocytes, thrombin-stimulated activation of MAPKs was involved in COX-2 induction, which was attenuated by their respective MAPKs inhibitors or siRNA, consistent with previous reports indicating that MAPKs are crucial to thrombin-induced responses in several other cell types (Lo et al., 2009; Sekiguchi et al., 2011). Moreover, phosphorylation of MAPKs by thrombin was attenuated by pretreatment with PPACK and SCH79797, indicating that thrombin stimulated activation of MAPKs via a PAR1-mediated manner. These results are consistent with earlier data showing PAR1 stimulated MAPK pathways leading to PGE2 formation in RGM1 cells (Sekiguchi et al., 2007).
The transcription factor AP-1, a complex of c-Jun, c-Fos and ATF subunits, regulates gene expression in response to various stimuli, such as cytokines, stress or growth factors. Previous studies demonstrate that AP-1 is engaged in endoplasmic reticulum stress-induced heart failure and participates in oxidant-induced cardiac hypertrophy (Tu et al., 2003; Sawada et al., 2010). Our data showed that pretreatment with tanshinone IIA reduced thrombin-induced c-Jun phosphorylation, COX-2 protein and mRNA expression, and PGE2 generation, suggesting that AP-1 was required for thrombin-induced COX-2 expression. These results are consistent with earlier work demonstrating that thrombin-induced COX-2 expression was mediated through activation of AP-1 binding to COX-2 promoter in vascular smooth muscle cells (Hsieh et al., 2008). Moreover, we showed that phosphorylation of c-Jun/AP-1 was attenuated by various inhibitors used in this study, suggesting that thrombin activates c-Jun/AP-1 via a PAR1-dependent MAPK activation pathway in human cardiomyocytes.
In the heart, the major form of NF-κB is the p50/p65 heterodimer associated with I-κB and, in a resting state, this remains in the cytoplasm which can be triggered by a variety of stimuli-related to inflammatory responses, mainly via transcription activation (Norman et al., 1998). Moreover, NF-κB activation has been shown to aggravate cardiac diseases through activation of pro-inflammatory pathways and enhancing heart failure (Kawamura et al., 2005). Here, we used an NF-κB inhibitor, helenalin to investigate its role in COX-2 induction, indicating that NF-κB is required for thrombin-induced COX-2 expression and PGE2 generation in human cardiomyocytes. Moreover, thrombin-stimulated p65 phosphorylation was mediated through PAR1, Erk1/2 and JNK1/2 in human cardiomyocytes, consistent with the results indicating that thrombin and PAR1 induce COX-2 expression via coordination of MAPKs and NF-κB in human umbilical vascular endothelial cells (Syeda et al., 2006).
Thrombin can induce cardiofibroblast proliferation via PAR-dependent signalling pathways (Sabri et al., 2002). However, proliferation of cardiomyocytes still remains controversial. As it is believed that cardiomyocytes are incapable of proliferating and are terminally undifferentiated, it is unlikely that adult cardiomyocytes have proliferating capability. A previous study demonstrated that cardiomyocytes in human failing hearts showed dividing cells by ki67 staining, implying that cardiomyocytes may be able to form new cells (Nadal-Ginard et al., 2003). In our study, we found that the pro-proliferation effects of thrombin on human cardiomyocytes occurred within 36–48 h, consistent with increased PCNA and cyclin D1 expression during embryonic heart development and disease states. The mitochondria-formazan used as a proliferation index still remains controversial. Therefore, cell counting was also performed to show that thrombin and PAR1 agonist did increase cell number, which was significantly but not completely attenuated by SCH79797 and AH6809, implying that PAR1-independent pathways may be also involved in proliferation in our model. Herber et al. demonstrated that endothelial cell growth was mediated through proteolytic- and non-proteolytic-dependent paths (Herbert et al., 1994). Synthetic thrombin receptor peptides activated the GPCR but were incapable of inducing mitogenesis (Vouret-Craviari et al., 1992). These different results may be due to cell specificity and various experimental conditions. During a variety of heart diseases, measurement of mitotic index revealed cell proliferation, even under electron micrograph showing mild mitosis (Nadal-Ginard et al., 2003; Chang et al., 2010). Although injuries were observed for the loss of pre-existing cells, the intact left ventricle recovered within 6 months, suggesting that human cardiomyocyte proliferation could be activated by various factors involved in pathological progression, and one of potential candidates may be thrombin.
The compensatory responses in failing hearts comprise not only hypertrophy but also the growth responses induced by thrombin. Here, we demonstrated that thrombin induced cell proliferating events via PAR1-dependent MAPKs/AP-1 and NF-κB linking to induction of the COX-2/PGE2 system. EP receptors have been identified in cardiomyocytes in mice, in contrast to our finding that human cardiomyocytes only possess EP2 receptors (Xiao et al., 2004). These receptors are known for their proliferative effects in many types of cells through downstream signalling pathways (Guo et al., 2011; Liu et al., 2012; Lee et al., 2013). In this study, we demonstrated that the EP2 receptor antagonist, AH6809, inhibited thrombin-induced cell proliferation and cell cycle-dependent protein expression in human cardiomyocytes. Moreover, the cell counting and XTT assays showed that PGE2 alone promoted cell proliferation, which was also observed in other cell types as well (Jang et al., 2012; Lee et al., 2013). In a post-infarction model, PGE2 promotes replenishment of newly formed cardiomyocytes through both direct and indirect EP2 receptor pathways (Hsueh et al., 2014). Collectively, these observations provide a novel insight into COX-2/PGE2 regulation in heart. As there is a loss of proliferating ability of cardiomyocytes following myocardial infarction, exploration of the factors driving cardiomyocyte regeneration is essential. There is evidence that proliferation of cardiomyocytes is triggered by various exogenous factors, such as fibroblast growth factor, PDGF or IL-6 (Kardami et al., 2003; Hinrichsen et al., 2007; Lu et al., 2008). The increased cell number in our neonatal cell model is likely due to transitory capacity being maintained. Optimization of thrombin given in an appropriate time and dose could be beneficial for tissue repair. However, the origin of increased cells could not rule out the effects of cardiac progenitor cells, which need to be clarified. These results merit further investigation as the proliferation could serve as a strategy to maintain or protect the loss of cardiomyocytes following a pathological loss of cells.
In conclusion, we demonstrated that thrombin induced COX-2 expression in human cardiomyocytes. Thrombin/PAR1-dependent COX-2 up-regulation was mediated through activation of MAPKs, AP-1 and NF-κB, and resulted in increased cell proliferation. Based on published data and our findings, Figure 7F provides a model for the molecular mechanisms underlying thrombin-induced COX-2 expression and cell proliferation in human cardiomyocytes. These findings highlight the critical role of COX-2/PGE2/EP2 system in PAR-mediated interventions in heart disease. Pharmacological approaches suggest that targeting the COX-2/PGE2 system or PAR-dependent downstream signalling components may yield useful therapeutic targets for heart failure.
Acknowledgments
This work was supported by the Ministry of Education, Taiwan, Grant number: EMRPD1D0231 and EMRPD1D0241; National Science Council, Taiwan, Grant number: NSC102-2321-B-182-011, NSC101-2320-B-182-039-MY3 and NSC102-2320-B-255-005-MY3; Chang Gung Medical Research Foundation, Grant number: CMRPD1B0383, CMRPD1C0102, CMRPD1C0561, CMRPD1C0562, CMRPF1C0191 and CMRPF1A0063. We thank Mr Li-Der Hsiao for his technical assistance.
Glossary
- ANF
atrial natriuretic factor
- PAR
protease-activated receptor
- PCNA
proliferating cell nuclear antigen
Author contributions
P. T-Y. C., H-L. H., P-L. C. and C-M. Y. developed and designed experiments, and analysed and interpreted data. P. T-Y. C. and P-L. C. conducted the experiments and collected data. This article was drafted by P. T-Y. C., H-L. H., P-L. C. and C-M. Y.
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site: http://dx.doi.org/10.1111/bph.12794
Figure S1 Effects of TFLLR-NH2 in human cardiomyocytes. (A) Cells were pretreated with SCH79797 (1 μM) and incubated with TFLLR-NH2 (50 μM) for the indicated time intervals. The cell lysates were subjected to Western blot analysis. (B) Cells were pretreated wth SCH79797 (10 μM) and incubated with TFLLR-NH2 (50 μM) for the indicated times. The COX-2 mRNA and promoter luciferase activity were detected by real-time PCR (open bar) and promoter assay (black bar). (C) Cells were pretreated with SCH79797 (10 μM) and then stimulated with TFLLR-NH2 (50 μM) for indicated time intervals. The cell lysates were analysed by Western blot using an anti-phospho-p38, phospho-Erk1/2, phospho-JNK1/2, phospho-p65, phospho-c-Jun, or GAPDH (as an internal control) antibody. (D) The proliferating effects of thrombin on human cardiomyocytes were analysed by a XTT kit. Cells were pretreated with SCH79797 (SCH, 1 μM) for 1 h and then incubated with TFLLR-NH2 (50 μM) for 48 h. Data are expressed as mean ± SEM (n = 3). #P < 0.01, significantly different from TFLLR-NH2 alone (C).
Figure S2 Thrombin, TFLLR-NH2 and PGE2 induce cell proliferation. (A) Cells were treated with thrombin (3 U·mL−1) or TFLLR-NH2 (50 μM) in the presence or absence of AH6809 (10 μM) and the cell number of human cardiomyocytes were counted by cell counting assay. *P < 0.05, as compared with thrombin or TFLLR-NH2 stimulation alone. (B) Cells were stimulated with PGE2 (10 μM) for the indicated times. After stimulation, the cell number of human cardiomyocytes was counted by cell counting assay. *P < 0.05; #P < 0.01, significantly different from vehicle (B) or thrombin and TFLLR-NH2 (A) alone (n = 3).
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariens RA. Fibrin(ogen) and thrombotic disease. J Thromb Haemost. 2013;11(Suppl. 1):294–305. doi: 10.1111/jth.12229. [DOI] [PubMed] [Google Scholar]
- Barnes JA, Singh S, Gomes AV. Protease activated receptors in cardiovascular function and disease. Mol Cell Biochem. 2004;263:227–239. doi: 10.1023/B:MCBI.0000041864.14092.5b. [DOI] [PubMed] [Google Scholar]
- Chang KT, Taylor GP, Meschino WS, Kantor PF, Cutz E. Mitogenic cardiomyopathy: a lethal neonatal familial dilated cardiomyopathy characterized by myocyte hyperplasia and proliferation. Hum Pathol. 2010;41:1002–1008. doi: 10.1016/j.humpath.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999;96:11023–11027. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- Duvivier J, Wolf D, Heusghem C. Enzymatic properties of prostaglandin synthetase from bovine seminal vesicles. Biochimie. 1975;57:521–528. doi: 10.1016/s0300-9084(75)80131-3. [DOI] [PubMed] [Google Scholar]
- Glembotski CC, Irons CE, Krown KA, Murray SF, Sprenkle AB, Sei CA. Myocardial alpha-thrombin receptor activation induces hypertrophy and increases atrial natriuretic factor gene expression. J Biol Chem. 1993;268:20646–20652. [PubMed] [Google Scholar]
- Guo D, Chen NN, Hou LB, Lei LS. [Prostaglandin E2 promotes hepatocellular carcinoma cell proliferation through EP2 prostanoid receptor] Nan Fang Yi Ke Da Xue Xue Bao. 2011;31:1564–1567. [PubMed] [Google Scholar]
- Harding P, Murray DB. The contribution of prostaglandins versus prostacyclin in ventricular remodeling during heart failure. Life Sci. 2011;89:671–676. doi: 10.1016/j.lfs.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Herbert JM, Dupuy E, Laplace MC, Zini JM, Bar Shavit R, Tobelem G. Thrombin induces endothelial cell growth via both a proteolytic and a non-proteolytic pathway. Biochem J. 1994;303:227–231. doi: 10.1042/bj3030227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichsen R, Haunso S, Busk PK. Different regulation of p27 and Akt during cardiomyocyte proliferation and hypertrophy. Growth Factors. 2007;25:132–140. doi: 10.1080/08977190701549835. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Sun CC, Wang TS, Yang CM. PKC-delta/c-Src-mediated EGF receptor transactivation regulates thrombin-induced COX-2 expression and PGE2 production in rat vascular smooth muscle cells. Biochim Biophys Acta. 2008;1783:1563–1575. doi: 10.1016/j.bbamcr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Hsueh YC, Wu JM, Yu CK, Wu KK, Hsieh PC. Prostaglandin E2 promotes post-infarction cardiomyocyte replenishment by endogenous stem cells. EMBO Mol Med. 2014;6:496–503. doi: 10.1002/emmm.201303687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenovic ER, Soskic S, Trpkovic A, Dobutovic B, Popovic M, Gluvic Z, et al. Insulin, thrombine, ERK1/2 kinase and vascular smooth muscle cells proliferation. Curr Pharm Des. 2010;16:3895–3902. doi: 10.2174/138161210794454987. [DOI] [PubMed] [Google Scholar]
- Ito K, Date T, Ikegami M, Hongo K, Fujisaki M, Katoh D, et al. An immunohistochemical analysis of tissue thrombin expression in the human atria. PLoS ONE. 2013;8:e65817. doi: 10.1371/journal.pone.0065817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffre F, Friedman AE, Hu Z, Mackman N, Blaxall BC. beta-adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation. 2012;125:2993–3003. doi: 10.1161/CIRCULATIONAHA.111.066787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MW, Yun SP, Park JH, Ryu JM, Lee JH, Han HJ. Cooperation of Epac1/Rap1/Akt and PKA in prostaglandin E2-induced proliferation of human umbilical cord blood derived mesenchymal stem cells: involvement of c-Myc and VEGF expression. J Cell Physiol. 2012;227:3756–3767. doi: 10.1002/jcp.24084. [DOI] [PubMed] [Google Scholar]
- Jug B, Vene N, Salobir BG, Sebestjen M, Sabovic M, Keber I. Procoagulant state in heart failure with preserved left ventricular ejection fraction. Int Heart J. 2009;50:591–600. doi: 10.1536/ihj.50.591. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Mbonye UR, DeLong CJ, Wada M, Smith WL. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog Lipid Res. 2007;46:108–125. doi: 10.1016/j.plipres.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardami E, Banerji S, Doble BW, Dang X, Fandrich RR, Jin Y, et al. PKC-dependent phosphorylation may regulate the ability of connexin43 to inhibit DNA synthesis. Cell Commun Adhes. 2003;10:293–297. doi: 10.1080/cac.10.4-6.293.297. [DOI] [PubMed] [Google Scholar]
- Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, et al. Blockade of NF-kappaB improves cardiac function and survival without affecting inflammation in TNF-alpha-induced cardiomyopathy. Cardiovasc Res. 2005;66:520–529. doi: 10.1016/j.cardiores.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Kawao N, Nagataki M, Nagasawa K, Kubo S, Cushing K, Wada T, et al. Signal transduction for proteinase-activated receptor-2-triggered prostaglandin E2 formation in human lung epithelial cells. J Pharmacol Exp Ther. 2005;315:576–589. doi: 10.1124/jpet.105.089490. [DOI] [PubMed] [Google Scholar]
- Kotlyar E, Vita JA, Winter MR, Awtry EH, Siwik DA, Keaney JF, Jr, et al. The relationship between aldosterone, oxidative stress, and inflammation in chronic, stable human heart failure. J Card Fail. 2006;12:122–127. doi: 10.1016/j.cardfail.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Kreda SM, Seminario-Vidal L, van Heusden CA, O’Neal W, Jones L, Boucher RC, et al. Receptor-promoted exocytosis of airway epithelial mucin granules containing a spectrum of adenine nucleotides. J Physiol. 2010;588:2255–2267. doi: 10.1113/jphysiol.2009.186643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisch E, Jansen A, Kojima F, Loffler I, Kapoor M, Kawai S, et al. Prostaglandin E2 differentially modulates proinflammatory/prodestructive effects of TNF-alpha on synovial fibroblasts via specific E prostanoid receptors/cAMP. J Immunol. 2009;183:1328–1336. doi: 10.4049/jimmunol.0900801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IT, Lin CC, Wang CH, Cherng WJ, Wang JS, Yang CM. ATP stimulates PGE2/cyclin D1-dependent VSMCs proliferation via STAT3 activation: role of PKCs-dependent NADPH oxidase/ROS generation. Biochem Pharmacol. 2013;85:954–964. doi: 10.1016/j.bcp.2012.12.016. [DOI] [PubMed] [Google Scholar]
- Li W, Olshansky B. Inflammatory cytokines and nitric oxide in heart failure and potential modulation by vagus nerve stimulation. Heart Fail Rev. 2011;16:137–145. doi: 10.1007/s10741-010-9184-4. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rajagopal M, Lee K, Battini L, Flores D, Gusella GL, et al. Prostaglandin E2 mediates proliferation and chloride secretion in ADPKD cystic renal epithelia. Am J Physiol Renal Physiol. 2012;303:F1425–F1434. doi: 10.1152/ajprenal.00010.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HM, Chen CL, Tsai YJ, Wu PH, Wu WB. Thrombin induces cyclooxygenase-2 expression and prostaglandin E2 release via PAR1 activation and ERK1/2- and p38 MAPK-dependent pathway in murine macrophages. J Cell Biochem. 2009;108:1143–1152. doi: 10.1002/jcb.22341. [DOI] [PubMed] [Google Scholar]
- Lu SY, Sontag DP, Detillieux KA, Cattini PA. FGF-16 is released from neonatal cardiac myocytes and alters growth-related signaling: a possible role in postnatal development. Am J Physiol Cell Physiol. 2008;294:C1242–C1249. doi: 10.1152/ajpcell.00529.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshal KS, Tyagi N, Henderson B, Ovechkin AV, Tyagi SC. Protease-activated receptor and endothelial-myocyte uncoupling in chronic heart failure. Am J Physiol Heart Circ Physiol. 2005a;288:H2770–H2777. doi: 10.1152/ajpheart.01146.2004. [DOI] [PubMed] [Google Scholar]
- Moshal KS, Tyagi N, Moss V, Henderson B, Steed M, Ovechkin A, et al. Early induction of matrix metalloproteinase-9 transduces signaling in human heart end stage failure. J Cell Mol Med. 2005b;9:704–713. doi: 10.1111/j.1582-4934.2005.tb00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92:139–150. doi: 10.1161/01.res.0000053618.86362.df. [DOI] [PubMed] [Google Scholar]
- Norman DA, Yacoub MH, Barton PJ. Nuclear factor NF-kappa B in myocardium: developmental expression of subunits and activation by interleukin-1 beta in cardiac myocytes in vitro. Cardiovasc Res. 1998;39:434–441. doi: 10.1016/s0008-6363(98)00118-7. [DOI] [PubMed] [Google Scholar]
- Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, et al. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116:2298–2306. doi: 10.1161/CIRCULATIONAHA.107.692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Research. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravingerova T, Barancik M, Strniskova M. Mitogen-activated protein kinases: a new therapeutic target in cardiac pathology. Mol Cell Biochem. 2003;247:127–138. doi: 10.1023/a:1024119224033. [DOI] [PubMed] [Google Scholar]
- Ritter O, Neyses L. The molecular basis of myocardial hypertrophy and heart failure. Trends Mol Med. 2003;9:313–321. doi: 10.1016/s1471-4914(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Rolin S, Hanson J, Vastersaegher C, Cherdon C, Pratico D, Masereel B, et al. BM-520, an original TXA2 modulator, inhibits the action of thromboxane A2 and 8-iso-prostaglandin F2alphain vitro and in vivo on human and rodent platelets, and aortic vascular smooth muscles from rodents. Prostaglandins Other Lipid Mediat. 2007;84:14–23. doi: 10.1016/j.prostaglandins.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Sabri A, Muske G, Zhang H, Pak E, Darrow A, Andrade-Gordon P, et al. Signaling properties and functions of two distinct cardiomyocyte protease-activated receptors. Circ Res. 2000;86:1054–1061. doi: 10.1161/01.res.86.10.1054. [DOI] [PubMed] [Google Scholar]
- Sabri A, Short J, Guo J, Steinberg SF. Protease-activated receptor-1-mediated DNA synthesis in cardiac fibroblast is via epidermal growth factor receptor transactivation: distinct PAR1 signaling pathways in cardiac fibroblasts and cardiomyocytes. Circ Res. 2002;91:532–539. doi: 10.1161/01.res.0000035242.96310.45. [DOI] [PubMed] [Google Scholar]
- Sabri A, Alcott SG, Elouardighi H, Pak E, Derian C, Andrade-Gordon P, et al. Neutrophil cathepsin G promotes detachment-induced cardiomyocyte apoptosis via a protease-activated receptor-independent mechanism. J Biol Chem. 2003;278:23944–23954. doi: 10.1074/jbc.M302718200. [DOI] [PubMed] [Google Scholar]
- Saito T, Giaid A. Cyclooxygenase-2 and nuclear factor-kappaB in myocardium of end stage human heart failure. Congest Heart Fail. 1999;5:222–227. [PubMed] [Google Scholar]
- Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T, et al. X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes. J Mol Cell Cardiol. 2010;48:1280–1289. doi: 10.1016/j.yjmcc.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Sekiguchi F, Saito S, Takaoka K, Hayashi H, Nagataki M, Nagasawa K, et al. Mechanisms for prostaglandin E2 formation caused by proteinase-activated receptor-1 activation in rat gastric mucosal epithelial cells. Biochem Pharmacol. 2007;73:103–114. doi: 10.1016/j.bcp.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Sekiguchi F, Ohi A, Maeda Y, Takaoka K, Sekimoto T, Nishikawa H, et al. Delayed production of arachidonic acid contributes to the delay of proteinase-activated receptor-1 (PAR1)-triggered prostaglandin E2 release in rat gastric epithelial RGM1 cells. J Cell Biochem. 2011;112:909–915. doi: 10.1002/jcb.23005. [DOI] [PubMed] [Google Scholar]
- Seminario-Vidal L, Kreda S, Jones L, O’Neal W, Trejo J, Boucher RC, et al. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J Biol Chem. 2009;284:20638–20648. doi: 10.1074/jbc.M109.004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah R. Protease-activated receptors in cardiovascular health and diseases. Am Heart J. 2009;157:253–262. doi: 10.1016/j.ahj.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol. 2010;160:191–203. doi: 10.1111/j.1476-5381.2010.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicher JM, Wang Y. The role of COX-2 in heart pathology. Cardiovasc Hematol Agents Med Chem. 2008;6:69–79. doi: 10.2174/187152508783329948. [DOI] [PubMed] [Google Scholar]
- Streicher JM, Kamei K, Ishikawa TO, Herschman H, Wang Y. Compensatory hypertrophy induced by ventricular cardiomyocyte-specific COX-2 expression in mice. J Mol Cell Cardiol. 2010;49:88–94. doi: 10.1016/j.yjmcc.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syeda F, Grosjean J, Houliston RA, Keogh RJ, Carter TD, Paleolog E, et al. Cyclooxygenase-2 induction and prostacyclin release by protease-activated receptors in endothelial cells require cooperation between mitogen-activated protein kinase and NF-kappaB pathways. J Biol Chem. 2006;281:11792–11804. doi: 10.1074/jbc.M509292200. [DOI] [PubMed] [Google Scholar]
- Tu VC, Bahl JJ, Chen QM. Distinct roles of p42/p44(ERK) and p38 MAPK in oxidant-induced AP-1 activation and cardiomyocyte hypertrophy. Cardiovasc Toxicol. 2003;3:119–133. doi: 10.1385/ct:3:2:119. [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari V, Van Obberghen-Schilling E, Rasmussen UB, Pavirani A, Lecocq JP, Pouyssegur J. Synthetic alpha-thrombin receptor peptides activate G protein-coupled signaling pathways but are unable to induce mitogenesis. Mol Biol Cell. 1992;3:95–102. doi: 10.1091/mbc.3.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wen S, Bunnett NW, Leduc R, Hollenberg MD, MacNaughton WK. Proteinase-activated receptor-2 induces cyclooxygenase-2 expression through beta-catenin and cyclic AMP-response element-binding protein. J Biol Chem. 2008;283:809–815. doi: 10.1074/jbc.M703021200. [DOI] [PubMed] [Google Scholar]
- Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116:1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation. 2004;109:2462–2468. doi: 10.1161/01.CIR.0000128046.54681.97. [DOI] [PubMed] [Google Scholar]
- Yang CM, Lee IT, Lin CC, Wang CH, Cherng WJ, Hsiao LD. c-Src-dependent MAPKs/AP-1 activation is involved in TNF-alpha-induced matrix metalloproteinase-9 expression in rat heart-derived H9c2 cells. Biochem Pharmacol. 2013;85:1115–1123. doi: 10.1016/j.bcp.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q, et al. Cyclooxygenase-2-derived prostaglandin E2 promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res. 2013;113:104–114. doi: 10.1161/CIRCRESAHA.113.301033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of TFLLR-NH2 in human cardiomyocytes. (A) Cells were pretreated with SCH79797 (1 μM) and incubated with TFLLR-NH2 (50 μM) for the indicated time intervals. The cell lysates were subjected to Western blot analysis. (B) Cells were pretreated wth SCH79797 (10 μM) and incubated with TFLLR-NH2 (50 μM) for the indicated times. The COX-2 mRNA and promoter luciferase activity were detected by real-time PCR (open bar) and promoter assay (black bar). (C) Cells were pretreated with SCH79797 (10 μM) and then stimulated with TFLLR-NH2 (50 μM) for indicated time intervals. The cell lysates were analysed by Western blot using an anti-phospho-p38, phospho-Erk1/2, phospho-JNK1/2, phospho-p65, phospho-c-Jun, or GAPDH (as an internal control) antibody. (D) The proliferating effects of thrombin on human cardiomyocytes were analysed by a XTT kit. Cells were pretreated with SCH79797 (SCH, 1 μM) for 1 h and then incubated with TFLLR-NH2 (50 μM) for 48 h. Data are expressed as mean ± SEM (n = 3). #P < 0.01, significantly different from TFLLR-NH2 alone (C).
Figure S2 Thrombin, TFLLR-NH2 and PGE2 induce cell proliferation. (A) Cells were treated with thrombin (3 U·mL−1) or TFLLR-NH2 (50 μM) in the presence or absence of AH6809 (10 μM) and the cell number of human cardiomyocytes were counted by cell counting assay. *P < 0.05, as compared with thrombin or TFLLR-NH2 stimulation alone. (B) Cells were stimulated with PGE2 (10 μM) for the indicated times. After stimulation, the cell number of human cardiomyocytes was counted by cell counting assay. *P < 0.05; #P < 0.01, significantly different from vehicle (B) or thrombin and TFLLR-NH2 (A) alone (n = 3).