

Abstract
Endothelin-1 (ET-1) is a known algogen that causes acute pain and sensitization in humans and spontaneous nociceptive behaviors when injected into the periphery in rats, and is elevated during vaso-occlusive episodes (VOEs) in sickle cell disease (SCD) patients. Previously, our lab has shown that a priming dose of ET-1 produces sensitization to capsaicin-induce secondary hyperalgesia. The goal of this study was to determine if the sensitization induced by ET-1 priming is occurring at the level of the primary afferent neuron. Calcium imaging in cultured dorsal root ganglion (DRG) neurons was utilized to examine the effects of ET-1 on primary afferent neurons. ET-1 induces [Ca2+]i transients in unprimed cells. ET-1 induced [Ca2+]i transients are attenuated by priming with ET-1. This priming effect occurs whether the priming dose is given 0-4 days prior to the challenge dose. Similarly, ET-1 priming decreases capsaicin-induced [Ca2+]i transients. At the level of the primary afferent neuron, ET-1 priming has a desensitizing effect on challenge exposures to ET-1 and capsaicin.
Keywords: Endothelin-1(ET-1), Capsaicin, Dorsal root ganglia, Calcium transients
1. Introduction
The hallmark feature of sickle cell disease (SCD) is painful vaso-occlusive episodes (VOE). Occlusion of the blood vessels leads to localized ischemia, which results in tissue damage, which then leads to pain through the release of certain molecular mediators [20]. The role of vasoactive regulators in VOEs is an active area of investigation. It has been speculated that occlusion in one part of the blood vessel leads to the release of certain vasoactive substances from damaged endothelial cells, such as vasoconstrictive peptides that promote vaso-occlusion downstream from the original site of vaso-occlusion [11]. It may also be possible that vasoactive humoral factors could be responsible for the extension of pain from the original site of painful crisis [11]. We speculate that it is the vasoactive and nociceptive actions of endothelin-1, a potent vasoconstrictive and nociceptive agent, that are some of the factors responsible for this phenomenon. Endothelin-1 (ET-1) has been found to be elevated during VOEs in SCD patients and stimulation of endothelial cells with sickled red blood cells from SCD patients induces ET-1 mRNA production and the release of ET-1 [9, 15, 18]. In addition to ET-1 being elevated during VOEs, higher levels of ET-1 have been found to be correlated with higher ratings of baseline pain in children with SCD, suggesting a relationship between ET-1 and vaso-occlusion, the most likely source of baseline pain in these patients [17].
Endothelin-1 is a 21 amino acid peptide that is both a vasoconstrictor and a potent algogen that has been shown to cause acute pain and sensitization in humans [3, 7, 10]. In rodent models, the administration of ET-1 produces spontaneous nociceptive behaviors [8] [4]. In adult male rats, repeated ET-1 administration to the sciatic nerve produces behavioral desensitization [5]. Previously our laboratory has shown that a priming exposure to ET-1 alters behavioral responses to a challenge dose of ET-1 at the same location in neonatal rats. This priming effect is sex dependent with sensitization in males and desensitization in females [13]. However, when the challenge dose of ET-1 was in the contralateral hindpaw both males and females exhibit sensitization. Furthermore, priming with ET-1 enhances capsaicin-induced secondary hyperalgesia in both males and females. Priming with ET-1 also enhances c-fos activation in dorsal horn neurons [19]. The goal of this study was to build upon our previous studies to determine if our finding of endothelin-induced sensitization in males is mediated at the primary afferent neuron level. To examine this, primary DRG cell cultures were kept in culture for four days to mimic the timeline (postnatal day 7-postnatal day 11) in our in vivo model and exposed to ET-1 similarly to our in vivo study. Calcium imaging was used to measure neuronal activity in these DRG cultures after exposure to ET-1.
2. Methods
2.1 Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of South Carolina. Efforts were made to limit the amount of distress and the number of animals used. Male Sprague-Dawley (Charles River Laboratories, MA) rats were kept on a 12 hour light/dark cycle with food and water available ad libitum.
2.2 Cell Culture
Dorsal root ganglia (DRG) from adult male rats were dissected and collected in ice cold Tyrode buffer (132 mM NaCl, 4.8 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM dextrose, 5 mM HEPES). DRG were digested using a solution of dispase (1 mg/mL, Gibco 171050-041) and collagenase (2 mg/mL, Roche 1088831) in Tyrode buffer and incubation in a shaking water bath at 37°C for 45 minutes. Following digestion, the DRGs were triturated with a fire polished Pasteur pipet followed by separation using an OptiPrep (Sigma D1556) gradient. Cells were centrifuged at 1900 rpm for 20 minutes then the bottom layer was isolated and the cells were counted.
The DRG cells were seeded in 96 well black clear bottom plates (Costar, Corning Inc.) coated with poly-D-lysine at a density of approximately 60,000-80,000 cells per well (day 0). Cells were maintained in Minimum Essential Medium Eagle media (Sigma Aldrich M0643) (supplemented with 20% glucose, 10% FBS (Invitrogen 16000-036), 1% penicillin-streptomycin solution (Invitrogen 15070-063), and 10 μg/ml of NGF (Millipore 01-125).
2.3 Calcium imaging for ET-1 experiment
Cells were maintained in culture for 4 days and on the 5th day, calcium imaging was performed. On day 0 (the day of culture), cells were either treated with 1.1 nmole of ET-1 (22 μM, Enzo Life Sciences) or vehicle (sterilized deionized water). On day 4, cells were first washed with either Ca2+-containing NaCl-based extracellular (EC) buffer (130 mM NaCl, 2.5 mM KCl, 1mM MgCl2, 4 mM CaCl2, 10 mM HEPES, 5mM glucose) with 2.5 mM probenecid (Molecular Probes P36400) or Ca2+-free NaCl-based EC buffer then incubated with Fluo-4AM solution (1 μM, Molecular Probes, Invitrogen F14201) containing pluronic acid at 37°C for 1 hour. After incubation with the dye solution, cells were washed twice with the appropriate NaCl-based EC buffer, then 190 μl of the appropriate buffer was added to each well. After incubation at 37°C for 1 hour, the plate was loaded into the BioTek Synergy 2 plate reader and the BioTek Gen5™ Data Analysis Software was used to perform reader control. After a 5 minute delay, the cells were baselined every 5 seconds for 30 seconds. Following the baseline measures, either 10 μL of ET-1 (0.55μM) or 10 μL of vehicle (sterile filtered deionized water) was added to the appropriate wells. The change in fluorescence was then measured every 3 seconds for 3 minutes. Fluorescence was excited at 485 nm and emission was measured at 528 nm. Temperature was maintained at 37°C. A total of 6 wells were measured for each treatment group.
2.4 Drug treatment and Ca2+ Imaging for ET-1 time course
In a separate experiment, cultured cells were treated with either 0.11 nmole of ET-1 (2.2 μM, Alexis Biochemicals) or vehicle over a period of 4 days. On day 0, one set of cells was treated with ET-1 and the others treated with vehicle. On day 1, a different set of cells was treated with ET-1 and the others treated with vehicle. This pattern was repeated on days 2 and 3 with a different set of cells being treated with ET-1. During the course of the experiment, different sets of cells were pre-treated with ET-1 only once on either day 0, 1, 2, or 3. There were a total of 6 wells for each treatment group.
On day 4, calcium imaging was performed. Cells were loaded with the dye solution as mentioned previously. The plate reader was programmed to have a 5 minute delay, after which, baseline measurements were taken every 5 seconds for 30 seconds for the first set of wells. ET-1 (10 μl, of either 5.5 μM or 0.55 μM) or vehicle (10 μl) was added to the appropriate wells, then readings were made continuously every 3 seconds for 3 minutes.
2.5 Calcium imaging for capsaicin experiment
The cells were treated with either vehicle (sterile filtered deionized water) or 1.1 nmole of ET-1 (22 μM, American Peptide, Sunnyvale, CA on day 0 (the day of cell culture). On day 4, calcium imaging was performed. Following the baseline measures, either 10 μL of capsaicin (1 μM) or 10 μL of vehicle (DMSO in NaCl-based EC buffer) was added to the appropriate wells (5 wells/treatment group) and the change in fluorescence measured every 3 seconds for 5 minutes.
3. Results
3.1 Effect of ET-1 on [Ca2+]i in afferent neurons
On day 4 in culture, application of 0.55 μM of ET-1 onto naïve cells significantly increased intracellular Ca2+ compared to vehicle treated cells (p<0.001) (Figure 1A). However, priming with 22 μM of ET-1 on day 0 followed by a challenge with 0.55 μM of ET-1 on day 4 did not produce a significant influx of Ca2+ into the cultured afferent neurons (Figure 1A). The increase in intracellular Ca2+ due to ET-1 is dependent on extracellular Ca2+, as removal of Ca2+ from the extracellular buffer prevents ET-1-induced Ca2+ transients (Figure 1A inset). Similarly, a reduction in area under the curve (AUC) (Fig 1B) and maximum peak height (Fig 1C) is seen in ET-1 primed cells compared to unprimed cells exposed to ET-1 for the first time at the time of imaging (Fig 1B). The time to the calcium peak is similar between treatments with a trend to a more rapid peak in cells exposed to ET-1 compared to vehicle (Fig 1D).
Figure 1. ET-1-induced increase in Ca2+ due to influx of Ca2+ not from release of intracellular stores and priming with ET-1 prevents ET-1-induced Ca2+ influx.

Cells incubated in buffer containing Ca2+ exhibited a significant increase in intracellular Ca2+ upon treatment with ET-1 compared to cells treated with vehicle (p<0.001) when measuring the change in fluorescence (A), the area under the curve (B), and the maximum peak height (C). Cells primed with ET-1 on day 0 then treated with ET-1 at the time of imaging show a significant decrease in Ca2+ influx compared to vehicle primed cells treated with ET-1 at the time of imaging (p<0.001). Cells incubated in Ca2+ buffer did not show an increase in intracellular Ca2+ upon treatment with ET-1 (A inset).
3.2 Time course of priming effect of ET-1
Application of low dose (0.55 μM, Fig 2A) or high dose (5.5 μM, Fig 2B) of ET-1 produced a statistically significant rise in [Ca2+]i. Priming of the cells with 2.2 μM of ET-1 on Day 1, 2, or 3 of culture produced a time-dependent blockade of the ET-1 stimulated rise in [Ca2+]i when a low challenge dose of ET-1 was used (Fig 2A, C). In contrast, priming on any day suppressed calcium transients when a high challenge dose of ET-1 was used (Fig 2B, C). Measurement of the maximum peak height (Fig 2E) showed similar time dependent priming with low but not high dose ET-1 as the area under the curve measurements (Fig 2C). The time to peak was similar in all treatments using a low dose ET-1 challenge (Fig 2D). In contrast, the time to peak was delayed by priming on days 1 and 3 when a high dose of ET-1 was used to challenge (Fig 2D). This data suggests that both the day of priming and the challenge dose of ET-1 have a significant impact on ET-1 induced desensitization in primary afferent neurons.
Figure 2. Time course for prevention of ET-1-induced Ca2+ influx due to priming effect of ET-1.

Reducing the priming dose of ET-1 to 2.2 μM reveals a particular time course for the decrease in [Ca2+]i. (A, C) Cells primed with 2.2 μM of ET-1 on day 0 and challenged with 0.55 μM of ET-1 on day 4 show a significantly smaller increase in [Ca2+]i compared to cells treated with ET-1 only at the time of imaging (***p<0.001) and this level of increase in [Ca2+]i is significantly different from vehicle treated cells (**p<0.01). (B, C) Cells primed with 2.2 μM of ET-1 on day 0 and challenged with 5.5 μM of ET-1 on day 4 show a significantly smaller increase in [Ca2+]i compared to cells only treated with ET-1 at the time of imaging (***p<0.001) and this level of [Ca2+]i is not significantly different from vehicle treated cells. (D) The time to calcium peak appeared to be more rapid for cells treated with ET-1 for the first time on the day of imaging with 5.5 μM ET-1 compared to cells challenged with vehicle (*p<0.05). This was also true for cells primed with ET-1 on days 0 and 2 and challenged with 5.5 μM ET-1 (**p<0.01). (E) Maximum peak height for unprimed ET-1 treated cells and cells primed on day 0 and challenged with 0.55 μM ET-1 had significantly greater maximum peak heights compared to unprimed vehicle treated cells (***p<0.001 and **p<0.01, respectively).
3.3 Effect of ET-1 priming on capsaicin-induced increase in [Ca2+]i
Previous results from our lab have shown that prior exposure to ET-1 can modify subsequent behavioral nociceptive responses to capsaicin [19]. The purpose of this experiment was to determine whether that effect of ET-1 priming on capsaicin responses might be mediated at the level of the afferent neuron. Treatment of cells with capsaicin on day 4 causes a significant increase in [Ca2+]i compared to cells treated with vehicle on day 4 (p<0.001) (Figure 3). Priming with ET-1 on day 0 then challenging with capsaicin causes significantly lower increase in [Ca2+]i compared to cells treated with vehicle on day 0 then given capsaicin on day 4 (p<0.001) (Figure 3).
Figure 3. Pre-treatment with ET-1 causes decrease in capsaicin-induced influx of Ca2+.
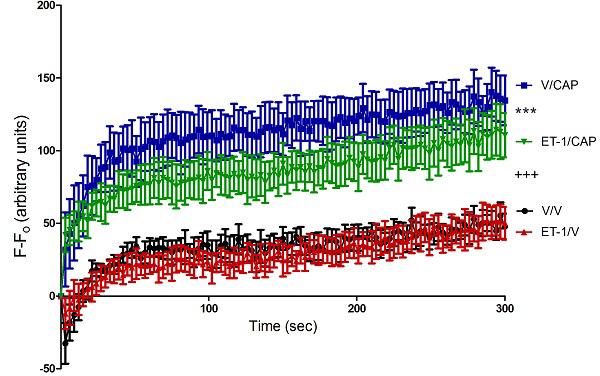
Capsaicin caused a significant increase in [Ca2+]i compared to cells treated with vehicle on day 4 (+++ p<0.001) There is a significantly greater difference from baseline in capsaicin treated cells primed with vehicle on day 0 compared to capsaicin treated cells primed with ET-1 on day 4 (***p<0.001). There was no significant difference between cells vehicle treated cells primed with vehicle on day 0 and vehicle treated cells primed with ET-1 on day 0.
4. Discussion
In this study, we examined the endothelin system in an in vitro model correlate of our animal model of localized acute VOEs. We found that in our in vitro model in primary afferent neurons from DRGs isolated from adult male rats, ET-1 causes a large influx of Ca2+ in naïve cells, but a “desensitized” response in ET-1 primed cells, which parallels the desensitized behavioral response to subsequent ET-1 exposure in female neonatal rats. The ET-1 priming effect is dependent upon the timing of the priming dose as well as on the dose of the ET-1 challenge. Similarly, ET-1 priming desensitizes primary afferent neurons to subsequent challenge with capsaicin.
The present calcium imaging results demonstrating ET-1 priming and desensitization is supported by previous studies which showed that ET-1 causes an increase in [Ca2+]i in neuronal cells, while repeated administration of ET-1 onto mouse neuroblastoma-rat DRG hybrid cells reduces [Ca2+]i transients [23, 24]. These studies also found that the ET-1-induced increase in [Ca2+]i is mediated by activation of the ETA receptor [24]. However, in these previous studies, they found the increase in [Ca2+]i to come from intracellular stores, while this study found that this increase was due to the influx from extracellular Ca2+. The difference in the source of [Ca2+]i increase may be explained by the much larger dose of ET-1 used in our study to prime the cells as well as a potential species difference in DRG neurons.
Similar to the results shown in our current in vitro study, repeated administration of ET-1 onto the sciatic nerve leads to reduced paw flinching behavior, or a “desensitized” response, compared to a single administration and this effect lasts for at least 24 hours [5]. It has also previously been shown that intrathecal administration of ET-1 has an analgesic effect in a postoperative pain model which is accompanied by a decrease in incision-induced ERK phosphorylation in DRG neurons [2]. The desensitization we observed in DRG cells after repeated administration of ET-1 may be due to a decrease in ERK phosphorylation of DRG neurons. The greater increase in ET-1 induced Ca2+ transients in cells primed with ET-1 on day 0 compared to cells primed on days 1, 2, or 3 could be due to a time dependent recovery of receptors or other components of the signaling cascade, such as ERK.
Depending on the cell type and species investigated, ET-1-induced influx of extracellular Ca2+ can be due to direct or indirect regulation of voltage gated Ca2+ channels as well as non-selective cation channel and store-operated Ca2+ channel activation [21]. In mammalian parasympathetic neurons, ET-1 causes an increase in [Ca2+]I by activating receptor-operated Ca2+ channels [14]. In many cell types, ET-1-induced Ca2+ mobilization is caused by a mixture of voltage-dependent Ca2+ influx, voltage-independent Ca2+ influx, and the release of Ca2+ from intracellular stores [21]. One possible explanation for voltage-dependent Ca2+ influx due to ET-1 may be the effect of ET-1 on sodium channels. ET-1, through the ETA receptor, lowers the threshold for activation of tetrodotoxin-resistant Na+ channels in DRG neurons thereby increasing the excitability of the neurons [25]. In DRG neurons, ET-1 also enhances neuronal excitability by suppressing delayed-rectifier type of K+ currents [6]. An explanation for voltage-independent Ca2+ influx due to ET-1 may be its effects on the opening of cation channels. In rat glioma cells, the major source of ET-1-induced increase in [Ca2+]I was found to be from an influx of extracellular Ca2+ possibly through the opening of non-selective cation channels such as TRPV1 channels [12].
Priming with ET-1 also desensitized the [Ca2+]i response to capsaicin, which is the chemical found in chili peppers that produces a burning sensation when applied externally or ingested. Capsaicin activates the TRPV1 ion channel on DRG neurons, more specifically on small diameter neurons, which leads to an influx of Ca2+ [1]. In DRG neurons, which mainly express ETA receptors, ET-1 potentiates the activity of TRPV1 channels in response to capsaicin [16]. It has been shown that application of capsaicin onto DRG neurons causes an increase in [Ca2+]i , while repeated applications of capsaicin leads to desensitized response of a reduction in [Ca2+]i [22, 23]. Our result of a decreased response to capsaicin in ET-1 primed cells is somewhat in conflict with a previous study showing an enhancement of the increase in [Ca2+]i after application of ET-1 followed by capsaicin [23]. However, this difference may be explained by the fact that the neurons used in our study were incubated with ET-1 for hours before being removed and challenged with capsaicin days later, while the previous published study applied ET-1 seconds before adding capsaicin looking at an acute modulator role versus a long-term modulatory role.
The results of this study suggest that the endothelin-induced sensitization upon repeated administration we observed in our in vivo model may not be occurring at the primary afferent level. It should be noted, however, that our in vivo model involves neonatal animals while this study utilized cells from adult animals. We cannot rule out a developmental difference in the Ca2+ response to repeated exposure to ET-1 in DRG neurons, which may explain why we observed a desensitized response rather than a sensitized response in these neurons from male rats. Furthermore, priming with ET-1 produces a long lasting desensitization of afferent neurons to subsequent ET-1 and the alternative algogen capsaicin. This suggests that behavioral sensitization to repeat ET-1 exposure may be occurring in the skin, in the spinal cord, or in the brain. Future studies are needed to begin to understand how the different levels of the pain neuroaxis respond to repeated exposure to ET-1.
Highlights.
ET-1 causes increase in Ca2+ transients in unprimed dorsal root ganglion neurons.
ET-1 priming has a desensitizing effect on subsequent exposures to ET-1.
ET-1 priming effect is dependent on timing of the priming dose and on the challenge dose.
Priming with ET-1 also causes a desensitized response to capsaicin.
Acknowledgements
This work was funded by National Institutes of Health grants R01 DA023593 and NS26363, a South Carolina Post-Baccalaureate Research Education Programs for Minorities funded by grants R25 GM066526 and R25 GM076277 from the National Institutes of Health, and the Alfred P Sloan Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 2.Chen G, Tanabe K, Yanagidate F, Kawasaki Y, Zhang L, Dohi S, Iida H. Intrathecal endothelin-1 has antinociceptive effects in rat model of postoperative pain. Eur J Pharmacol. 2012;697:40–46. doi: 10.1016/j.ejphar.2012.09.035. [DOI] [PubMed] [Google Scholar]
- 3.Dahlof B, Gustafsson D, Hedner T, Jern S, Hansson L. Regional haemodynamic effects of endothelin-1 in rat and man: unexpected adverse reaction. J Hypertens. 1990;8:811–817. doi: 10.1097/00004872-199009000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Davar G, Hans G, Fareed MU, Sinnott C, Strichartz G. Behavioral signs of acute pain produced by application of endothelin-1 to rat sciatic nerve. Neuroreport. 1998;9:2279–2283. doi: 10.1097/00001756-199807130-00025. [DOI] [PubMed] [Google Scholar]
- 5.Fareed MU, Hans G, Atanda AG, Jr., Strichartz G. Davar, Pharmacological characterization of acute pain behavior produced by application of endothelin-1 to rat sciatic nerve. J Pain. 2000;1:46–53. [Google Scholar]
- 6.Feng B, Strichartz G. Endothelin-1 raises excitability and reduces potassium currents in sensory neurons. Brain Res Bull. 2009;79:345–350. doi: 10.1016/j.brainresbull.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira SH, Romitelli M, de Nucci G. Endothelin-1 participation in overt and inflammatory pain. J Cardiovasc Pharmacol. 1989;13(Suppl 5):S220–222. doi: 10.1097/00005344-198900135-00065. [DOI] [PubMed] [Google Scholar]
- 8.Gokin AP, Fareed MU, Pan HL, Hans G, Strichartz GR, Davar G. Local injection of endothelin-1 produces pain-like behavior and excitation of nociceptors in rats. J Neurosci. 2001;21:5358–5366. doi: 10.1523/JNEUROSCI.21-14-05358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M, McMillen MA. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood. 1998;92:2551–2555. [PubMed] [Google Scholar]
- 10.Hans G, Deseure K, Robert D, De Hert S. Neurosensory changes in a human model of endothelin-1 induced pain: a behavioral study. Neurosci Lett. 2007;418:117–121. doi: 10.1016/j.neulet.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Kaul DK, Fabry ME, Nagel RL. The pathophysiology of vascular obstruction in the sickle syndromes. Blood reviews. 1996;10:29–44. doi: 10.1016/s0268-960x(96)90018-1. [DOI] [PubMed] [Google Scholar]
- 12.Lin WW, Kiang JG, Chuang DM. Pharmacological characterization of endothelin-stimulated phosphoinositide breakdown and cytosolic free Ca2+ rise in rat C6 glioma cells. J Neurosci. 1992;12:1077–1085. doi: 10.1523/JNEUROSCI.12-03-01077.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKelvy AD, Sweitzer SM. Endothelin-1 exposure on postnatal day 7 alters expression of the endothelin B receptor and behavioral sensitivity to endothelin-1 on postnatal day 11. Neurosci Lett. 2009;451:89–93. doi: 10.1016/j.neulet.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura T, Akasu T, Krier J. Endothelin modulates calcium channel current in neurones of rabbit pelvic parasympathetic ganglia. Br J Pharmacol. 1991;103:1242–1250. doi: 10.1111/j.1476-5381.1991.tb12331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. 1995;96:1145–1151. doi: 10.1172/JCI118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plant TD, Zollner C, Mousa SA, Oksche A. Endothelin-1 potentiates capsaicin-induced TRPV1 currents via the endothelin A receptor. Exp Biol Med (Maywood) 2006;231:1161–1164. [PubMed] [Google Scholar]
- 17.Schlenz AM, McClellan CB, Mark TR, McKelvy AD, Puffer E, Roberts CW, Sweitzer SM, Schatz JC. Sensitization to acute procedural pain in pediatric sickle cell disease: modulation by painful vaso-occlusive episodes, age, and endothelin-1. J Pain. 2012;13:656–665. doi: 10.1016/j.jpain.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiu YT, McIntire LV, Udden MM. Sickle erythrocytes increase prostacyclin and endothelin-1 production by cultured human endothelial cells under flow conditions. Eur J Haematol. 2002;68:163–169. doi: 10.1034/j.1600-0609.2002.01494.x. [DOI] [PubMed] [Google Scholar]
- 19.Smith T, Beasley S, Smith S, Mark I, Sweitzer SM. Endothelin-1-induced priming to capsaicin in young animals. Neurosci Lett. 2014 doi: 10.1016/j.neulet.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stinson J, Naser B. Pain management in children with sickle cell disease. Paediatr Drugs. 2003;5:229–241. doi: 10.2165/00128072-200305040-00003. [DOI] [PubMed] [Google Scholar]
- 21.Tykocki NR, Watts SW. The interdependence of endothelin-1 and calcium: a review. Clinical science. 2010;119:361–372. doi: 10.1042/CS20100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. The Journal of physiology. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto H, Kawamata T, Ninomiya T, Omote K, Namiki A. Endothelin-1 enhances capsaicin-evoked intracellular Ca2+ response via activation of endothelin a receptor in a protein kinase Cepsilon-dependent manner in dorsal root ganglion neurons. Neuroscience. 2006;137:949–960. doi: 10.1016/j.neuroscience.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 24.Zhou QL, Strichartz G, Davar G. Endothelin-1 activates ET(A) receptors to increase intracellular calcium in model sensory neurons. Neuroreport. 2001;12:3853–3857. doi: 10.1097/00001756-200112040-00050. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Z, Davar G, Strichartz G. Endothelin-1 (ET-1) selectively enhances the activation gating of slowly inactivating tetrodotoxin-resistant sodium currents in rat sensory neurons: a mechanism for the pain-inducing actions of ET-1. J Neurosci. 2002;22:6325–6330. doi: 10.1523/JNEUROSCI.22-15-06325.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]