

Abstract
Total or near-total loss of insulin-producing β-cells is a situation found in diabetes (Type 1, T1D) 1,2. Restoration of insulin production in T1D is thus a major medical challenge. We previously observed in mice in which β-cells are completely ablated that the pancreas reconstitutes new insulin-producing cells in absence of autoimmunity 3. The process involves the contribution of islet non-β-cells; specifically, glucagon-producing α-cells begin producing insulin by a process of reprogramming (transdifferentiation) without proliferation 3. Here we studied the influence of age on β-cell reconstitution from heterologous islet cells after near-total β-cell loss. We found that senescence does not alter α-cell plasticity: α-cells can reprogram to produce insulin from puberty through adulthood, and also in aged individuals, even a long-time after β-cell loss. In contrast, prior to puberty there is no detectable α-cell conversion, although β-cell reconstitution after injury is more efficient, always leading to diabetes recovery; it occurs through a newly discovered mechanism: the spontaneous en masse reprogramming of somatostatin-producing δ-cells. The younglings display “somatostatin-to-insulin” δ-cell conversion, involving de-differentiation, proliferation and re-expression of islet developmental regulators. This juvenile adaptability relies, at least in part, upon combined action of FoxO1 and downstream effectors. Restoration of insulin producing-cells from non-β-cell origins is thus enabled throughout life via δ- or α-cell spontaneous reprogramming. A landscape with multiple intra-islet cell interconversion events is emerging, thus offering new perspectives.
To determine how ageing affects the mode and efficiency of β-cell reconstitution after β-cell loss, we administered diphtheria toxin (DT) to adult (2-month-old) or aged (1-and 1.5-year-old) RIP-DTR mice, whose β-cells bear DT receptors 3, and followed them for up to 14 months. Collectively, we found that α-to-β cell conversion is the main mechanism of insulin cell generation after massive β-cell loss in adult post-pubertal mice, whether middle-aged or very old, and α-cells are progressively recruited into insulin production with time (Extended Data Fig.1; Supp. Tables S1-5).
In this study we focused on the regeneration potential during early postnatal life by inducing β-cell ablation before weaning, at 2 weeks of age (Fig. 1a). We found that prepubescent mice rapidly recover from diabetes after near-total β-cell loss: four months later all younglings were almost normoglycemic, thus displaying a faster recovery relative to adults (Fig. 1b and Extended Data Fig.2a,b; see Extended Data Fig.1a).
Figure 1. β-cell ablation before puberty and diabetes recovery.

a) Experimental designs depicting the ages at DT-administration and the various analyses (“mpa”, months post-ablation). b) Comparative evolution of glycemia in β-cell-ablated younglings (n=5) and middle-aged adults (n=4); 2.5 months after β-cell ablation, insulin administration was stopped (Mann-Whitney [p=0.0014]). c) Islets from 2-week-old (“control”), 0.5 mpa and 4 mpa (Supp. Table S6). d) α-cell tracing in pups. Scale bars: 20μm.
Histologically, 99% of the β-cells were lost at 2 weeks following DT administration (Fig. 1c). The β-cell number increased by 45-fold 4 months after ablation, representing 23% of the normal age-matched β-cell mass (Fig. 1c; Supp. Table S6) and correlating with normoglycemia recovery 1.
All animals remained normoglycemic during the rest of their life (Supp. Table S6). Mice were neither intolerant to glucose nor insulin resistant during the period of analysis, up to 15 months after injury (Extended Data Fig. 2c-e).
We investigated whether the new insulin+ cells were reprogrammed α-cells, as in adults, using Glucagon-rtTA; TetO-Cre; R26-YFP; RIP-DTR pups (Fig. 1d). We observed that almost no insulin+ cell co-expressed YFP or glucagon (Supp. Table S7), indicating that α-cells do not reprogram in younglings.
We further explored the age-dependency of rescue after near-total β-cell loss. To this aim, normoglycemic 5-month-old mice, which had recovered from β-cell loss at 2 weeks of age, were re-administered DT to ablate the regenerated insulin+ cells. One month following the second ablation, 30% of the insulin-containing cells also contained glucagon (Extended Data Fig.2f; Supp. Table S8), like β-cell-ablated adults (Extended Data Fig. 1k), confirming that the pre-pubertal regeneration mechanism is restricted temporally.
We measured proliferation rates at different time-points during 2 months of regeneration. The proportion of Ki67-labeled insulin+ cells was very low (Extended Data Fig.2g; Supp. Table S9), indicating that neither escaping β-cells nor regenerated insulin+ cells proliferate during this period. However, there was a transient 3.5-fold increase in the number of insular Ki67+ cells 2 weeks after ablation, unlike in adult animals (Extended Data Fig.2h; Supp. Table S10). Replicating cells were hormone-negative, chromogranin A-negative, and were not lineage-traced to either α- or escaping β-cells (Extended Data Fig.2i,j).
Coincident with the peak of islet cell proliferation we noticed in pups a 4.5-fold decrease in the number of somatostatin-producing δ-cells (from 13 to 3 δ-cells/islet section; Extended Data Fig.3a; Supp. Table S11) and a 76-fold decrease of somatostatin transcripts (Extended Data Fig.3b), without indication of increased islet cell death. We therefore lineage-traced δ-cells and observed that regenerated insulin-producing cells were dedifferentiated δ-cells. At 2 months of age in Somatostatin-Cre; R26-YFP; RIP-DTR mice, about 81% of δ-cells were YFP+ in the absence of β-cell ablation, whereas α- and β-cells were labeled at background levels (0.9% for β-cells and 0.2% for α-cells; Extended Data Fig.3c,d, Supp. Table S12). During β-cell reconstitution in pups, 2 weeks after β-cell ablation, 80% of YFP+ cells were proliferating (Ki67+) and somatostatin-negative (Fig. 2a,b; Supp. Table S13), while most Ki67+ cells were YFP-labeled (85%; Supp. Table S14).
Figure 2. δ-cells dedifferentiate, proliferate and reprogram into insulin production after extreme β-cell loss in Somatostatin-Cre; R26-YFP; RIP-DTR juvenile mice.

a) Immunofluorescence for YFP and Ki67 at 0.5 mpa. b) 80% of somatostatin-traced YFP+ cells are Ki67+ after β-cell-ablation (controls: n=6, 2,754 YFP+-cells scored; DT: n=6; 3,146 YFP+-cells scored; Welch’s test [p<0.0001], Mann-Whitney [p=0.0022]). c, d) At 1.5 mpa 90% of insulin+ cells coexpress YFP (controls: n=3, 6,480 insulin+-cells scored;DT: n=7, 1,592 insulin+-cells scored; Welch’s test [p<0.0001], Mann-Whitney [p=0.0167]). Arrow: YFP+/somatostatin+ cells; arrowhead: YFP+/insulin+ cells;. e) In controls, 99.9% of the YFP+ cells are somatostatin+ (n=3, 1,673 YFP+-cells scored). In contrast, at 1.5 mpa only 55% of the YFP+ cells are somatostatin+, while 45% of the YFP+ cells are insulin+ (n=5, 2,295 YFP+-cells scored; Welch’s test [p<0.0001], Mann-Whitney [p=0.0357]). f) Comparative evolution of glycemia after β-cell (n=5), δ-cell (n=4) and β- & δ-cell co-ablation (n=5) in younglings. g) δ-cell conversion sequence. Scale bars: 20 μm. Error bars: s.d.
These observations suggest that in β-cell-ablated pre-pubertal mice most δ-cells undergo a loss of somatostatin expression and enter the cell cycle.
We further investigated the fate of proliferating dedifferentiated δ-cells. At 1.5 months post-ablation, most insulin+ cells expressed YFP (90%), indicating their δ-cell origin (Fig. 2c,d; Supp. Table S15). Furthermore, in contrast to non-ablated age-matched controls, where all YFP+ cells were somatostatin+ (>99%), about half of YFP+ cells were insulin+ after 1.5 months of regeneration (45%; Fig. 2e; Supp. Table S16). This reveals that half of the progeny of dedifferentiated δ-cells becomes insulin expressers. Bihormonal somatostatin+/insulin+ cells were rare (Supp. Table S17).
Combined, these observations show that at the cell population level, each dedifferentiated δ-cell yields one insulin expresser cell and one somatostatin+ cell(Extended Data Fig.4).
We confirmed with two other assays that regeneration and diabetes recovery in juvenile mice are δ-cell-dependent: by inducing β-cell destruction with streptozotocin (STZ) instead of DT (Extended Data Fig.5a-c), and by co-ablating β- and δ-cells simultaneously in Somatostatin-Cre; R26-YFP; R26-iDTR; RIP-DTR pups. In absence of δ-cells there was no insulin+ cell regeneration, and no recovery (Fig. 2f).
In adults, δ-cells neither de-differentiated nor proliferated following β-cell ablation (Extended Data. Fig.5d,e; Supp. Table S20). Nevertheless, like α-cells, a few δ-cells reprogrammed into insulin production, so that after 1.5 month of regeneration 17% of the rare insulin-producing cells were YFP+, i.e. δ-cell-derived (Extended Data Fig. 5f-h; Supp. Tables S21, S22).
By transplanting Somatostatin-Cre; R26-YFP; RIP-DTR juvenile islets into adult wild-type mice we observed that, following β-cell ablation, the newly formed insulin+ cells were reprogrammed δ-cells, thus showing that the pup-specific regeneration is intrinsic to islets (Extended Data Fig.6).
Contrary to β-cells in age-matched adult mice, δ-cell-derived insulin+ cells replicated transiently (Extended Data Fig.7a; Supp. Table S23); the β-cell mass thus reached between 30% to 69% of the normal values, and remained stable for life (above, Supp. Table S6).
We characterized the δ-cell-derived insulin+ cells at the gene expression level by qPCR. We first compared islets isolated 2 weeks after β-cell ablation or after recovery (4 months post-DT), with age-matched control islets. Expression of all the β-cell-specific markers tested was robustly increased in recovered mice (Extended Data Fig.7b). We also compared regenerated insulin+ cells with native β-cells using sorted mCherry+ cells obtained from either recovered or unablated age-matched (4.5-month-old) insulin-mCherry; RIP-DTR mice (Extended Data Fig.7c). The two cell populations were very similar (Extended Data Fig.7d), yet the δ-cell-derived replicating β-cells displayed apotent downregulation of cyclin-dependent kinase inhibitors and regulators (Extended Data Fig.7e,f). This suggests that reconstituted insulin+ cells are like β-cells with transient proliferation capacity. Future studies will establish whether reconstituted (δ)β-like cells are true equivalents to native β-cells.
qPCR and lineage-tracing analyses on islets isolated from pups at different regeneration time-points, together with Ngn3 KO induction after β-cell ablation, revealed that Ngn3transcription is required for the δ-to-insulin+ cell conversion to occur (Extended Data Fig.8a-k, Supp. Tables S24-S29). Of note, brief expression of Ngn3 is a feature of islet precursor cells in the embryonic pancreas 4. Together, these observations are compatible with a model in which β-cell reconstitution after ablation in younglings occurs following a sequence of events: δ-cells dedifferentiate, replicate once and then half of the progeny activates Ngn3 expression before insulin production (Fig. 2g). This was tested in a combined double lineage-tracing experiment using Somatostatin-Cre; R26-Tomato; Ngn3-YFP; RIP-DTR mice. Six weeks post-β-cell ablation, insulin+ cells in younglings were Tomato+/YFP+ (Extended Data Fig.8k).
One key reprogramming and cell cycle entry player is FoxO1, a transcription factor whose downregulation triggers Ngn3 expression in human fetal pancreatic explants 5 and favors insulin production in Ngn3+ entero-endocrine progenitors 6. FoxO1, usually in cooperation with TGFβ/SMAD signaling 7,8, inhibits cell proliferation through the transcriptional regulation of cell cycle inhibitors and activators 18, and is involved in cellular senescence 7 (Extended Data Fig.9a). We then explored the FoxO1 molecular network in purified adult or juvenile δ-cells before and after (1-week) β-cell ablation, using Somatostatin-Cre; R26-YFP; RIP-DTR mice.
δ-cells displayed a divergent regulation of FoxO1 in injured juvenile and adult mice. Consistent with FoxO1 downregulation in juvenile δ-cells, PDK1 and AKT levels were increased, cdkn1a/p21 and cdkn2b/p15Ink4b were downregulated, and CKS1b, CDK2 and SKP were upregulated (Fig. 3a), which is compatible with the proliferative capacity of juvenile δ-cells after β-cell ablation. The opposite was found in δ-cells of ablated adults (Fig. 3a; Extended Data Fig.9b).
Figure 3. Age-dependent effect of β-cell loss on δ-cells.

a, b) Transcriptional variation of cell cycle regulators, PI3K/AKT/FoxO1 network genes (a), and TGFβ and BMP components and effectors (b) in juvenile and adult δ cells 1 week after ablation, as compared with age-matched controls. c) β-cell loss before puberty triggers FoxO1 downregulation in δ-cells, while the opposite occurs in adults (see Extended Data Fig.9b). d) Experimental design to transiently inhibit FoxO1 in β-cell-ablated adult mice. e) Induction of δ-to-insulin cell conversion in diabetic adult mice. Scale bars: 20 μm. f,g) Insulin+ cells are 11-fold more abundant in FoxO1 inhibitor-treated mice (treated: n=190 islets, 4 mice; untreated: n=95 islets, 3 mice (Welch’s test [inter-islet p<0.0001, inter-individual p=0.0065], Mann-Whitney [p<0.0001]) (f), and they are YFP+ (93%) (treated: n=4, 894 insulin+-cells scored; untreated: n=6, 370 insulin+-cells scored, Welch’s test [p<0.0001], Mann-Whitney [p=0.0095]) (g). h) One fourth of δ-(YFP+) cells in adult β-cell-ablated FoxO1-inhibited mice dedifferentiate and become insulin expressers (treated: n=4, 3,358 YFP+-cells scored; untreated: n=6, 2,559 YFP+-cells scored). Error bars: s.d.
Moreover, in δ-cells of younglings, but not in adults, there was a robust upregulation of BMP1/4 downstream effectors (Fig. 3b) 9,10. Inversely, TGFβ pathway genes were upregulated in δ-cells of regenerating adults (Fig. 3b), which is compatible with the senescence scenario 7 involving PI3K/FoxO1 and TGFβ/SMAD cooperation to maintain differentiation and cycle arrest (Extended Data Fig9a,b).
In summary, PI3K/AKT and SKP2/SCF pathways potentially cooperate to downregulate FoxO1 in δ-cells of regenerating younglings. Also, upregulation of BMP effectors (ID1 and ID2) could contribute to δ-cell dedifferentiation and proliferation, as observed in other systems 9,10 (Fig. 3c). Conversely, the PI3K/AKT pathway remained downregulated in δ-cells of ablated adults, which would allow FoxO1 to impede proliferation and dedifferentiation, probably through partnership with previously described SMADs 11 (Extended Data Fig.9b).
We then checked whether a transient FoxO1 inhibition in adult mice would lead to a juvenile-like δ-to-β cell conversion. Indeed, inactivation of FoxO1 in β-cells causes their dedifferentiation 12. Here, Somatostatin-Cre; R26-YFP; RIP-DTR β-cell-ablated adult mice were given a FoxO1 inhibitor (AS1842856) for 1 week, either immediately following ablation (Fig. 3d) or 1 month later (Extended Data Fig.10f; Supp. Tables S37-S39) 13,14. While FoxO1 inhibition in non-ablated controls had minimal effect on insulin expression (Extended Data Fig.10a-d; Supp. Tables S30-S32), regeneration in diabetic mice was improved: insulin+ cells were more abundant (11-fold; Fig. 3e,f; Supp. Table S33), and were reprogrammed δ-cells (93% were YFP+, Fig. 3g; Supp. Table S34). One-fourth of the YFP+ cells expressed insulin only (Fig. 3h; Extended Data Fig.10e; Supp. Tables S35, S36), revealing that, like in younglings, an important fraction of δ-cells had converted to insulin production.
These results support the involvement of a regenerative FoxO1 network and confirm that δ-cell conversion can be pharmacologically induced in diabetic adults. FoxO1 blockade has a pleiotropic effect: inhibition of hepatic gluconeogenesis 13,14 and promotion of δ-cell reprogramming (this study).
A century ago Morgan coined the terms epimorphosis and morphallaxis to designate, respectively, regeneration involving either cell dedifferentiation and proliferation or direct conversion from one cell type into another without proliferation 15. Here we report in mammals an age-dependent switch (“adult transition”) between epimorphic regeneration during youth, and a less efficient yet persistent throughout life proliferation-independent morphallactic mechanism.
Our findings uncover a novel role for δ-cells; perhaps somatostatin+ cells in the stomach, intestine or hypothalamus share the same capabilities. Intra-islet cell plasticity triggered by the disappearance of β-cells is influenced by age: the proliferation decline in ageing cells 16 would explain the need of an “adult transition”. Although less efficient, α-cell plasticity remains long-time after β-cell loss since it is proliferation-independent.
These phenomena might be translatable to humans, for there is efficient β-cell regeneration in children with T1D or after pancreatectomy 3,17-19, and glucagon/insulin bihormonal human cells have been described upon epigenetic manipulation ex vivo 20, and in diabetic patients 21,22. Knowing also that only a small fraction of the α-cell population is sufficient to maintain glucagon signaling 23, understanding the nature of the diverse forms of intra-islet cell conversion might provide new opportunities to fostering the formation of (α)β-like and (δ)β-like cells.
Extended Data
Extended Data Figure 1. Maintenance of α-cell plasticity in diabetic aged mice.

a) Evolution of glycemia in β-cell-ablated adults (middle-aged) and aged mice. The “area under the curve” (AuC) in middle-aged (2-month-old, n=4) and aged (1- and 1.5-year-old, n=5 and n=3) mice before and after stopping insulin administration revealed no statistical difference between groups (Welch’s test [p[0-45mpa]=0.1029, 0.3321; p[4.5-7mpa]=0.1748, 0.5007], one-way Anova [p=0.1161; p=0.2681], and Mann Whitney [p=0.1640, 0.4519]). b) Evolution of glycemia in 14 aged mice during 14 months post-ablation (“mpa”). Mice were treated with insulin for 4.5 months; most of them (5/7 in each group) subsequently recovered from diabetes. c-e) Pancreatic islets before (c) and after (d, e) β-cell ablation in 1.5-year-old mice; β-cell mass increases 3.5-fold between 0.5 and 1 mpa, 12-fold at 7 mpa and 32-fold at 14 mpa, in all age groups. 0.3% and 4.4% indicate β-cell mass relative to unablated controls (Supp. Table S1, 2-month-old: n(0.5mpa)=4, n(1mpa)=4, n(7mpa)=4; 1-year-old: n(0.5mpa)=5, n(1mpa)=5, n(7mpa)=5, n(14mpa)=8; 1.5-year-old: n(0.5mpa)=3, n(1mpa)=3, n(7mpa)=3 , n(14mpa)=8). f) β-cell proliferation is very low in aged mice, whether control (1.5%, n=8, 39,790 insulin+-cells scored) or ablated (0.2%, n=6, 938 790 insulin+-cells scored) (Supp. Table S2). g) Proportion of insulin+ cells also containing glucagon after DT is not different between groups (Supp. Table S3; control: n[2-month-old]=3, n[1-year-old]=3, n[1.5-year-old]=3; 0.5mpa: n[2-month-old]=5, n[1-year-old]=5, n[1.5-year-old]=6; 1mpa: n[2-month-old]=4, n[1-year-old]=6, n[1.5-year-old]=4; 7mpa: n[2-month-old]=5, n[1-year-old]=5, n[1.5-year-old]=6; one-way Anova [p=0.6796, 0.4297, 0.9266, 0.2411]); note that 40% of the cells containing insulin at 1 mpa also contained glucagon. The proportion of glucagon+/insulin+ cells remains constant between 0.5 and 7 mpa, while the number of insulin+ cells increases with time (e, Supp. Table S1), suggesting that there is acumulative recruitment of α-cells into insulin production. h) Islet with YFP+/glucagon+/insulin+ cells in 1-year-old Glucagon-rtTA; TetO-Cre; R26-YFP; RIP-DTR mice, 7 mpa; rtTA expression allows the selective irreversible YFP-labeling of adult α-cells upon administration of doxycycline (DOX) before β-cell ablation. i) Proportion of YFP-labeled insulin-expressing cells in DOX-treated mice. 80% of insulin+ cells are YFP+ after 7 mpa, in all age groups (Supp. Table S4; control: n[2-month-old]=3, n[1-year-old]=3, n[1.5-year-old]=3; 1mpa: n[2-month-old]=5, n[1-year-old]=3, n[1.5-year-old]=3; 7mpa: n[2-month-old]=5, n[1-year-old]=5, n[1.5-year-old]=5; one-way Anova [p=0.9417, 0.8910, 0.9641]). j,k) YFP+/glucagon+/insulin+ cells at 7 mpa, following a DOX pulse-labeling at 5.5 months after β-cell loss (Supp. Table S5, control: n[1-year-old]=5, n[1.5-year-old]=5; 7mpa: n[1-year-old]=5, n[1.5-year-old]=5; Welch’s correction [p=0.8272,0.8926], Mann-Whitney [p=0.9444]. On average, 15% of the insulin+ cells found were YFP-labeled, some of which no longer contained glucagon as in (j), lower row. Note the decreased proportion of YFP-labeled insulin+ cells when α-cells are tagged late after ablation (from 80% to 15%; compare (i) and (k)), and the presence of YFP-labeled insulin+/glucagon-negative cells in the latter situation (j), suggesting that bihormonal α-cells slowly but gradually lose glucagon gene activity. Scale bars are 20 μm. Error bars: standard deviation (s.d.).
Extended Data Figure 2. Diabetes recovery in pre-pubertal mice.

a) Evolution of glycemia (“AuC”) between 2.5 and 4 mpa, in pups and adults (see Fig 1b, Welch’s test [p=0.0188]). b) qPCR of insulin2 mRNA after β-cell ablation; insulin2 transcripts are 25-fold more abundant in pups than in adults at 2 mpa (n=3 mice/group, each individual sample was run in triplicate in each reaction, for a total of 3 independent reactions); built-in Welch’s test [p=0.0134, 0.0049], error bars: s.d.). c) Glucose tolerance tests (IPGTT) for DT-treated (4.5 mpa, n=4) and age-matched controls (n=4); note the fold-increase between glucose injection and the glycemic peak during IPGTT for each animal, and fold-decrease between glycemic peak and T120 (two-tailed unpaired t-test, [p(I)=0.5836, p(II)=0.4937]). d) Plasma insulin at T0, T15 and T30 during the IPGTT (control: n=4; DT: n=4; two-tailed paired t-test [p=0.0008]). e) Insulin tolerance tests (ITT) performed 1,5 year after β-cell ablation at 2 weeks of age (controls: n=7; DT: n=10). f) 4.5 months after β-cell ablation (at 2 weeks), 3 mice became normoglycemic and received a second treatment with DT. Ablation of regenerated insulin+-cells in recovered mice leads to the appearance of glucagon+/insulin+ cells, corresponding to the type of “α-cell-dependent” regeneration observed in adults (31% of insulin+ cells also contained glucagon, Supp. Table S8). Arrow: glucagon+/insulin+ bihormonal cell; error bars: s.e.m. g) β-cell proliferation is very low in regenerating pups (Supp. Table S9, control: n[1-mo-old]=3, 6,006 insulin+-cells scored, n[2-mo-old]=3, 6,358 insulin+-cells scored; DT: n[0.5mpa]=5, 412 insulin+-cells scored; n[1.5mpa]=3, 675 insulin+-cells scored; Welch’s test [p=0.1197, p=0.0688], error bars: s.e.m.). h) Islet cell proliferation is increased (3.5-fold; Ki67+ cells) in islets of DT-treated pups at 0.5 mpa (control: n(1-month-old)=3, 95 islets scored; n(1.5-month-old)=3, 94 islets scored; n(2-month-old)=3, 90 islets scored; n(2.5-month-old)=3, 89 islets scored; n(3-month-old ctrl)=3, 91 islets scored; n(3.5-month-old)=3, 93islets scored; n(18.5-month-old)=3, 83 islets scored; 19(19-month-old ctrl)=3, 83 islets scored; n(19.5-month-old)=3, 88 islets scored; DT(2-week-old): n(0.5mpa)=6, 333 islets scored; n(1mpa)=3, 91 islets scored; n(1.5mpa)=3, 90 islets scored; DT(2-month-old): n(0.5mpa)=3, 76 islets scored; n(1mpa)=3, 77 islets scored; n(1.5mpa)=3, 81 islets scored; DT(1.5-year-old): n(0.5mpa)=3, 74 islets scored; n(1mpa)=3, 81 islets scored; n(1.5mpa)=3, 77 islets scored; error bars: s.d. Welch’s test, one-way Anova [p<0.001] Mann-Whitney [p=0.0238]). i) Ki67+ cells are hormone, chromogranin A-negative; lineage-traced α- and DT-spared β-cells are Ki67-negative. Scale bars: 20 μm.
Extended Data Figure 3. δ-cell labeling and tracing in transgenic mice.
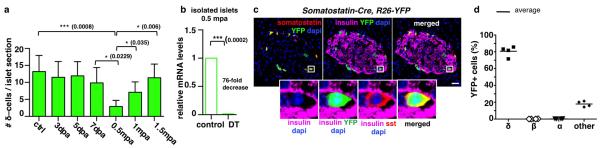
a) The number of somatostatin+ cells transiently decreases by 80% during the 2nd week after ablation (n[control]=255 islets, 7 mice; n[3dpa]=240 islets, 5 mice; n[5dpa]=228 islets, 5 mice; n[7dpa]=251 islets, 5 mice; n[0.5mpa]=267 islets, 6 mice; n[1mpa]=266 islets, 5 mice; n[1.5mpa]=206 islets, 5 mice; error bars: s.d.; Welch’s test [p=0.0008, 0.0229, 0.006, 0.035], one-way Anova [p<0.0001], Mann-Whitney [p=0.0043]). b) Relative somatostatin gene expression sharply decreases 2 weeks after β-cell ablation in 2-week-old mice (n=3 mice/group, each individual sample of each experimental group was run in triplicate, in 3 independent reactions); built-in Welch’s test [p=0.0002], error bars: s.d.). c) Somatostatin-Cre; R26-YFP mice. Cre activity efficiently and specifically occurs in δ-cells (box: enlarged cell). Scale bar: 20 μm. d) Quantitative values of reporter gene expression in islet cells (n=4, 1,263 YFP+-cells scored).
Extended Data Figure 4. δ-cells dedifferentiate, proliferate and reprogram into insulin production after extreme β-cell loss in juvenile mice.

Observed and expected numbers of somatostatin+ and insulin+ cells per islet section, before and after β-cell ablation. Cells scored after 6 weeks (Extended Data Fig. 3a) correspond (χ2 test) with estimates made assuming that dedifferentiated proliferating δ-cells yield 2 types of progeny (as deduced from Fig. 2c,e). Dashed arrows, phenotypic stability; plain arrows, dynamic behavior (dedifferentiation and replication).
Extended Data Figure 5. Regeneration in streptozotocin-treated pups and DT-treated adults.

a) Immunofluorescence showing YFP-labeled insulin+ cells at 1.5 month following streptozotocin (STZ)-induced ablation of β-cells in 2-week-old mice. Arrows: YFP+/insulin+ cells; arrowhead: YFP+/somatostatin+ cell; asterisks: escaping β-cells. b) Number of remaining β-cells per islet section at 2 weeks after streptozotocin or DT treatment in pups, reflecting difference in ablation efficiency of the 2 methods (Supp. Table S18)(n[STZ]=87 islets, 3 mice; n[DT]= 361 islets, 4 mice; Welch’s test [inter-islet p<0.0001; inter-individual p=0.0109], Mann-Whitney [p<0.001]). c) The number of YFP+/insulin+ cells per islet section at 1.5 mpa is not significantly different between the two β-cell ablation methods (Supp. Table S19)(n[STZ]=88 islets, 3 mice; n[DT]=193 islets, 7 mice; Welch’s test [p=0.4786]). d) δ-cell numbers per islet section in controls (n=3, 174 islets scored), 0.5 mpa (n=4, 140 islets scored) and 1 mpa (n=3, 86 islets scored) (unpaired t-test, two-tailed, [p=0.6386; p=0.5406]). e) Immunofluorescence for YFP and Ki67 2 weeks (0.5 mpa) after DT, in Somatostatin-Cre; R26-YFP; RIP-DTR mice. f) Experimental design for δ-cell tracing in β-cell-ablated Somatostatin-Cre; R26-YFP; RIP-DTR mice at 2 months of age, and immunofluorescence for somatostatin, YFP and insulin at 1.5 mpa. Arrow: YFP+/insulin+/somatostatin− cell. g) At 1.5 mpa, 17% of insulin+ cells coexpress YFP vs. almost 100% in ablated prepubescent mice (control: n=4; DT: n=8; unpaired t-test, two-tailed [p=0.0462]). h) At 1.5 mpa, 98% of the YFP+ cells are somatostatin+, and 1% are insulin+ cells (vs. 44% in mice ablated before puberty; n=8, unpaired t-test, two-tailed). Scale bars: 20 μm. Error bars: s.d.
Extended Data Figure 6. δ-to-β cell conversion after β-cell ablation is maintained in young islets ablated underneath the kidney capsule of adult hosts.

a) Islet transplantation design: 400-600 islets isolated from 2-week-old Somatostatin-Cre; R26-YFP; RIP-DTR transgenics were transferred under the kidney capsule of 2-month-old immunodeficient (SCID) mice (n=3). b) Experimental design: after one week of engraftment, adult host mice were DT-treated and left to regenerate for 6 weeks. c) δ- to-β conversion was observed in β-cell-ablated engrafted islets, like in the pancreas of juvenile mice. Scale bars: 20 μm
Extended Data Figure 7. Characterization of δ-cell-derived regenerated insulin+ cells.

Once differentiated from δ-cells (YFP+), the newly formed β-cells re-enter the cell cycle (Ki67+ cells). Two waves of massive replication occur, at 3 and 4 months after injury, respectively (Supp. Table S23). b) qPCR for β-cell-specific genes using RNA extracted from islets isolated from control and DT-treated mice, either 2 weeks or 4 months following DT administration (“0.5 mpa” and “4 mpa”). Note that after an initial extreme downregulation of all the β-cell-specific markers explored, their levels significantly recover after 4 months, which correlates with the observed robust regeneration and diabetes recovery. Values represent the ratio between each regeneration time-point and its age-matched control. c) Experimental design. d) qPCR comparison between regenerated cherry+/insulin+ cells isolated from mice 4 months after β-cell ablation, and cherry+ β-cells obtained from age-matched controls (4.5-month-old). All markers tested are expressed at identical levels in both groups; non-β-cell markers are expressed at extremely reduced levels (CT ranging from 28 to 31), showing the same degree of purity in both types of cell preparations. e, f) Interestingly, in contrast to bona fide β-cells isolated from 4.5-month-old controls, regenerated insulin+ cells have lower levels of cyclin-dependent kinase inhibitors, FoxO1 and Smad3. This correlates with their increased proliferative capacity at this specific time-point. Scale bars: 20 μm; qPCRs: n=3 mice/group, each individual sample of each experimental group was run in triplicate, in 3 independent reactions); built-in Welch’s test; error bars: s.d.
Extended Data Figure 8. Ngn3 activation is required for insulin expression in dedifferentiated δ-cells.

a) qPCR for Ngn3 mRNA after β-cell ablation reveals a transitory 5-fold upregulation of Ngn3 transcripts 6 weeks after β-cell ablation when β-cell ablation is performed before puberty, but not in adult mice. (controls: n[1-month-old]=3, n[1.5-month-old]=3, n[2-month-old]=6, n[2.5-month-old]=3, n[2.5-month-old]=3, n[3-month-old]=3, n[3.5-month-old]=3, n[4-month-old]=3; DT(2-week-old): n[0.5mpa]=3, n[1mpa]=3, n[1.5mpa]=6, n[2mpa]=3; DT(2-month-old): n[0.5mpa]=3, n[1mpa]=3, n[1.5mpa]=3, n[2mpa]=3; each individual sample (mouse) was run in triplicate, in each one of 3 independent reactions; built-in Welch’s test [p=0.0112, 0.0178]). b) Ngn3 transcriptional activity can be monitored in Ngn3-YFP knock-add-on mice because Ngn3 promoter activity results in YFP expression. In non-ablated age-matched control pups, or in ablated adults, no islet YFP+ cells were found (not shown), yet when β-cells are ablated at 2 weeks of age, 86% of insulin+ cells also express YFP+ at 1.5 mpa (control: n=3, 6,358 insulin+-cells scored DT: n=3, 675 insulin+-cells scored; Welch’s test [p=0.0010]); c) At 1.5 mpa, 81% of YFP+ cells co-express insulin, but no glucagon, somatostatin or PP (not shown). Two weeks later, YFP+ cells are almost absent, reflecting the downregulation of Ngn3 expression reported in a), and suggesting that insulin+ cells originate from cells transiently activating Ngn3 expression after ablation (control: n[1-month-old]=3; n[1.5-month-old]=3; n[2-month-old]=3; n[2.5-month-old]=3; absent YFP+-cells in all control conditions; DT: n[0.5mpa]=3, 31 YFP+-cells; n[1mpa]=3, 123 YFP+-cells; n[1.5mpa]=3, 729 YFP+-cells; n[2mpa]=3; 47 YFP+-cells; Welch’s test and Anova [p<0.0001]). d) Irreversible lineage-tracing of Ngn3-expressing cells at 1 and 1.5 mpa upon tamoxifen (TAM) administration in Ngn3-CreERT; R26-YFP; RIP-DTR mice; immunofluorescence analyses reveal that in absence of β-cell ablation, there is no YFP induction (controls). In ablated mice, nearly all insulin+ cells are YFP+ with time (arrows). At early time-points (1 mpa), YFP+/hormone-negative cells are found: these are likely differentiating cells before insulin expression. e, f) In β-cell-ablated Ngn3-CreERT; R26-YFP; RIP-DTR pups, 91% of insulin+ cells coexpress YFP+ (control: n=3, 3,472 insulin+-cells scored, DT: n=3, 489 insulin+-cells scored) (e) and inversely, 93% of the YFP+ cells are insulin+ (f) (control: n=3; absent YFP+-cells in all control conditions, DT: n=3, 478 YFP+-cells scored). g) Experimental design to block Ngn3 upregulation in β-cell-ablated prepubescent mice, by administrating DOX to mice bearing 5 mutant alleles: Ngn3-tTA+/+; TRE-Ngn3+/+; RIP-DTR. In these mice the Ngn3 coding region is replaced by a DOX-sensitive transactivator gene (tTA); the endocrine pancreas develops normally because Ngn3 expression is allowed in absence of DOX by the binding of tTA to the promoter of TRE-Ngn3 transgene. Pups were given DT at 2 weeks of age and then DOX 2 weeks later, to block Ngn3 upregulation. They were euthanized when Ngn3 peaks after ablation (2-month-old).h) Islets from non-ablated (“no DT”) and ablated (“DT”) mice, exposed (Ngn3 inhibition) or not (normal Ngn3 expression) to DOX treatment from 4 weeks of age. β-cell regeneration is efficient in absence of DOX (as previously shown), but decreases after Ngn3 blockade, resulting in the appearance of glucagon/insulin bihormonal cells. i) Sharply decreased regeneration by blocking Ngn3 expression in DOX-treated mice reveals the requirement of Ngn3 for efficient β-cell regeneration in pups (DT: n=266 islets scored, 3 mice; DT+DOX: n=167, 4 mice; Welch’s test [inter-islet p<0.0001; inter-animal p=0.0352], Mann-Whitney [p<0.0001]). j) glucagon+/insulin+ bihormonal cells appear in DOX-treated β-cell-ablated pups (Ngn3 inhibition), suggesting a switch to an “adult-like”, less efficient, mechanism of regeneration (control+DOX: n=3, 9233 insulin+-cells scored; DT: n=3 1385 insulin+-cells scored; DT+DOX: n=4, 141 insulin+-cells scored; Welch’s test [p=0.0081], Anova [p<0.0001]). k) Combined double lineage tracing of δ-cells (Tomato+) and Ngn3-expressing cells (YFP+) show by immunofluorescence that nearly all insulin+ cells express both reporters, but no somatostatin (arrows). Somatostatin+ cells (arrowheads) are YFP- and insulin-negative. Scale bars: 20 μm. Error bars: s.d.
Extended Data Figure 9. FoxO1 regulatory network.

a) Cartoon depicting the FoxO1 network involved in the regulation of cell cycle progression and cellular senescence: FoxO1 arrests the cell cycle by repressing activators (cyclinD1, cyclinD2) and inducing inhibitors (cdkn1a/p21, cdkn1b/p27, cdkn2b/p15Ink4b, cdkn1c/p57) [PMID: 10102273; PMID: 17873901]. cdkn1a/p21 and cdkn2b/p15Ink4b activation, a sign of cellular senescence [PMID: 17667954], is regulated by FoxO1 through direct interaction with Skp2 protein. In turn, Skp2 blocks FoxO1 and, together with CKS1b, CDK1 and CDK2, triggers the direct degradation of cdkn1a/p21 and cdkn1b/p27, thus promoting proliferation [PMID: 15668399]. FoxO proteins are inhibited mainly through PI3K/AKT-mediated phosphorylation [PMID: 10102273, PMID: 12621150, PMID: 21708191, PMID: 10217147, PMID: 17604717]: PDK1, the master kinase of the pathway, stimulates cell proliferation and survival by directly activating AKT, which phosphorylates (inhibits) the FoxOs [PMID: 10698680, PMID: 19635472]. PI3K/AKT/FoxO1 circuit requires active TGFβ/SMAD signaling [PMID: 24238962, PMID: 15084259] in order to co-regulate cdkn1a/p21-dependent cell senescence. Active TGFβ signaling downregulates the BMP pathway downstream effectors ID1 and ID2, known to promote dedifferentiation and proliferation during embryogenesis and cancer progression, probably through cdkn2b/p15Ink4b regulation [PMID: 11840321, PMID: 16034366]. b) β-cell ablation in adults triggers FoxO1 upregulation and the subsequent cell cycle arrest in δ-cells.
Extended Data Figure 10. δ-cell dedifferentiation in adult mice upon transient FoxO1 inhibition.

a-d) The 1 week FoxO1 inhibition with the compound AS1842856 in control unablated adult mice (a) results in dedifferentiation of one-fourth of the δ-cell population (b; Supp. Table S30, treated: n=3, 1,347 YFP+-cells scored; untreated: n=4, 1,224, YFP+-cells scored; error bars: s.d.), without leading to insulin (c; Supp. Table S31, treated: n=3, 3,249 insulin+-cells scored; untreated: n=4, 9,562 insulin+-cells scored; error bars: s.d.; Welch’s test [p=0.1590]) or glucagon (d; Supp. Table S32, treated: n=2, 728 YFP+-cells scored; error bars: s.e.m.) expression. e) One month following FoxO1 transient inhibition in β-cell-ablated adults, dedifferentiated δ-cells do not express glucagon (Supp. Table S36, treated: n=2, 986 YFP+-cells scored; error bars: s.e.m.). f) Transient FoxO1 inhibition long-time (1 month) after β-cell ablation also leads to the appearance of lineage-traced dedifferentiated δ-cells that express insulin (Supp. Table S37-39, treated: n=3, 71 islets scored; 300 insulin+-cells scored; 1216 YFP+-cells scored; error bars: s.d.). Scale bars: 20 μm.
Supplementary Material
Acknowledgements
We are grateful to Dominique Belin, Pierre Vassalli, Roland Stein, Alan Cookson, Ariel Ruiz i Altaba, Marcos González Gaitán, Brigitte Galliot and Iván Rodríguez for comments, support and discussions, and to G. Gallardo, O. Fazio, K. Hammad and B. Polat for the technical help. We thank G. Gradwohl for the Ngn3-YFP mice. F.M.G. and F.R. were funded by Wellcome Trust grants WT088357/Z/09/Z and WT084210/Z/07/Z, respectively. Work was funded with grants from the NIH/NIDDK (“Beta Cell Biology Consortium”), JDRF (Juvenile Diabetes Research Foundation) and Swiss National Science Foundation (“NRP63”) to P.L.H.
Footnotes
Author contributions. S.C. conceived and performed the experiments and analyses, and wrote the manuscript. F.M.G. and F.R. generated the Somatostatin-Cre line, and G.G. and J.N.J. the Ngn3-CreERT, Ngn3-tTA and TRE-Ngn3 lines. D.B. characterized the pancreatic expression of the Somatostatin-Cre line and performed the adult analysis. L.G. performed experiments and analyses. V.C. profiled sorted fluorescent adult islet cells. K.F., F.T. performed immunofluorescence microscopy. P.L.H. conceived the experiments and wrote the manuscript.
References
- 1.Matveyenko AV, Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes, obesity & metabolism. 2008;10(Suppl 4):23–31. doi: 10.1111/j.1463-1326.2008.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harbor perspectives in medicine. 2012;2 doi: 10.1101/cshperspect.a007641. doi:10.1101/cshperspect.a007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thorel F, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desgraz R, Herrera PL. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development. 2009;136:3567–3574. doi: 10.1242/dev.039214. doi:10.1242/dev.039214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Masri M, et al. Effect of forkhead box O1 (FOXO1) on beta cell development in the human fetal pancreas. Diabetologia. 2010;53:699–711. doi: 10.1007/s00125-009-1632-0. doi:10.1007/s00125-009-1632-0. [DOI] [PubMed] [Google Scholar]
- 6.Talchai C, Xuan S, Kitamura T, DePinho RA, Accili D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nature genetics. 2012;44:406–412. S401. doi: 10.1038/ng.2215. doi:10.1038/ng.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munoz-Espin D, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. doi: 10.1016/j.cell.2013.10.019. doi:10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 8.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 9.Yokota Y. Id and development. Oncogene. 2001;20:8290–8298. doi: 10.1038/sj.onc.1205090. doi:10.1038/sj.onc.1205090. [DOI] [PubMed] [Google Scholar]
- 10.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nature reviews. Cancer. 2005;5:603–614. doi: 10.1038/nrc1673. doi:10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 11.van der Vos KE, Coffer PJ. FOXO-binding partners: it takes two to tango. Oncogene. 2008;27:2289–2299. doi: 10.1038/onc.2008.22. doi:10.1038/onc.2008.22. [DOI] [PubMed] [Google Scholar]
- 12.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. doi:10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagashima T, et al. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Molecular pharmacology. 2010;78:961–970. doi: 10.1124/mol.110.065714. doi:10.1124/mol.110.065714. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka H, et al. Effects of the novel Foxo1 inhibitor AS1708727 on plasma glucose and triglyceride levels in diabetic db/db mice. European journal of pharmacology. 2010;645:185–191. doi: 10.1016/j.ejphar.2010.07.018. doi:10.1016/j.ejphar.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 15.Morgan TH. Regeneration and Liability to Injury. Science (New York, N.Y. 1901;14:235–248. doi: 10.1126/science.14.346.235. doi:14/346/235 [pii]10.1126/science.14.346.235. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, et al. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. 2011;478:349–355. doi: 10.1038/nature10502. doi:10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karges B, et al. Complete long-term recovery of beta-cell function in autoimmune type 1 diabetes after insulin treatment. Diabetes care. 2004;27:1207–1208. doi: 10.2337/diacare.27.5.1207. [DOI] [PubMed] [Google Scholar]
- 18.Karges B, et al. Immunological mechanisms associated with long-term remission of human type 1 diabetes. Diabetes/metabolism research and reviews. 2006;22:184–189. doi: 10.1002/dmrr.600. doi:10.1002/dmrr.600. [DOI] [PubMed] [Google Scholar]
- 19.Desgraz R, Bonal C, Herrera PL. beta-cell regeneration: the pancreatic intrinsic faculty. Trends in endocrinology and metabolism: TEM. 2011;22:34–43. doi: 10.1016/j.tem.2010.09.004. doi:10.1016/j.tem.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Bramswig NC, et al. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. The Journal of clinical investigation. 2013;123:1275–1284. doi: 10.1172/JCI66514. doi:10.1172/JCI66514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler AE, et al. Marked Expansion of Exocrine and Endocrine Pancreas with Incretin Therapy in Humans with increased Exocrine Pancreas Dysplasia and the potential for Glucagon-producing Neuroendocrine Tumors. Diabetes. 2013 doi: 10.2337/db12-1686. doi:10.2337/db12-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoneda S, et al. Predominance of beta-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. The Journal of clinical endocrinology and metabolism. 2013;98:2053–2061. doi: 10.1210/jc.2012-3832. doi:10.1210/jc.2012-3832. [DOI] [PubMed] [Google Scholar]
- 23.Thorel F, et al. Normal glucagon signaling and beta-cell function after near-total alpha-cell ablation in adult mice. Diabetes. 2011;60:2872–2882. doi: 10.2337/db11-0876. doi:10.2337/db11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.