

Abstract
Rationale
Cholesterol esters (CE), especially cholesterol oleate, generated by hepatic and intestinal sterol O-acyltransferase 2 (SOAT2) play a critical role in cholesterol homeostasis. However, it is unknown if the contribution of intestine-derived CE from SOAT2 would have similar effects in promoting atherosclerosis progression as for liver-derived CE.
Objective
To test whether, in low-density lipoprotein receptor null (LDLr−/−) mice, the conditional knockout of intestinal SOAT2 (SOAT2SI-/SI-) or hepatic SOAT2 (SOAT2L-/L-) would equally limit atherosclerosis development when compared to the global deletion of SOAT2 (SOAT2−/−).
Methods and Results
SOAT2 conditional knockout mice were bred with LDLr−/− mice creating LDLr−/− mice with each of the specific SOAT2 gene deletions. All mice then were fed an atherogenic diet for 16 weeks. SOAT2SI-/SI-LDLr−/− and SOAT2−/− LDLr−/− mice had significantly lower levels of intestinal cholesterol absorption, more fecal sterol excretion, and lower biliary cholesterol levels. Analysis of plasma LDL showed that all mice with SOAT2 gene deletions had LDL CE with reduced percentages of cholesterol palmitate and cholesterol oleate. Each of the LDLr−/− mice with SOAT2 gene deletions had lower accumulations of total cholesterol and CE in the liver compared with control mice. Finally, aortic atherosclerosis development was significantly lower in all mice with global or tissue-restricted SOAT2 gene deletions. Nevertheless, SOAT2−/− LDLr−/− and SOAT2L-/L-LDLr−/− mice had less aortic CE accumulation and smaller aortic lesions than SOAT2SI-/SI-LDLr−/− mice.
Conclusions
SOAT2-derived CE from both the intestine and liver significantly contribute to the development of atherosclerosis, although the CE from the hepatic enzyme appeared to promote more atherosclerosis development.
Keywords: SOAT2, cholesterol esters, lipids and lipoproteins, atherosclerosis, lipid and lipoprotein metabolism, molecular biology, physiology/pathophysiology
INTRODUCTION
Steroyl O-acyltransferases (SOATs), are microsomal proteins responsible for catalyzing intracellular cholesterol ester (CE) synthesis using free sterol and acyl CoA as substrates1. SOAT subtype1 (SOAT1) is expressed in a variety of tissues while SOAT subtype2 (SOAT2) is expressed preferentially in enterocytes of the intestine and hepatocytes of the liver2, where one of its roles is to generate CE for packaging into chylomicrons and very low-density lipoproteins (VLDL), respectively3, 4. Cholesterol esters, especially cholesterol oleate, are major components of atherogenic lipoproteins, appearing to play a critical role in the pathogenesis of atherosclerosis5, 6.
Previous studies have shown that SOAT2 deficient animals had delayed atherosclerosis development. Global deletion of SOAT2 in low-density lipoprotein receptor null (LDLr−/−) or apolipoprotein E null (apoE−/−) mice resulted in highly significant reductions of aortic atherosclerosis lesion area with a reduction in CE deposited in atherosclerotic plaques7, 8. These observed phenotypes are attributed to the combined effects of the decreased capability of cholesterol esterification and secretion by both the liver and the intestine, leading to a less atherogenic lipoprotein profile. However, some evidence has suggested that hepatic SOAT2 expression and activity in humans are different from that in rodents and nonhuman primates and might be expressed at a relatively lower level9–11. Relevant to the human condition, it is uncertain whether blocking cholesterol absorption alone through inhibiting intestinal SOAT2 would be sufficient to delay atherosclerosis progression.
Through LoxP-Cre recombinase technologies, our group previously generated two novel SOAT2 conditional knockout (KO) mice, the liver-specific (SOAT2L-/L-) and the intestine-specific (SOAT2SI-/SI-) knockouts12. In response to increased dietary cholesterol, plasma and hepatic lipid changes of SOAT2SI-/SI- mice, indicated by low plasma VLDL-cholesterol, low hepatic total cholesterol and CE concentrations12, were essentially the same as occurred in SOAT2L-/L- mice. The data suggest that elimination of intestinal cholesterol esterification with subsequent absorption was sufficient to alleviate many of the dietary cholesterol induced changes in cholesterol transport in the circulation and tissues.
Although the blunted cholesterol absorption that occurred via intestinal SOAT2 inhibition was hypocholesterolemic, the specific contribution made by intestine-derived CE to atherosclerosis development is still uncertain. LDLr−/− mice are known to develop a spectrum of cholesterol enriched apoB-containing lipoproteins most of which apparently promote progression of atherosclerosis on high fat/cholesterol-enriched diet13. The mass and distribution of plasma lipids of LDLr−/− mice are marked and distinct from the relatively mild elevation of lipids following high cholesterol intake by wild type mice. It is unknown whether, after cholesterol feeding, SOAT2SI-/SI-LDLr−/− mice would be protected from atherosclerosis progression to the same extent as that of LDLr−/− mice with gene deletions of either hepatic SOAT2 or whole body SOAT214.
We believe it is critically important to understand the tissue-specific roles for SOAT2 in atherosclerosis development if effective therapeutic strategies targeting SOAT2-driven cholesterol esterification are to be designed. This prompted us to cross tissue-specific SOAT2 knockouts with LDLr−/− mice. Here we tested whether conditional knockout of hepatic SOAT2 versus intestinal SOAT2 similarly limit atherosclerosis development as compared to the global deletion of SOAT2.
METHODS
Mice, diet and study design
Generation of tissue-specific SOAT2 knockouts was described in detail previously12. Floxed mice (LoxP sites flanked exons 11 through 13 of the SOAT2 gene on chromosome 15) are designated as SOAT2fl/fl. After introducing Cre recombinase driven either by the albumin or villin promoter, SOAT2 was specifically deleted in the liver (SOAT2fl/flAlbCre+) or small intestine (SOAT2fl/flVilCre+), which are designated as SOAT2L-/L- or SOAT2SI-/SI-. SOAT2fl/fl mice were maintained on a mixed background (strains of C57BL/6, 129S6, 129SvEv). To create the conditional knockouts on LDLr−/− background, SOAT2L-/L- and SOAT2SI-/SI- mice were bred with SOAT2+/+LDLr−/− (strain of C57BL/6) or SOAT2−/− LDLr−/− (strain of C57BL/6). Genotypes of litters were screened by PCR. Animals used in the study were from breeders set up as follows: heterozygotes of SOAT2fl/+LDLr−/− with or without AlbCre (or VilCre), SOAT2fl/-LDLr−/− with or without AlbCre (or VilCre). Both male and female mice were included in the study. At the age of 8 to 9 weeks, mice were fed a semi-synthetic diet containing fat (as lard) at 20% energy, protein % energy, carbohydrate 63% energy, and cholesterol 0.1% (wt/wt) for a total of 16 weeks. Blood was drawn at baseline, 8 weeks, and 16 weeks from mice that were fasted at 9AM with blood collection from the superficial temporal vein starting at 1 PM. After consuming the diet for eight weeks, mice were housed individually on wire bottom cages for 3 days to allow fecal collections for measurement of cholesterol absorption and fecal sterol loss16. For necropsy at the end of the study, mice were fasted from 9 AM with terminations starting at 1 PM. All mice used in the studies were housed in a pathogen-free barrier facility at Wake Forest University School of Medicine approved by the American Association for Accreditation of Laboratory Animal Care. The Institutional Animal Care and Use Committee approved all protocols for use of animals prior to execution of the studies.
Plasma lipid and lipoprotein analysis
Total plasma cholesterol (TPC) and triglyceride (TG) concentrations were measured using colorimetric enzymatic assays as previously described7, 15, 16. For lipoprotein measurement, an aliquot of plasma containing about twenty μg of total plasma cholesterol (TPC) was diluted in phosphate buffered saline (PBS) into a final volume of 400 μL. After centrifugation to remove any protein precipitates, samples were injected onto a Superose 6 HR 10/30 (Amersham Pharmacia) chromatography column, which was subsequently run at 0.4 mL/min. The signal was integrated using Chrom Perfect Spirit Software (Justice Laboratory Software). VLDL-, LDL-, and HDL-cholesterol were determined by multiplying the TPC concentration by the cholesterol percentage within the elution region for each lipoprotein class.
LDL isolation and cholesterol ester analysis
For isolated LDL, a narrow center of the LDL peak window (26 to 34 min) was collected for each sample. Total lipid of this LDL fraction was extracted with 6 mL chloroform/methanol (2:1) and phases were split with 0.7 mL H2O. After vortexing and centrifugation, the chloroform phase was recovered and dried down under nitrogen and then dissolved in 1 mL chloroform/methanol (1:1) and stored at −20°C until analysis. Fifty μL of LDL lipid extract (~15 μg/μL per sample) was diluted in 500 μL methanol containing 500 pg/μL of cholesterol heptadecanoate (Nu-Chek Prep) as an internal standard and 1 ng/μL of sodium formate. After standing for 30 min, the solution was analyzed by direct infusion into a Waters Quattro II tandem mass spectrometer operated at a flow rate of 10 μL/min in the positive ion mode. Cholesterol ester species were quantified with a response curve against 0.78 μM internal standard as described elsewhere17.
Cholesterol absorption measurement by a fecal dual-isotope technique
The method was essentially as reported earlier by Temel et al16. After eight-weeks of diet consumption, all mice were gavaged with 50 μL soybean oil containing 0.055 μCi of [14C]-cholesterol (American Radiolabeled Chemicals) and 0.135 μCi of β-[3H]-sitosterol (New England Nuclear) and were subsequently individually housed in wired-bottom cages with free access to food and water. Feces were collected for a total of three days and dried overnight in a vacuum oven at 70°C. Dried fecal pellets were crushed into fine powder and approximately 50 mg of fecal samples, along with 100μg 5-alpha cholestane (as an internal standard), were saponified and extracted with hexane. For fractional cholesterol absorption, an aliquot of hexane phase was dried under nitrogen. The radioactivity was quantified in a liquid scintillation spectrometer (Beckman Coulter LS 6500) and percentage of cholesterol absorption was calculated as . Another aliquot of hexane phase was transferred to a GLC vial and fecal neutral sterol (FNS) was quantified by gas-liquid chromatography (GLC). The mass of FNS represented the sum of cholesterol and coprostanol, and results were expressed as mg sterol per day per 100g body weight.
Hepatic and biliary lipid analysis
Extraction of liver and biliary lipids for enzymatic quantification of total triglyceride, cholesterol esters, free cholesterol, phospholipids and bile acids were performed as previously described7, 16. Briefly, for analysis of the liver lipids about 50 to 100 mg of liver sample was minced and thawed in a glass tube. Lipids were extracted in chloroform/methanol (2:1) at room temperature overnight. Lipid extract was dried down under nitrogen and redissolved in a measured volume of 2:1 chloroform/methanol. Following addition of 0.05% H2SO4 and centrifugation, the aqueous upper phase was aspirated and discarded, and an aliquot of the bottom phase was transferred and dried down; 1% Triton X-100 in chloroform was then added, and the solvent was evaporated. Deionized water was then added to each tube and vortexed until the solution was clear. Lipids were quantified using available enzymatic assay kits. For analysis of biliary lipid concentrations, a measured volume (5 to 10 uL) of bile was placed into a glass tube and the lipids were extracted in chloroform/methanol (2:1). An aliquot of chloroform phase was used for quantification of biliary cholesterol and phospholipids, similarly as described for liver lipid measurement. An aliquot of the aqueous phase of the extraction was analyzed for total bile acid content using hydroxysteroid dehydrogenase-based enzymatic assay.
Quantification (En Face) of aortic lesion
At the time of necropsy, the entire length of aorta was excised and submersion fixed in 10% formalin for at least 24 hrs. Adherent adipose and connective tissue were removed before the measurement. The aortas were opened longitudinally along the ventral midline and then pinned flat. Images of the aortas were captured using a Sony digital camera (Model DXC-S500) and evaluated for lesion extent using Scion Image software (Version 1.62). Percent of surface area occupied by lesion was calculated and expressed as percentage of surface area with visible atherosclerotic lesion7.
Quantification of cholesterol in the aortic plaques
Lipids of the fixed entire aorta were then extracted in chloroform/methanol (2:1) overnight together with addition of 20 μg of 5-alpha cholestane as an internal standard7. Aortic protein was then washed twice with chloroform/methanol (2:1) and pooled solvent with all lipids was evaporated under nitrogen. Dried lipids were dissolved in 250μL hexane and 1 μL of hexane phase was injected onto ZB-50 GLC column to measure free cholesterol (FC). After the FC quantification, the remaining samples were saponified and total cholesterol (TC) of aortas was also determined by GLC. Aortic CE was calculated using the equation .
Statistical analyses
All graphs were plotted using GraphPad Prism (version 5.05) or Microsoft Excel for Mac (version 14.4.3). Data were analyzed by one-way or two-way ANOVA with Tukey post-hoc test using GraphPad Prism (version 5.05) or JMP statistical software (version 5.0.1.2). Relationships between lipid and atherosclerosis parameters were analyzed by regression analysis with associations shown with least-square best-fit regression lines. Regression coefficients are given in the figures. Statistically significant differences were considered at p<0.05.
RESULTS
Intestinal SOAT2, but not liver SOAT2, is a critical determinant of cholesterol absorption
SOAT2 wild type control mice (SOAT2+/+LDLr−/−) and SOAT2 floxed mice (SOAT2fl/flLDLr−/−) had similar levels of cholesterol absorption (figure 1A). In agreement with our previous findings13, SOAT2 total body knockout mice (SOAT2−/− LDLr−/−) and intestinal SOAT2 knockout mice (SOAT2SI-/SI-LDLr−/−) had significantly decreased levels of cholesterol absorption, compared with SOAT2+/+LDLr−/− control mice with their level of decrease being 33% and 35%, respectively (figure 1A). Cholesterol absorption in liver-specific SOAT2 knockout mice (SOAT2L-/L-LDLr−/−) did not differ significantly from that of SOAT2+/+LDLr−/− mice (figure 1A). SOAT2SI-/SI-LDLr−/− mice had two-fold more fecal neutral sterol loss, when compared with control mice (figure 1B). Total body SOAT2−/− LDLr−/− mice also showed significantly more fecal neutral sterol excretion (2.9 fold), compared with SOAT2+/+LDLr−/− control mice (figure 1B). The amount of fecal neutral sterol loss of SOAT2L-/L-LDLr−/− mice was similar to that of floxed control mice, and this outcome was the same as for fecal neutral sterol excretion in the LDLr−/− mice with SOAT2 intact in the enterocytes of the small intestine (figure 1B).
Figure 1. Whole body SOAT2 knockout and intestine-specific, but not liver-specific SOAT2 knockout reduces cholesterol absorption.

(A) Fractional cholesterol absorption was determined by the dual isotope method. After consuming diet for a total of 8 weeks, each mouse was gavaged with 50 μL soybean oil containing 0.055 μCi of [14C]-cholesterol and 0.135 μCi of β-[3H]-sitosterol. Mice were then housed individually in wired-bottom cages for three days. Fecal samples were collected and fractional absorption was calculated as . (B) About 50 mg of grounded feces were saponified for an hour. Lipids were extracted with hexane. Fecal neutral sterol loss was quantified by GLC using 103 μg of 5-alpha cholestane as an internal standard. +/+: SOAT2+/+LDLr−/−; fl/fl: SOAT2fl/flLDLr−/−; −/−: SOAT2−/− LDLr−/−; L-/L-: SOAT2L-/L-LDLr−/−; SI-/SI-: SOAT2SI-/SI-LDLr−/−. Data represent the mean ± SEM from 14 to 16 mice per genotype. Bars not sharing common letters differ with P < 0.05.
Hepatic SOAT2, but not Intestinal SOAT2, determines LDL-cholesterol levels in LDLr−/− mice
Total plasma cholesterol (TPC) concentrations were similar among the various genotypes at the beginning of the study (week 0, Table 1). LDLr−/− mice with SOAT2 intact had significant time-dependent increases of TPC after 8 and 16 weeks of consumption of the cholesterol enriched diet (Table 1). SOAT2fl/flLDLr−/− and SOAT2SI-/SI-LDLr−/− mice had similar levels of TPC as the LDLr−/− mice with SOAT2 intact at each time point. By contrast, at the end of weeks 8 and 16, TPC concentrations were significantly lower in LDLr−/− mice with SOAT2−/− and SOAT2L-/L- genotypes (Table 1).
Table 1.
Plasma total cholesterol and triglycerides
| Lipid | Time | +/+ | fl/fl | −/− | L-/L- | SI-/SI- |
|---|---|---|---|---|---|---|
| TC | wk 0 | 214 ± 7a | 206 ± 9a | 119 ± 4a | 153 ± 8a | 163 ± 7a |
| wk 8 | 633 ± 15cd | 604 ± 14c | 333 ± 20b | 413 ± 23b | 534 ± 29c | |
| wk16 | 741 ± 31e | 689 ± 25ce | 443 ± 26b | 478 ± 31b | 685 ± 44de | |
| TG | wk 0 | 84 ± 5a | 67 ± 5a | 87 ± 3a | 85 ± 5a | 86 ± 7a |
| wk 8 | 160 ± 8b | 133 ± 7b | 216 ± 25b | 204 ± 13b | 303 ± 40c | |
| wk16 | 213 ± 21b | 155 ± 16b | 412 ± 39c | 360 ± 33c | 532 ± 59d |
+/+: SOAT2 LDLr; −/−: SOAT2 LDLr; fl/fl: SOAT2 LDLr; L-/L-: SOAT2 LDLr; SI-/SI-: SOAT2SI-/SI-LDLr−/−. Data represent the mean (mg/dL) ± SEM of 19 to 20 mice per genotype. TC: total cholesterol; TG: triglycerides. Two-way ANOVA and Tukey post-hoc analyses were applied to TC and separately, TG, for all groups at all times to determine the difference in genotype, time and genotype by time interaction for either lipid. Superscripts within each lipid class (TC or TG) not sharing common letters differ with P < 0.05 with all data included in the comparison.
All groups of LDLr−/− mice had similar levels of plasma TG at week 0. Plasma TG of SOAT2+/+LDLr−/− and SOAT2fl/flLDLr−/− mice increased by 2 to 2.5 fold after 16-weeks of being fed the elevated diet cholesterol level (Table 1). All mice with a SOAT2 gene deletion had time-dependent elevations of plasma TG. Relative to the level at week 0, plasma TG was significantly increased by 4.6 fold, 4 fold and 6 fold in SOAT2−/− LDLr−/−, SOAT2L-/L-LDLr−/− and SOAT2SI-/SI-LDLr−/− mice, respectively, at the end of week16 (Table 1). The increased level of plasma TG that occurred together with cholesterol feeding was most pronounced in SOAT2SI-/SI-LDLr−/− mice but the explanation for this outcome is unknown.
To examine possible genetic responses that could be related to the hypertriglyceridemia in the SI-/SI- mice, we examined gene expression levels for several genes in liver and intestine of animals fed for 16 weeks. The data are in online supplement Table 1 and show that the only significant differences observed were in SCD-1 which was elevated over 10 fold in the intestine of whole body and intestine specific SOAT2 gene deleted animals. Significant differences were not seen in the livers of these animals. Understanding of relationship between intestinal SCD-1 and hypertriglyceridemia in mice with intestinal SOAT2 deficiency will require further study.
SOAT2 knockout mice have lower plasma VLDL and LDL cholesterol concentrations
Plasma lipoprotein cholesterol concentrations were measured in all groups of mice after 16 weeks of consumption of the cholesterol-enriched diet (figure 2A). Compared with LDLr−/− mice with SOAT2 intact, all LDLr−/− mice with SOAT2 gene deletions had significantly lower levels of plasma VLDL-cholesterol although this measurement in the mice with the intestine specific SOAT2 deletion was as high as that in the floxed mice. SOAT2−/− LDLr−/− and SOAT2L-/L-LDLr−/− mice but not SOAT2SI-/SI-LDLr−/− mice had significantly lower LDL-cholesterol concentrations, compared with the SOAT2 intact mice (figure 2A). HDL-cholesterol was not different among genotypes. Representative HPLC chromatographic profiles of lipoprotein cholesterol from animals representing each study group are shown in online supplement Figure I.
Figure 2. SOAT2 knockouts affect cholesterol distribution and fatty acid composition of cholesterol esters isolated from plasma LDL.

(A) Plasma cholesterol distribution quantified by FPLC. VLDL-C: very low-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol. N = 17 to 18 per group. (B) Percentage of saturated, monounsaturated, polyunsaturated fatty acids to total fatty acids found in LDL-CE. Plasma LDL fraction was identified by FPLC and corresponding elution was collected. Total lipids were extracted with chloroform/methanol. CE species were quantified by LC-MS. Vales are expressed as percentage of the mass of CE containing saturated, monounsaturated or polyunsaturated fatty acids to total mass of CE recovered in LDL. Data represent the mean ± SEM from 9 to 10 samples per group. Statistical analyses were performed in each subclass of lipoproteins or each fatty acid species. Bars not sharing common letters differ with P < 0.05.
Cholesterol ester compositions were also measured in plasma LDL of all LDLr−/− mouse groups of the study (figure 2B and Table 2). Groups with SOAT2 gene deletions had significantly lower percentages of saturated and monounsaturated fatty acids and higher percentages of polyunsaturated fatty acids (figure 2B). These differences were most remarkable in mice with no hepatic SOAT2, i.e. SOAT2−/− LDLr−/− and SOAT2L-/L-LDLr−/− mice. Further analysis indicated that the percentages of palmitic acid (C16:0), stearic acid (C18:0), palmitoleic acid (C16:1), and oleic acid (C18:1) all were significantly decreased in LDL cholesterol esters (Table 2) while percentages of linoleic acid (C18:2), arachidonic acid (C20:4), eicosapentaenoic acid (C20:5) and docosahexaenoic acid (C22:6) all were increased in LDL cholesterol esters of mice with SOAT2 gene deletions, as compared with LDL CE in LDLr−/− mice having intact SOAT2 (Table 2). The extent of the decrease in the proportion of monounsaturated CE (and increase in polyunsaturated CE) was intermediate in the LDL of SOAT2SI-/SI- mice.
Table 2.
Percentage fatty acid composition of LDL CE
| Fatty acid species | SOAT2 | ||||
|---|---|---|---|---|---|
| +/+ | fl/fl | −/− | L-/L- | SI-/SI- | |
| C16:0 | 9.1 ± 0.3a | 8.9 ± 0.5a | 3.4 ± 0.1b | 4.9 ± 0.2c | 6.2 ± 0.3c |
| C18:0 | 2.1 ± 0.2a | 1.9 ± 0.3a | 0.3 ± 0.1b | 0.7 ± 0.1b | 0.9 ± 0.2b |
| C16:1 | 12.8 ± 1.0a | 13.3 ± 1.2a | 3.6 ± 0.5b | 5.3 ± 0.5bc | 7.5 ± 0.8c |
| C18:1 | 39.0 ± 3.0a | 37.0 ± 3.5a | 7.0 ± 0.2b | 10.0 ± 0.5b | 24.5 ± 1.5c |
| C18:2 | 18.5 ± 1.5a | 20.0 ± 2.3a | 36.5 ± 1.0b | 35.2 ± 1.4b | 23.3 ± 0.7a |
| C18:3 | 1.7 ± 0.3a | 1.4 ± 0.3a | 2.0 ± 0.4a | 2.0 ± 0.4a | 1.6 ± 0.2a |
| C20:4 | 13.5 ± 1.0a | 13.8 ± 1.3a | 37.8 ± 1.6b | 33.4 ± 1.7bc | 30.0 ± 1.5c |
| C20:5 | 1.4 ± 0.2a | 1.5 ± 0.3a | 3.0 ± 0.2b | 3.3 ± 0.2b | 2.0 ± 0.1a |
| C22:6 | 2.0 ± 0.2a | 2.0 ± 0.4a | 5.3 ± 0.1b | 4.9 ± 0.2bc | 4.0 ± 0.4c |
Fatty acid species of LDL CE were quantified by LC-MS. Vales are expressed as percentage of the mass of CE containing individual fatty acid to total mass of CE recovered in LDL. Data represent the mean ± SEM from 9 to 10 samples per genotype. Superscripts not sharing common letters within each row indicate difference from SOAT2+/+LDLr−/− control mice with P < 0.05. C18:0 stearic acid; C16:0 palmitic acid. C16:1 palmitioleic acid; C18:1 oleic acid; C18:2 linoleic acid; C18:3 linolenic acid; C20:4 arachidonic acid; C20:5 eicosapentaenoic acid; C22:6 docosahexaenoic acid.
Both intestine- and liver-specific SOAT2 knockout mice are protected from diet-induced hepatic cholesterol accumulation
Liver weight and body weight were not different among genotypes at the time of necropsy (online supplement Figure II). SOAT2+/+LDLr−/− and SOAT2fl/flLDLr−/− mice had similar concentrations of each of the hepatic lipids including TC, FC, CE, and TG (figure 3). Compared with SOAT2+/+LDLr−/− mice, all SOAT2 knockouts had significant lower concentrations of liver TC and CE (figure 3A, B). SOAT2−/− LDLr−/− and SOAT2L-/L-LDLr−/− mice had less TC and CE accumulated in the liver than SOAT2SI-/SI-LDLr−/− mice (figure 3A, B). SOAT2−/− LDLr−/− mice also had significant less FC and TG in the liver while SOAT2 conditional knockouts had similar hepatic FC and TG as control mice (figure 3C, D). The concentration of PL was not significantly different among any of the genotypes (data not shown).
Figure 3. Deletion of SOAT2 inhibits diet-induced cholesterol ester accumulation in the liver.

Lipids of pre-weighed liver sample (50 to 100 mg) were extracted with CHCl3/MeOH (2:1). Quantification of hepatic lipids was determined by commercial enzymatic assay kits. (A) Hepatic total cholesterol (TC); (B) cholesterol ester (CE); (C) free cholesterol (FC); (D) triglycerides (TG). Data represent the mean ± SEM from 19 to 20 mice per group and are expressed as mg lipids per wet weight. Bars not sharing common letters differ with P < 0.05.
Intestinal SOAT2, but not liver SOAT2, is a critical determinant of biliary cholesterol levels
SOAT2+/+LDLr−/− and SOAT2fl/flLDLr−/− mice had similar levels of biliary cholesterol (online supplement figure III). By contrast, SOAT2−/− LDLr−/− and SOAT2SI-/SI-LDLr−/− mice had significant less biliary cholesterol (online supplement figure III). Biliary cholesterol of SOAT2L-/L-LDLr−/− mice was similar to that of control mice. So that, only in mice without SOAT2 in the intestine was biliary cholesterol significantly lower than in mice with intact SOAT2. There was no difference in biliary PL or total bile acid among any of the genotypes.
Both intestine- and liver-specific SOAT2 knockout mice are protected against atherosclerosis
Surface lesions were predominantly seen in the ascending aorta and aortic arch (figure 4). When atherosclerosis extent was expressed as the percentage of aortic surface area occupied by lesion, the amount of atherosclerosis in SOAT2+/+LDLr−/− and SOAT2fl/flLDLr−/− mice was equivalent [means of 8% and 9%, respectively (figure 5A)]. SOAT2−/− LDLr−/− mice had essentially undetectable atherosclerosis (figure 5A). Both SOAT2L-/L-LDLr−/− and SOAT2SI-/SI-LDLr−/− mice had significantly less aortic surface atherosclerosis than SOAT2+/+LDLr−/− and floxed control mice. Aortic cholesterol quantification indicated that SOAT2+/+LDLr−/− and SOAT2fl/flLDLr−/− mice had the highest (and similar) concentrations of TC, FC, and CE (figure 5B). All groups of mice with a SOAT2 gene deletion had significantly reduced TC, FC and CE concentrations in the aorta (figure 5B). The reduction of aortic cholesterol accumulation was equivalent and more pronounced in mice without liver SOAT2 (SOAT2−/− LDLr−/− and SOAT2L-/L-LDLr−/−) than was found in mice without intestinal SOAT2 (SOAT2SI-/SI-LDLr−/− mice, figure 5B). Regression analysis showed that the percentage of aortic surface area as lesion was significantly associated with aortic CE concentration (r = 0.86, P<0.0001; data not shown). Furthermore, the percentage of monounsaturated cholesterol esters in LDL was significantly correlated with aortic CE concentration (r = 0.74, P<0.0001; online supplement Figure IV) and with aortic surface area occupied with lesion (r = 0.58, P<0.0001; Figure 5C). The importance of LDL CE composition as a contributor to atherogenesis in addition to LDL cholesterol concentration can be appreciated when comparisons between the regressions of LDL cholesterol and LDL CE composition with atherogenesis (in this case expressed as aortic CE concentration) were made (Online supplement Figure IV). Both endpoints for LDL are significantly related to the extent of atherosclerosis but the CE composition of LDL seems to be the more highly associated.
Figure 4. Images of aorta isolated from all SOAT2 knockouts.
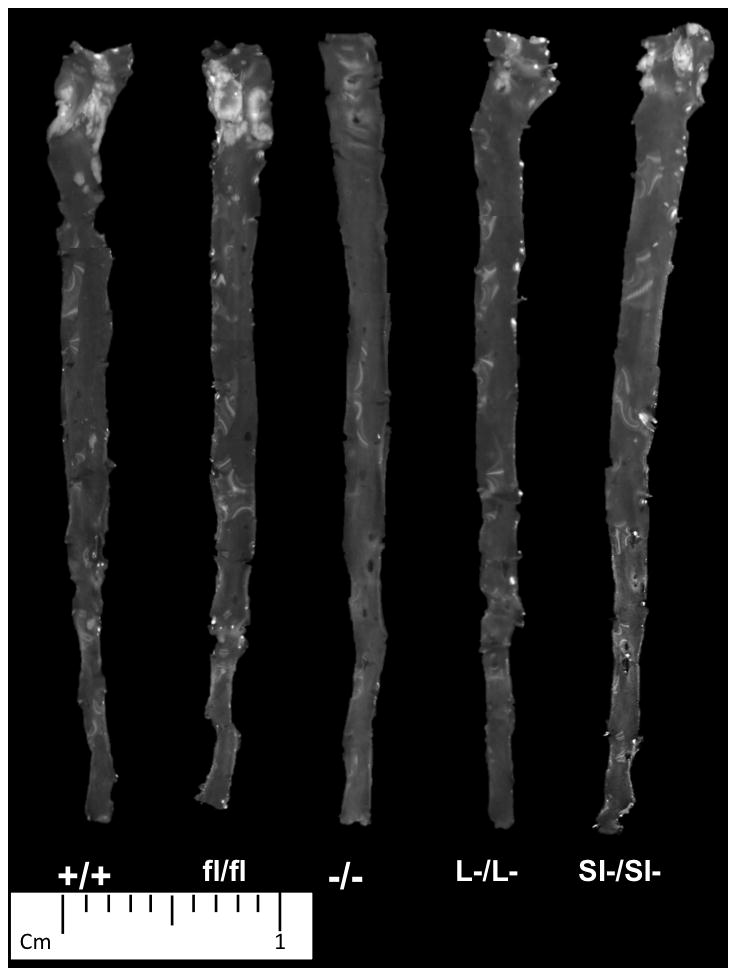
At the time of necropsy, the entire length of aorta was excised. Atherosclerotic lesions were noticeable as opaque areas mainly in the areas of ascending aorta and aortic arch. Representative image of each genotype was presented. N = 10 to 12 per genotype.
Figure 5. All SOAT2 knockouts are protected from atherosclerosis progression.
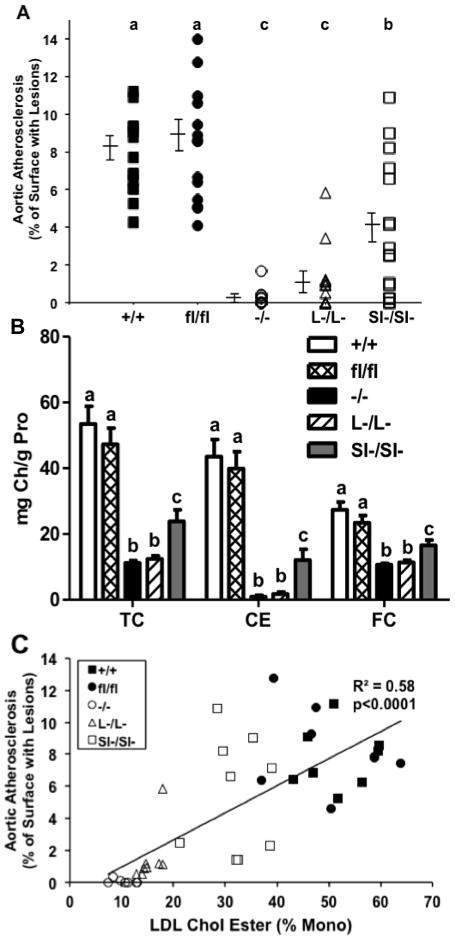
(A) En face analysis of aortic surface lesion area. Data were expressed as percentage of lesion surface area to total aorta surface area. N = 10 to 12 per genotype. Scatterplots not sharing common letters differ with P < 0.05. (B) Lipids of entire aorta were extracted with CHCl3/MeOH (2:1). Cholesterol was quantified by GLC. Data represent the mean ± SEM from 14 to 16 mice per genotype. Statistical analyses were performed for each subclass of aortic lipids. Bars not sharing common letters differ with P < 0.05. (C) Correlation was calculated between percentage of cholesterol oleate of LDL-CE and aortic CE using least-square regression model.
DISCUSSION
Previous work from our laboratory has consistently shown that mice with whole body SOAT2 gene deletions are protected against atherosclerosis development7, 8. Since SOAT2 is selectively expressed in only two tissues, the liver and the intestine2, 11, both of which are lipoprotein cholesterol ester secreting organs, we felt that it would be informative to determine if there were differences in the effect on atherosclerosis of having SOAT2 deletions separately in either of these tissues. To achieve this, the SOAT2 deletions need to be expressed in an atherosclerosis-susceptible mouse model. Given the lipoprotein profile of LDL receptor deficient vs. apoE deficient mice, the two main mouse models of atherosclerosis susceptibility, we studied the LDL receptor deficient mouse since the lipoprotein profile is typically more similar to that seen in humans, and is the mouse model that we have most often used previously. Our data show that while there is a somewhat higher level (~2X) of SOAT2 activity in the intestine than in the liver in mice, the cholesterol esters secreted into lipoproteins by the liver are expected to more readily distribute into the LDL, which are the lipoproteins that appear to circulate longer at higher concentrations in plasma during atherogenesis. Thus, given that this study was done in LDL receptor deficient animals the outcome will be characteristic of mice with higher LDL levels. We have studied the responses to dietary cholesterol in SOAT2 liver and intestine specific knockout mice with LDL receptors intact and have published our findings12. In this case, the deficiency of SOAT2 in either tissue resulted in a similar plasma phenotype although the percentage of intestinal cholesterol absorption was significantly less in the SOAT2SI-/SI- mice while the hepatic cholesterol ester concentrations were lower in the SOAT2L-/L- mice.
Excessive circulating apoB-containing lipoproteins, especially LDL, would be retained in the arterial intima6 and may initiate atherogenesis. SOAT2 knockout animals have delayed atherosclerosis development that is presumably due to the loss of capability of esterification of cholesterol oleate and packaging of those cholesterol esters into potentially atherogenic lipoprotein particles. Interestingly, intestine SOAT2 knockouts in the study had significantly less aortic lesion area and cholesterol deposition regardless of similar total mass of plasma LDL as that found in control mice (Figure 2). The small but significant decrease in VLDL cholesterol (and possibly reduced VLDL cholesteryl oleate) in the SOAT2Si/Si mice may contribute to the decrease in atherosclerosis in these animals. One recent study suggested that LDL core enrichment in cholesterol oleate led to intimal deposition in the aorta through LDL-proteogylcan binding6. LDL isolated from global SOAT2 knockouts had minimal cholesterol oleate in the core and had a lower affinity to bind to proteoglycan6, which could contribute to the delayed atherosclerosis development. The current study pinpoints the critical importance of LDL cholesterol oleate, in addition to LDL cholesterol mass (online supplement Figure IV), as a potential marker to predict the severity of the atherosclerosis. As discovered by Miller et al17, plasma cholesterol oleate and other SOAT2 derived CE, are valuable in predicting a patient’s possibility of having acute coronary syndrome during an attack of sudden chest pain. Furthermore, SOAT2-derived cholesterol esters have recently been identified as predictors of cardiovascular disease risk in a large unbiased human lipidomic study18. These results suggest that our current findings have broad implications in human disease. Our current study also greatly extends our understanding of distinct roles of liver- and intestine-derived CE in atherosclerosis progression. Although intestine-specific SOAT2 knockouts were protected from atherosclerosis development, protection was not to the extent seen in liver-specific SOAT2 and total body SOAT2 knockouts. The difference could be explained by the fact that intestinal knockouts still efficiently esterify cholesterol in the liver despite the reduction in intestinal cholesterol absorption. This speculation is supported by the fact that more cholesterol oleate was recovered in LDL-CE of SOAT2SI-/SI-LDLr−/− mice than other two knockouts (Table 2).
One of the characteristic changes in plasma lipids was the significant increase of TG in whole body SOAT2 knockout, anti-sense oligonucleotide-mediated hepatic SOAT2 knockdown, and liver-specific SOAT2 knockouts8, 14, 15, 19–21. Liver perfusion studies suggested that TG is more available for secretion in the absence of hepatic SOAT2, resulting in a higher rate of mobilization of hepatic TG15, 19. Interestingly, SOAT2SI-/SI-.
LDLr−/− mice appeared to have even higher plasma TG than liver-specific SOAT2 knockouts. Absence of intestinal SOAT2 would generate the CE-poor chylomicrons4 and remnants delivered to the liver could possibly lead to a similar effect to increase TG mobilization and packaging of more TG into the newly synthesized VLDL and circulating LDL. Furthermore, it is uncertain whether the absence of SOAT2 in the intestine or liver would affect the activities of lipoprotein lipase and/or its activator apolipoprotein CII, or interaction with apolipoprotein E, which all contribute to apoB-containing lipoprotein clearance and atherosclerosis progression. It is important to emphasize that the hypertriglyceridemia in SOAT2 knockout animals does not seem to contribute significantly to the development of atherosclerosis as all of the SOAT2 knockouts have elevated circulating triglyceride yet delayed atherosclerosis progression. The aforementioned mechanisms would warrant future studies, such as liver perfusion assay, for clarification. When we looked at gene expression differences (online supplement Table I), a 10 fold higher level of SCD-1 mRNA was detected in the intestine of the SOAT2SI-/SI- mice suggesting that this effect is related to the hypertriglyceridemia of these animals although the mechanism for such an outcome is not clear.
Our previous publication12 and current findings clearly show that liver and intestine-derived CEs both contribute to cholesterol accumulation in the circulation and tissues. We have found that human hepatic SOAT2 is expressed in much lower abundance relative to human intestinal SOAT2 among surgical samples collected from bariatric patients (unpublished data, Rudel LL, Fernandez, AZ, and Davis MA, 2011). This suggests that pharmaceutical strategies for targeting intestinal SOAT2 possibly could achieve a modest cardioprotective effect. A recent study showed that a non-specific SOAT1 and SOAT2 inhibitor not only limited aortic lesion size but modified the composition in pre-existing atherosclerotic plaque of apoE−/− mice during atherosclerosis regression22. However, it is noted that non-specific SOAT inhibitors also reduce SOAT1 activity, which can lead to free cholesterol accumulation across all cell types, potentially causing cytotoxicity to the cells. In fact, SOAT1 deficiency did not prevent the development of atherosclerosis lesion in either apoE−/− or LDLr−/− mice that had massive xanthomatosis23, 24. Discouraging findings of A-PLUS and ACTIVATE studies25, 26 also suggested that non-specific SOAT inhibitors may not benefit patients by slowing atherosclerosis progression although efficacy was not clearly apparent in either of these studies. The limited amount of data suggests that future studies to investigate advantages of developing highly selective SOAT2 inhibitors during atherosclerosis progression and/or regression could be informative. Furthermore, a sensitive and reliable marker, such as plasma cholesterol oleate percentage and concentration, would appear useful as a surrogate endpoint to evaluate disease severity and potential for treatment of atherosclerosis in patients with premature CHD17. Given the lower levels of SOAT2 in human liver relative to intestine, species differences need to be considered.
In conclusion, our data clearly demonstrate that both hepatic and intestinal SOAT2-derived cholesterol esters can promote the potentially atherogenic CE accumulation that occurs during atherosclerosis progression. The data suggest that SOAT2 in either liver or intestine could possibly be a pharmaceutical target for treatment of atherogenesis.
Supplementary Material
Novelty and Significance.
What Is Known?
Sterol O-acyltransferase2 (SOAT2) derived cholesterol esters (CE) contribute significantly to cholesterol homeostasis.
The importance of SOAT2 derived CE in atherogenesis has been apparent from the limited amount of atherosclerosis that develops in whole body SOAT2 knock- out mice.
What New Information Does This Article Contribute?
Conditional knockout of hepatic and intestinal SOAT2 showed that CE derived from both tissues significantly contribute to the progression of atherosclerosis.
Liver-specific and whole body SOAT2 knockout animals are more protected from atherosclerosis progression than intestine-specific SOAT2 knockouts.
Cholesterol oleate, the CE product from SOAT2, is a potential marker to predict the severity of atherosclerosis development.
SOAT2 is a microsomal enzyme that converts free cholesterol into cholesterol esters (CE) many of which get packaged into apoB-containing lipoproteins mainly in the liver and intestine. Whole body deletion of SOAT2 in mice has consistently been associated with reduced atherosclerosis. We generated liver specific and intestine-specific SOAT2 knockout mice to isolate the contribution in plasma of liver- and intestine-derived CEs in atherosclerosis development. The absence of SOAT2 in either the liver or small intestine resulted in a retarded atherosclerosis progression. However, two separate measurements of atherosclerosis, namely aortic lesion area and aortic cholesterol ester concentration, were significantly lower in liver-specific SOAT2 knockouts than in intestinal SOAT2 knockouts. Significant positive associations between atherosclerosis extent and plasma LDL-cholesterol concentration and LDL percentage of cholesterol oleate (mostly from SOAT2) were found for both intestine and liver SOAT2 knockouts, but higher percentages of cholesterol oleate in LDL and more atherosclerosis were found in intestine-specific knock out mice. The correlation with atherosclerosis of LDL percentage cholesterol oleate was stronger than the correlation of atherosclerosis with LDL cholesterol concentration suggesting that the presence of SOAT2 CE in plasma LDL is atherogenic.
Acknowledgments
SOURCES OF FUNDING
This work was supported by grants from the National Institute of Health (P01-HL49373 to L.L.R.) and the American Heart Association (Postdoctoral Fellowship 12POST11070006 to J.Z.).
Nonstandard Abbreviations and Acronyms
- APO
Apolipoprotein (apo)
- Chol
Cholesterol (Chol)
- CE
Cholesterol ester (CE)
- FC
Free (unesterified) cholesterol (FC)
- GLC
Gas-liquid chromatography (GLC)
- HDL
High density lipoproteins (HDL)
- LDL
Low density lipoproteins (LDL)
- LDLr
Low density lipoprotein receptor (LDLr)
- SEM
Standard error of the mean (SEM)
- SOAT
Sterol O- acyltransferase (SOAT)
- TC
Total cholesterol (TC)
- TPC
Total plasma cholesterol (TPC)
- T
Triglycerides (TG)
- VLDL
Very low density lipoproteins (VLDL)
Footnotes
DISCLOSURES
None.
References
- 1.Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme a:Cholesterol acyltransferases. American journal of physiology Endocrinology and metabolism. 2009;297:E1–9. doi: 10.1152/ajpendo.90926.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee RG, Willingham MC, Davis MA, Skinner KA, Rudel LL. Differential expression of acat1 and acat2 among cells within liver, intestine, kidney, and adrenal of nonhuman primates. Journal of lipid research. 2000;41:1991–2001. [PubMed] [Google Scholar]
- 3.Lee RG, Shah R, Sawyer JK, Hamilton RL, Parks JS, Rudel LL. Acat2 contributes cholesteryl esters to newly secreted vldl, whereas lcat adds cholesteryl ester to ldl in mice. Journal of lipid research. 2005;46:1205–1212. doi: 10.1194/jlr.M500018-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen TM, Sawyer JK, Kelley KL, Davis MA, Rudel LL. Cholesterol esterification by acat2 is essential for efficient intestinal cholesterol absorption: Evidence from thoracic lymph duct cannulation. Journal of lipid research. 2012;53:95–104. doi: 10.1194/jlr.M018820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Degirolamo C, Shelness GS, Rudel LL. Ldl cholesteryl oleate as a predictor for atherosclerosis: Evidence from human and animal studies on dietary fat. Journal of lipid research. 2009;50 (Suppl):S434–439. doi: 10.1194/jlr.R800076-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melchior JT, Sawyer JK, Kelley KL, Shah R, Wilson MD, Hantgan RR, Rudel LL. Ldl particle core enrichment in cholesteryl oleate increases proteoglycan binding and promotes atherosclerosis. Journal of lipid research. 2013;54:2495–2503. doi: 10.1194/jlr.M039644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee RG, Kelley KL, Sawyer JK, Farese RV, Jr, Parks JS, Rudel LL. Plasma cholesteryl esters provided by lecithin:Cholesterol acyltransferase and acyl-coenzyme a:Cholesterol acyltransferase 2 have opposite atherosclerotic potential. Circulation research. 2004;95:998–1004. doi: 10.1161/01.RES.0000147558.15554.67. [DOI] [PubMed] [Google Scholar]
- 8.Willner EL, Tow B, Buhman KK, Wilson M, Sanan DA, Rudel LL, Farese RV., Jr Deficiency of acyl coa:Cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein e-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1262–1267. doi: 10.1073/pnas.0336398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang CC, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, Lin S, Lee CY, Strom SC, Kashyap R, Fung JJ, Farese RV, Jr, Patoiseau JF, Delhon A, Chang TY. Immunological quantitation and localization of acat-1 and acat-2 in human liver and small intestine. The Journal of biological chemistry. 2000;275:28083–28092. doi: 10.1074/jbc.M003927200. [DOI] [PubMed] [Google Scholar]
- 10.Oelkers P, Behari A, Cromley D, Billheimer JT, Sturley SL. Characterization of two human genes encoding acyl coenzyme a:Cholesterol acyltransferase-related enzymes. The Journal of biological chemistry. 1998;273:26765–26771. doi: 10.1074/jbc.273.41.26765. [DOI] [PubMed] [Google Scholar]
- 11.Parini P, Davis M, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin B, Tomoda H, Omura S, Willingham MC, Rudel LL. Acat2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation. 2004;110:2017–2023. doi: 10.1161/01.CIR.0000143163.76212.0B. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Kelley KL, Marshall SM, Davis MA, Wilson MD, Sawyer JK, Farese RV, Jr, Brown JM, Rudel LL. Tissue-specific knockouts of acat2 reveal that intestinal depletion is sufficient to prevent diet-induced cholesterol accumulation in the liver and blood. Journal of lipid research. 2012;53:1144–1152. doi: 10.1194/jlr.M024356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veniant MM, Zlot CH, Walzem RL, Pierotti V, Driscoll R, Dichek D, Herz J, Young SG. Lipoprotein clearance mechanisms in ldl receptor-deficient “apo-b48-only” and “apo-b100-only” mice. The Journal of clinical investigation. 1998;102:1559–1568. doi: 10.1172/JCI4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell TA, 3rd, Brown JM, Graham MJ, Lemonidis KM, Crooke RM, Rudel LL. Liver-specific inhibition of acyl-coenzyme a:Cholesterol acyltransferase 2 with antisense oligonucleotides limits atherosclerosis development in apolipoprotein b100-only low-density lipoprotein receptor−/− mice. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1814–1820. doi: 10.1161/01.ATV.0000225289.30767.06. [DOI] [PubMed] [Google Scholar]
- 15.Brown JM, Bell TA, 3rd, Alger HM, Sawyer JK, Smith TL, Kelley K, Shah R, Wilson MD, Davis MA, Lee RG, Graham MJ, Crooke RM, Rudel LL. Targeted depletion of hepatic acat2-driven cholesterol esterification reveals a non-biliary route for fecal neutral sterol loss. The Journal of biological chemistry. 2008;283:10522–10534. doi: 10.1074/jbc.M707659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Temel RE, Lee RG, Kelley KL, Davis MA, Shah R, Sawyer JK, Wilson MD, Rudel LL. Intestinal cholesterol absorption is substantially reduced in mice deficient in both abca1 and acat2. Journal of lipid research. 2005;46:2423–2431. doi: 10.1194/jlr.M500232-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Miller CD, Thomas MJ, Hiestand B, Samuel MP, Wilson MD, Sawyer J, Rudel LL. Cholesteryl esters associated with acyl-coa:Cholesterol acyltransferase predict coronary artery disease in patients with symptoms of acute coronary syndrome. Academic emergency medicine : official journal of the Society for Academic Emergency Medicine. 2012;19:673–682. doi: 10.1111/j.1553-2712.2012.01378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stegemann C, Pechlaner R, Willeit P, Langley SR, Mangino M, Mayr U, Menni C, Moayyeri A, Santer P, Rungger G, Spector TD, Willeit J, Kiechl S, Mayr M. Lipidomics profiling and risk of cardiovascular disease in the prospective population-based bruneck study. Circulation. 2014;129:1821–1831. doi: 10.1161/CIRCULATIONAHA.113.002500. [DOI] [PubMed] [Google Scholar]
- 19.Alger HM, Brown JM, Sawyer JK, Kelley KL, Shah R, Wilson MD, Willingham MC, Rudel LL. Inhibition of acyl-coenzyme a:Cholesterol acyltransferase 2 (acat2) prevents dietary cholesterol-associated steatosis by enhancing hepatic triglyceride mobilization. The Journal of biological chemistry. 2010;285:14267–14274. doi: 10.1074/jbc.M110.118422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buhman KK, Accad M, Novak S, Choi RS, Wong JS, Hamilton RL, Turley S, Farese RV., Jr Resistance to diet-induced hypercholesterolemia and gallstone formation in acat2-deficient mice. Nature medicine. 2000;6:1341–1347. doi: 10.1038/82153. [DOI] [PubMed] [Google Scholar]
- 21.Repa JJ, Buhman KK, Farese RV, Jr, Dietschy JM, Turley SD. Acat2 deficiency limits cholesterol absorption in the cholesterol-fed mouse: Impact on hepatic cholesterol homeostasis. Hepatology. 2004;40:1088–1097. doi: 10.1002/hep.20439. [DOI] [PubMed] [Google Scholar]
- 22.Rong JX, Blachford C, Feig JE, Bander I, Mayne J, Kusunoki J, Miller C, Davis M, Wilson M, Dehn S, Thorp E, Tabas I, Taubman MB, Rudel LL, Fisher EA. Acat inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:4–12. doi: 10.1161/ATVBAHA.112.252056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr, Linton MF, Fazio S, Farese RV., Jr Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl coa:Cholesterol acyltransferase 1. The Journal of clinical investigation. 2000;105:711–719. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr Increased atherosclerosis in ldl receptor-null mice lacking acat1 in macrophages. The Journal of clinical investigation. 2001;107:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, Schoenhagen P, Waters DD, Pepine CJ, Crowe TD, Davidson MH, Deanfield JE, Wisniewski LM, Hanyok JJ, Kassalow LM. Effect of acat inhibition on the progression of coronary atherosclerosis. The New England journal of medicine. 2006;354:1253–1263. doi: 10.1056/NEJMoa054699. [DOI] [PubMed] [Google Scholar]
- 26.Tardif JC, Gregoire J, L’Allier PL, Anderson TJ, Bertrand O, Reeves F, Title LM, Alfonso F, Schampaert E, Hassan A, McLain R, Pressler ML, Ibrahim R, Lesperance J, Blue J, Heinonen T, Rodes-Cabau J. Effects of the acyl coenzyme a:Cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. 2004;110:3372–3377. doi: 10.1161/01.CIR.0000147777.12010.EF. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.