

Abstract
Furan is an abundant food and environmental contaminant that is a potent liver carcinogen in rodent models. To determine if furan is genotoxic in vivo, female B6C3F1 Big Blue transgenic mice were treated with 15 mg/kg bw furan by gavage 5 days a week for 6 weeks, or once weekly for 3 weeks. Liver cII trans-gene mutation-frequency and mutation spectra were determined. Furan did not increase the mutation frequency under either treatment condition. In the 6-week treatment regimen, there was a change in the cII transgene mutation-spectrum, with the fraction of GC to AT transitions significantly reduced. The only other significant change was an increase in GC to CG transversions; these represented a minor contribution to the overall mutation spectrum. A much larger furan-dependent shift was observed in the 3-week study. There was a significant increase in transversion mutations, predominantly GC to TA transversions as well as smaller non-significant changes in GC to CG and AT to TA transversions. To determine if these mutations were caused by cis-2-butene-1,4-dial (BDA), a reactive metabolite of furan, the mutagenic activity and the mutation spectrum of BDA was determined in vitro, in Big Blue mouse embryonic fibroblasts. This compound did not increase the cII gene mutation-frequency but caused a substantial increase in AT to CG transversions. This increase, however, lost statistical significance when adjusted for multiple comparisons. Together, these findings suggest that BDA may not be directly responsible for the in-vivo effects of furan on mutational spectra. Histopathological analysis of livers from furan-treated mice revealed that furan induced multifocal, hepatocellular necrosis admixed with reactive leukocytes and pigment-laden Kupffer cells, enhanced oval-cell hyperplasia, and increased hepatocyte mitoses, some of which were atypical. An indirect mechanism of genotoxicity is proposed in which chronic toxicity followed by inflammation and secondary cell proliferation triggers cancer development in furan-exposed rodents.
Keywords: Furan, Big Blue mouse, Aberrant mitosis, In-vivo mutagenesis
1. Introduction
Furan, a product of incomplete combustion, is found in cigarette smoke, engine exhaust, and processed food [1]. This volatile chemical induces liver tumors in F344 rats and B6C3F1 mice when administered orally [2]. Because of the potential for human exposure and its potency as a rodent carcinogen, furan is classified as a possible human carcinogen (Group 2b) [1,3]. To assist human cancer-risk assessment, the mechanism(s) of furan-induced tumorigenesis should be defined.
Oxidation catalyzed by cytochrome P450 is required for furan to exert its harmful effects [4–8]. The product of this reaction is cis-2-butene-1,4-dial (BDA) (Scheme 1). This metabolite readily alkylates protein and DNA nucleophiles [9–12]. It causes DNA damage [13–15] and is toxic and mutagenic in bacteria and mammalian cells [13,16], so it may play a role in furan-induced liver tumorigenesis.
Figure 3.
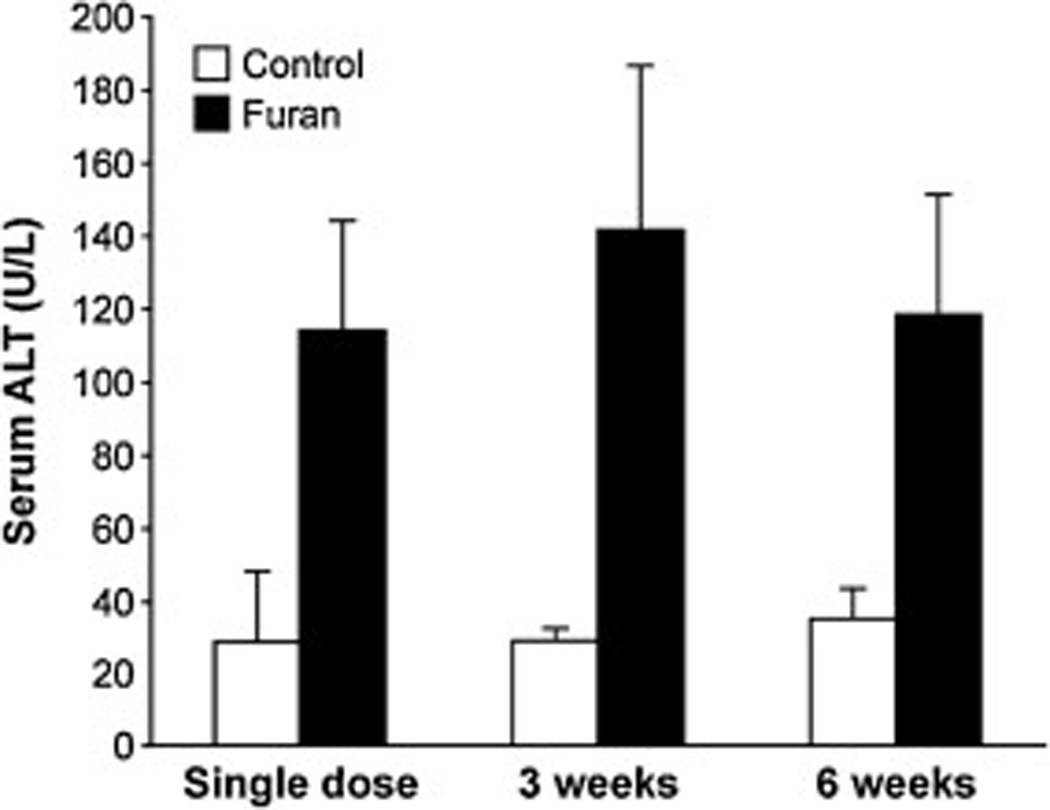
Serum alanine aminotransferase (ALT) concentrations at 24 h after the final dose Female B6C3F1 mice received either corn oil or furan (15 mg/kg bw) as a single dose, once a week for 3 weeks (three weekly doses), or 5 days per week for 6 weeks (6 weeks of five daily doses). Blood was collected via cardiac puncture and allowed to clot to collect serum. Data is displayed ±SD. The data from the full hepatic panel are displayed in Supplementary Table 13. The values for the treated animals are significantly different from those in the controls, p < 0.005, as determined by Student’s t-test.
Data indicate both non-genotoxic and genotoxic pathways for furan-induced tumorigenesis. In support of a non-genotoxic mechanism, furan does not elicit a DNA-repair response in rats [17] and furan-derived DNA damage in the liver is low [4]. Furan is a strong inducer of cell proliferation in mouse and rat liver [17–20]. Toxicity and proliferation precede furan-induced tumor development in rodents [19,21,22]. These studies support a mechanism of carcinogenesis in which chronic toxicity with secondary cell proliferation triggers cancer development in furan-exposed rodents.
Genotoxicity tests with furan have given mixed results, with many studies providing negative data [2,20,23–26]. However, furan exposure is associated with unique Hras1 mutations in liver tumors [27,28], strengthening support for at least a partial genotoxic mechanism for furan’s carcinogenic properties. Many of the in-vitro genotoxicity assays with furan were compromised by a number of factors, such as its low boiling point, the choice of cell line, the absence of appropriate activation enzymes, or the presence of detoxifying systems. The observation that the reactive metabolite BDA is mutagenic is consistent with a possible genotoxic component in furan-induced tumorigenesis [13,16]. However, the chemical reactivity of BDA complicates the proposed link between furan metabolism and a genotoxic event in the cell. In many test systems, furan was activated extracellularly [2,23]. Given the reactivity of BDA with protein nucleophiles, it is possible that insufficient quantities of the metabolite generated extracellularly were able to reach nuclear DNA to form mutagenic adducts.
An approach that overcomes most of these technical issues is to determine the ability of furan to cause mutations in the target tissue. Big Blue transgenic rodents harbor multiple copies of a bacteriophage lambda shuttle-vector in all cells. This shuttle vector contains the lac repressor gene (lacI) and the cII phage regulatory gene, so that mutagenesis can be readily measured in any tissue by isolating this DNA and quantifying the level of mutation in these genes by means of functional assays [29]. In addition, the genes can be sequenced to determine the mutation spectrum caused by the chemical treatment. This model has provided useful mechanistic information about a number of compounds thought to be non-genotoxic [30,31].
To test the hypothesis that furan is a genotoxic carcinogen, female Big Blue B6C3F1 mice were treated with carcinogenic doses of furan by gavage, and cII gene mutation-frequencies were determined in liver tissue. Since there are examples of compounds that shift the mutation spectrum without increasing the mutation frequency [31], the cII gene mutation-spectrum in these samples was also determined. To probe the role of BDA in any mutagenic effects observed, the mutation frequency and cII mutation spectrum were determined in vitro, in Big Blue transgenic mouse embryonic fibroblasts treated with BDA. Liver toxicity was determined by measurement of alanine transaminase (ALT) concentrations in serum and by assessment of alterations in histopathology in furan-treated wild-type female B6C3F1 mice, to help interpret the mutagenesis findings.
2. Materials and methods
2.1. Caution
Furan, dimethylnitrosamine (DMN) and N-nitrosomethylurethane (NMUR) are toxic and mutagenic in cells and carcinogenic in laboratory animals. They should be handled with proper safety equipment and precautions.
2.2. Materials
Furan was obtained from Sigma–Aldrich (St. Louis, MO) and distilled prior to use. DMN was purchased from Supelco Analytical (Bellefonte, PA). Solutions were prepared fresh on each day of treatment. NMUR was obtained from MRI Global Chemical Carcinogen Repository (Kansas City, MO). BDA was prepared from 2,5-diacetoxy-2,5-dihydrofuran as previously reported [11]. Its concentrations were determined by semicarbazide trapping according to published methods [11]. RecoverEase DNA Isolation Kits, the Select cII Mutation Detection System for Big Blue Rodents, and the Transpack packing extract were procured from Agilent Technologies (Santa Clara, CA). Phage-packaging extract was also prepared from bacterial strains Escherichia coli NM759 and BHB2688, generously supplied by Dr Peter Glazer (Yale, Univ. School of Medicine, New Haven, CT), according to published methods [32].
Dulbecco’s Modified Eagle Medium, fetal bovine serum, penicillin and streptomycin were purchased from Invitrogen (Carlsbad, CA). Trypsin was obtained from Gibco (Grand Island, NY).
2.3. Animals
Six-week-old female B6C3F1 Big Blue® mice hemizygous for the lambda LIZ shuttle vector containing the cII transgene were obtained from Agilent Technologies (Santa Clara, CA). Five-week-old female wild-type B6C3F1 mice were purchased from Charles River Laboratories (Kingston, NY). B6C3F1 mice were selected for these studies because they have been used in numerous short-term experiments and long-term cancer studies [2,17–19,33]. While male and female B6C3F1 mice have similar biochemical, proliferative and toxic responses to the hepatic effects of furan [17,18], female mice have a lower spontaneous tumor incidence than males [2,34]. Therefore, we used female mice in this study.
2.3.1. Animal treatments
All animal experiments were approved by the University of Minnesota Institutional Animal Care and Use Committee. The mice were housed five animals/cage according to IACUC and NIH guidelines, and given standard feed and tap water ad libitum. They were acclimated for 1 week prior to the start of the experiments.
2.3.2. Big Blue animal studies
Big Blue mice were treated with either corn oil (control) or furan dissolved in corn oil (15 mg/kg bw) at a volume of 5 mL/kg bw. When administered for 2 years, this dose yielded a 100% incidence of liver tumors in the cancer bioassay [2]. Solutions of furan in corn oil were prepared weekly and stored under nitrogen at −20 °C between treatments. Two different dosing regimens for furan were employed. In one study, five mice were treated with 15 mg/kg bw furan by gavage on 5 days/week for 6 weeks (the first two doses in the first week were 8 mg/kg bw). Five control mice received corn oil alone. Two control mice died before termination of the study due to damage caused by the gavage needle. Twenty-four hours after the final dose, mice were euthanized by CO2 inhalation and the livers were harvested, segmented by lobe and flash-frozen for storage at −80 °C for the transgene-mutation assay. In a second study, five mice were treated with 15 mg/kg bw by oral gavage once weekly for 3 weeks. Control animals received corn oil alone. The animals were euthanized by CO2 inhalation 1 week after the final dose. The livers were harvested as described above.
As a positive control, five Big Blue mice were given four daily doses of 4 mg/kg bw DMN in saline (5 mL/kg bw) by oral gavage [35]. A control group of five mice received saline alone. The animals were sacrificed 10 days after the last treatment by CO2 asphyxiation. The livers were harvested from each mouse, segmented by lobe, flash-frozen and stored at −80 °C for the transgene-mutation assay.
2.4. Histopathological studies
Groups of five female wild-type B6C3F1 mice were treated with 15 mg/kg bw furan in corn oil under one of the following treatment regimens: one single dose, one dose per week for 3 weeks, or five daily doses per week for 6 weeks. In all treatment regimens, the mice were euthanized by CO2 inhalation 24 h after the final dose. Blood was collected by cardiac puncture and left to clot at room temperature while other samples were collected. Serum was separated by centrifugation at 10,000 × g for 10 min. These samples were sent to Marshfield Labs (Marshfield, WI) for hepatic-panel analysis on a Beckman AU5800 Clinical Chemistry System. Livers were immediately removed by lobe and placed in 10% phosphate-buffered formalin (Fisher Scientific, Pittsburg, PA) at room temperature. After 24 h, fixed tissues were transferred to 70% ethanol.
Tissue processing and histopathological analyses were performed by the Comparative Pathology Shared Resource, University of Minnesota Masonic Cancer Center. Formalin-fixed sections of the left liver lobes (n = 3 per group) were processed into paraffin blocks by means of standard histology techniques, sectioned to 4-µm thickness, and stained with haematoxylin and eosin. Histology slides were evaluated with light microscopy, and diagnoses verified by two ACVP board-certified pathologists (R.C.K. and M.G.O’S.). Ki-67 protein was probed in tissues with anti-Ki-67 antibody (1:100 dilution, clone SP6, Biocare Medical, Concord, CA) followed by Rabbit EnVision Plus (Dako, Carpinteria, CA). Ki-67-staining was observed with light microscopy (Zeiss, Oberkochen, Germany). For each tissue, 20 pictures were taken at 400× magnification. Positive Ki-67 stains per field were counted blind as to treatment group by use of ImageJ [36].
2.5. Cell treatments
SV40 large T-antigen-transfected mouse embryonic fibroblasts, transgenic for lambda LIZ (Lac I/cII), were purchased from the American Type Culture Collection [CRL-2816] (Manassas, VA) and cultured in T75 flasks in high-glucose Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin, at 37 °C in 5% CO2.
Initial cytotoxicity assays were performed in 96-well plates seeded 24 h earlier with 700 cells/well. The cells were incubated with either 0–40 µM NMUR for 1 h in medium or 0–100 µM BDA in phosphate-buffered saline (PBS) for 0.5–1.5 h. Following treatment, the cells were given fresh medium. After 48 h, cell viability was determined with methylthiazol tetrazolium (MTT) according to manufacturer’s instructions (Promega, Madison, WI). The effect of the treatment was calculated as the absorbance of the treated cells/absorbance of control cells × 100. The highest concentration of NMUR reduced cell viability by less than 40%.
For the mutagenesis assay, cells were plated in 6-well plates at a density of 40,000 cells/well. The next day, they were treated for 1 h with 0–40 µM NMUR in medium or for 30 min with 0–10 µM BDA in PBS. The cells were given fresh medium and incubated at 37 °C for 48 h, trypsinized, collected by centrifugation and transferred in fresh medium to T-75 flasks. When the cells reached a concentration of 6–10 million cells per flask (~72 h), they were isolated by centrifugation for DNA isolation [37].
2.6. The Big Blue® cII mutation-detection assay
High molecular weight genomic DNA was extracted from either ~65 mg liver tissue from the right and left liver lobes or from approximately 6 million cells as described in the RecoverEase DNA Isolation Kit. The packaging of the phage, plating of the packaged DNA and determination of mutant frequency were performed according to the directions provided by Agilent Technologies (Santa Clara, CA). Briefly, the lambda LIZ shuttle vector was extracted and packaged into a virulent phage lambda and used to infect E. coli strain G1250 and plated. Plates were grown under selective (24.4 °C, 42 h) or non-selective (37 °C, 18 h) conditions. Mutant frequency was calculated by dividing the number of mutants (selective conditions) by total plaque forming units (non-selective conditions). In many instances, the assay was repeated to generate a minimum of 3 × 105 plaques from each sample or to obtain sufficient numbers of mutant clones for sequencing. A number of purported cII mutants were picked at random for verification of their mutant status by re-plating and growth at the selective temperature (24.4 °C) for 42 h. Verification plates were stored at 4 °C for up to 3 months.
2.7. Identification of cII mutations
Verified mutant clones were then used to characterize the types of cII gene mutations. The cII gene was PCR-amplified with Platinum pfx polymerase (Invitrogen, Carlsbad, CA), forward primer 5′-CTTGTCGCGACAGATTCCT-3′ and reverse primer 5′-CCTCTGCCGAAGTTGAGTAT-3′ [38]. Thermal cycling was as follows: 94 °C for 5 min, followed by 32 cycles of 94 °C for 15 s, 50 °C for 30 s, 68 °C for 1 min, a final extension time at 68 °C for 10 min. Protein, primers, and buffer were removed by use of MinElute PCR Purification Kit (Qiagen, Hilden, Germany). Purified PCR products (40 ng) were sequenced at the University of Minnesota BioMedical Genomics Center by means of the Sanger method on an ABI 3730xl sequencer. The primers were as follows: cII forward sequencing 5′-CCGCTCTTACACATTCCAGC-3′ and cII reverse sequencing 5′-CCTCTGCCGAAGTTGAGTAT-3′ [38]. Sequences were aligned to the cII gene (base-pairs -51 to 294) with Sequencher® 5.0 sequence-analysis software (Gene Codes Corporation, Ann Arbor, MI). Base pairs not unambiguously identified by the sequencer software were manually identified by analysis of the accompanying chromatogram. Mutants were counted once per position per animal. Mutants that appeared multiple times in the same animal, at the same base-pair, were thought to arise from clonal expansion and were eliminated from the final analysis by subtracting the percentage of clonal mutations from the mutant frequency. The clonal-corrected value was referred to as the mutation frequency. Mutants that did not contain a mutation in the cII gene were included in the calculation of mutant frequency, but excluded from any calculations in the mutational spectra. Approximately 12% of the mutants isolated from liver lobes from treated and control animals did not have mutations in the cII gene. These mutants may have resulted from alterations in the cI gene.
2.8. Statistics
For each mutation category, the mutation frequencies were computed by dividing the observed mutation count for each mutation category by total mutation count (sum of transition-, transversion- and frameshift-mutation counts). Recurrent mutations, in vivo or in vitro, were excluded from the analysis. To quantify the significance of the effect of exposure to furan or BDA on the cII mutation spectrum relative to the vehicle control, three different types of statistical approach were applied. Method 1, reported by Adams and Skopek, assesses the impact of chemical exposure on the overall mutation spectrum [39]. Two newer methods were employed to investigate the effect of chemical exposure on the individual mutation classes. Method 2 assumes dependence among non-recurrent mutations from the same mouse; in other words, the biological variation of the mice influences the mutation spectrum in each mouse [40]. Method 3 assumes that all the non-recurrent mutation events are independent [41]. We performed analyses with both Methods 2 and 3 using generalized estimating equations (GEE), and logistic regression, respectively. Method 2 provided greater power to distinguish between mutation classes, because there was a weak but measurable dependence of the mutation spectrum on the individual mouse. Therefore, all the significance values provided in Section 3 were derived with Method 2. A Bonferroni correction was applied for adjustment of multiple comparisons for each furan treatment-regimen or BDA concentration [42].
3. Results
3.1. In-vivo mutation studies
Two different treatment regimens were chosen to assess the mutagenic potential of furan in the transgenic mice. The first involved treatment of the mice with five daily doses of furan (15 mg furan/kg bw) for 6 weeks. This daily dose was employed in the 2- year tumorigenesis study performed by the National Toxicology Program [2]. Previously, a 6-week course of this dosing schedule induced cell proliferation in mouse liver [17], indicating that this treatment would result in sufficient cell turnover for the accumulation of mutations. The second dosing regimen was modeled after one used for the genotoxic carcinogen, 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) [43]. This protocol employed single weekly doses of the carcinogen with the sacrifice occurring 1 week after the last dose. The hypothesis was that the acute toxic effects of furan may interfere with the ability to observe any direct genotoxic effects of this chemical. Therefore, a reduction of the frequency of dosing might allow us to observe mutations triggered directly by a DNA-reactive metabolite of furan. To confirm that we were able to perform the mutagenesis assay appropriately, we also included a positive control where a group of mice were exposed to DMN under treatment conditions known to induce an increase in mutation frequency in these animals[35]. While this positive control DMN induced a significant three-fold increase in the mutant frequency in the cII transgene, no significant increase was observed in the any of the liver lobes from furan-treated mice in either treatment regimen (Table 1).
Table 1.
Mutant frequency in the cII gene in liver lobes of DMN- and furan-treated Big Blue B6C3F1 female micea.
| Treatment | Average (×10−5) ± SD | N | |
|---|---|---|---|
| Positive controlb | Control | 0.9 ± 0.5 | 5 |
| DMN | 3.0 ± 0.5c | 5 | |
| Six-week experimentd | Control | 1.2 ± 0.3 | 3 |
| Furan | 1.2 ± 0.2 | 5 | |
| Three-week experimente | Control | 1.8 ± 0.5 | 5 |
| Furan | 1.1 ± 0.2 | 5 |
Data for individual animals are displayed in Supplemental Tables 1–3 and were corrected for the percentage of clonal mutants, with the exception of the DMN-treated group and its control.
Groups of five female mice were given four doses of 0 or 4 mg/kg DMN in saline by oral gavage for 4 consecutive days. Mice were sacrificed 10 days after final treatment.
Statistically different from controls, p-value < 0.0003.
Groups of five mice were given 0 or 15 mg/kg bw furan in corn oil by oral gavage on 5 days per week for 6 weeks. Mice were sacrificed 24 h after the final dose.
Groups of five mice were given 0 or 15 mg/kg bw furan in corn oil by oral gavage once weekly for 3 weeks. Mice were sacrificed 1 week after the final dose.
Small shifts in the cII mutation spectra were observed in the livers from furan-treated animals compared with controls in the 6-week study (Fig. 1A, Table 2). The overall p-value was 0.495 (95% CI: 0.472, 0.517) using the method by Adams and Skopek [39]. For the analysis by mutation class, there was a reduction in the GC to AT transition mutations associated with furan exposure, which was statistically significant after adjustment for multiple comparisons (p < 0.0001). Treatment with furan also resulted in an increase in GC to CG transversion mutations (p = 0.02) which was non-significant after multiple comparison adjustment. A much larger shift in the mutation spectrum occurred in the livers of mice receiving three weekly doses of furan, with an approximately 50% reduction in GC to AT transition mutations, which was significant after adjustment for multiple comparisons (p = 0.001; Fig. 1B, Table 2). This was accompanied by a significant increase in overall transversion mutations (p = 0.005). The frequency of GC to TA mutations almost doubled; minor increases were also observed for GC to CG and AT to TA mutations. The overall p-value is 0.088 (95% CI: 0.077, 0.100), calculated with the method by Adams and Skopek [39].
Figure 1.
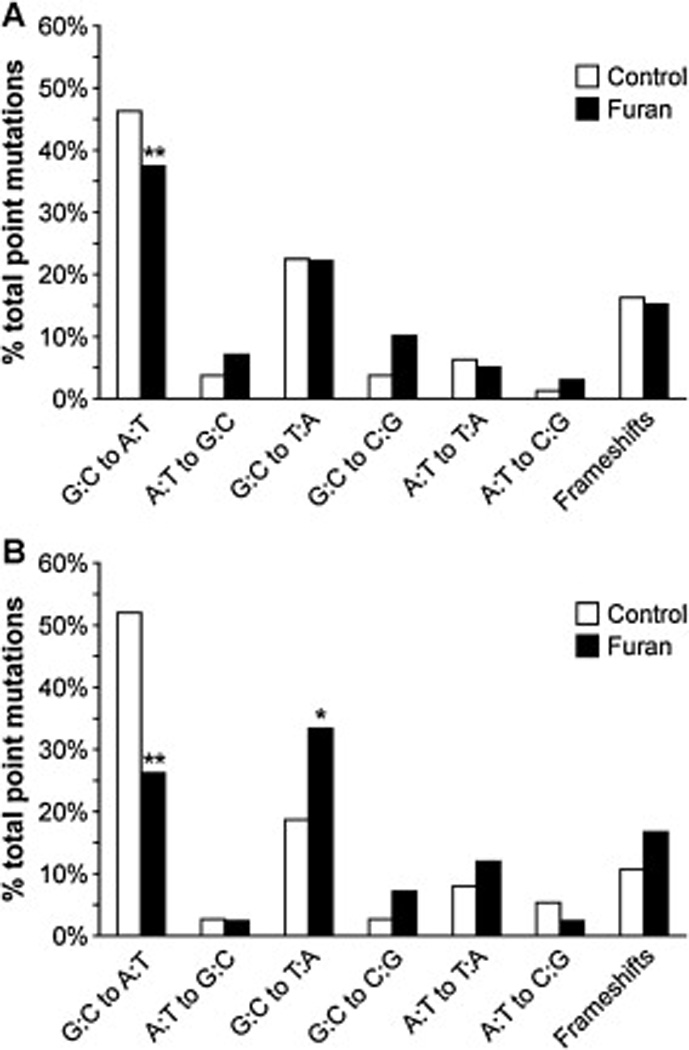
Distribution of mutation types in cII mutant clones isolated from A) livers of mice treated with furan for 6 weeks and B) livers of mice treated with furan for 3 weeks. *Different from control, p < 0.05; **Different from control, p ≤ 0.001.
Table 2.
Distribution of mutation types in cII mutant clones isolated from livers of Big Blue mice.
| Six-week studya | Three-week studyb | |||||||
|---|---|---|---|---|---|---|---|---|
| Controlc | Furan | Control | Furan | |||||
| No.d | %e | No. | % | No. | % | No. | % | |
| Transitions | 40 | 50% | 44 | 44% | 41 | 55% | 10 | 26%f |
| G:C to A:T | 37 | 46% | 37 | 37%f | 39 | 52% | 9 | 24%f |
| A:T to G:C | 3 | 4% | 7 | 7% | 2 | 3% | 1 | 3% |
| Transversions | 27 | 34% | 40 | 40% | 26 | 35% | 21 | 55%g |
| G:C to T:A | 18 | 23% | 22 | 22% | 14 | 19% | 13 | 34%h |
| G:C to C:G | 3 | 4% | 10 | 10%i | 2 | 3% | 2 | 5% |
| A:T to T:A | 5 | 6% | 5 | 5% | 6 | 8% | 5 | 13% |
| A:T to C:G | 1 | 1% | 3 | 3% | 4 | 5% | 1 | 3% |
| Frameshifts | 13 | 16% | 15 | 15% | 8 | 11% | 7 | 18% |
| Deletions | 10 | 13% | 8 | 8% | 4 | 5% | 2 | 5% |
| Insertions | 3 | 4% | 7 | 7% | 4 | 5% | 5 | 13% |
| Total cII mutants | 80 | 99 | 75 | 38 | ||||
| No. of mutations | 16 | 12 | 16 | 8 | ||||
| Clonal mutantsj | 12 | 11 | 3 | 6 | ||||
| Total mutants sequenced | 107 | 120 | 104 | 46 | ||||
Groups of five mice were given 0 or 15 mg/kg bw furan in corn oil by oral gavage on 5 days per week for 6 weeks. Mice were sacrificed 24 h after the final dose. The types and locations of the mutations detected in individual animals are displayed in Supplemental Tables 4 and 5.
Groups of five mice were given 0 or 15 mg/kg bw furan in corn oil by oral gavage once weekly for 3 weeks. Mice were sacrificed 1 week after the final dose. The types and locations of the mutations detected in individual animals are displayed in Supplemental Tables 6 and 7.
Includes one mutant clone that had a deletion of three base-pairs in the cII gene.
Number of clones.
Percent of total point mutations after correction for clonal mutants and colonies for which no cII mutant was detected.
Significantly decreased relative to control, p ≤ 0.001, with the GEE approach.
Increased relative to control, p < 0.01 with the GEE approach, but not significant after multiple comparison adjustment.
Increased relative to control, p = 0.5 with the GEE approach, but not significant after multiple comparison adjustment.
Increase relative to control, p < 0.03 with the GEE approach, but not significant after multiple comparison adjustment.
Mutants from the same animal that showed the identical mutation.
3.2. In vitro mutation studies
Since furan is metabolized to BDA and this metabolite is mutagenic in other test systems [13,16], the mutation spectrum of BDA was determined in Big Blue mouse embryonic fibroblasts. Initial studies indicated that complete cell-culture media neutralized the toxicity of BDA, presumably due to its reactivity with medium constituents (data not shown). Exploratory studies, in which cells were treated in PBS with and without BDA, indicated that incubations in PBS alone for longer than 30 min reduced the viability of the control cells (data not shown). In view of this, the cells were treated with BDA in PBS for only 30 min. This treatment regimen resulted in a concentration-dependent decrease in cell viability (Fig. 2), but did not alter the cII gene mutation-frequency compared with PBS-treated controls (Table 3). A known mutagen, NMUR, caused an increase in cII gene mutant-frequency in these cells (Table 3).
Figure 2.
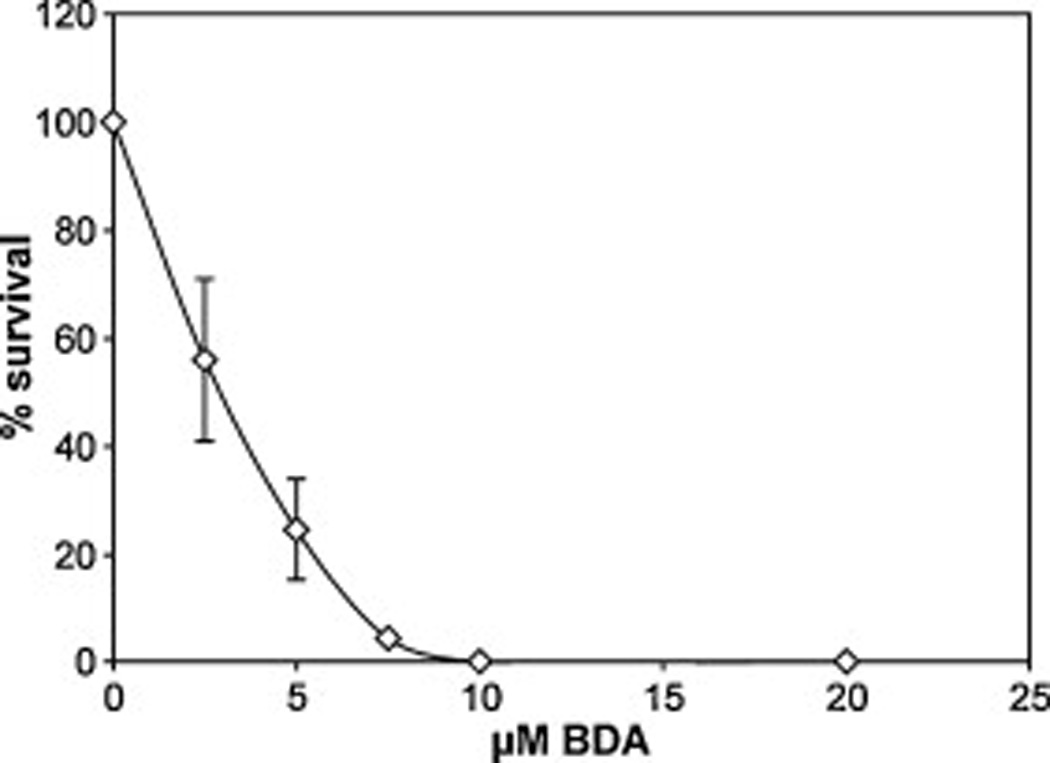
Cytotoxicity of BDA in mouse embryonic fibroblasts following a 0.5-h treatment with 0–20 µM BDA in PBS. Toxicity was measured by use of the MTT assay, at 48 h after treatment. Data points represent average of three replicate plates.
Table 3.
Mutant frequency in the cII gene in NMUR- or BDA-treated Big Blue transgenic mouse embryonic fibroblastsa.
| Treatment | Average (×10−5) ± SD | N |
|---|---|---|
| None | 10 ± 3 | 6 |
| 30 µM NMUR | 24 ± 4b | 3 |
| 40 µM NMUR | 38 ± 13b | 3 |
| PBS | 6.0 ± 2.0 | 8 |
| 2.5 µM BDA | 6.3 ± 3.8 | 7 |
| 5.0 µM BDA | 5.2 ± 2.2 | 5 |
Mouse embryonic fibroblasts were treated with 0–40 µM NMUR for 1 h in complete medium or with 0–5 µM BDA for 0.5 h in PBS. The cells were then incubated in fresh complete medium for 7 days at 37 °C prior to DNA extraction. N = number of replicate plates. Data for individual plates are displayed in Supplemental Tables 8 and 9. The data were corrected for the percentage of clonal mutants.
Statistically different from control with p = 0.002.
While there was no significant change in mutation frequency, BDA treatment led to an increase in AT to CG transversion mutations relative to control (p < 0.03; Table 4), the increase being larger in the cells exposed to 5 µM BDA than in those exposed to 2.5 µM BDA. These changes were not statistically significant after adjustment for multiple comparisons. The overall p-value according to Adams and Skopek’s approach was 0.287 (95% CI: 0.267, 0.307) [39].
Table 4.
Distribution of mutation types in cII mutant clones isolated from Big Blue mouse embryonic fibroblasts treated with BDAa.
| 0 µMb,c | 2.5 µMc | 5 µMc | ||||
|---|---|---|---|---|---|---|
| No.d | %e | No. | % | No. | % | |
| Transitions | 19 | 40% | 17 | 40% | 14 | 32% |
| G:C to A:T | 13 | 28% | 9 | 21% | 10 | 23% |
| A:T to G:C | 6 | 13% | 8 | 19%f | 4 | 9% |
| Transversions | 21 | 45% | 21 | 50% | 26 | 61% |
| G:C to T:A | 16 | 34% | 10 | 24% | 15 | 35% |
| G:C to C:G | 1 | 2% | 2 | 5% | 2 | 5% |
| A:T to T:A | 3 | 6% | 3 | 7% | 0 | 0% |
| A:T to C:G | 1 | 2% | 6 | 14%f | 9 | 21%f |
| Frameshifts | 7 | 15% | 4 | 10% | 3 | 7% |
| Deletions | 4 | 9% | 2 | 5% | 2 | 5% |
| Insertions | 3 | 6% | 2 | 5% | 1 | 2% |
| Total cII mutants | 47 | 42 | 43 | |||
| No mutations | 4 | 4 | 4 | |||
| Clonal mutationsg | 19 | 10 | 20 | |||
| Total mutants sequenced | 67 | 55 | 64 | |||
Mouse embryonic fibroblasts were treated with 0–5 µM BDA for 0.5 h in PBS. The cells were then incubated in fresh complete medium for 7 days at 37 °C prior to DNA extraction. The type and location of the mutations detected in individual replicates are displayed in Supplemental Tables 10–12.
Includes one mutant clone that had a deletion of three base-pairs in the cII gene.
Includes mutant clones that had two point mutations in the cII gene.
Number of clones.
Percent of total point mutations after correction for clonal mutants and colonies for which no cII mutant was detected.
Increased from control (p-value < 0.03) with the GEE approach, but not statistically significant after multiple comparison adjustment.
Mutants from the same treatment flask that showed the same mutation.
3.3. Determination of the hepatotoxic effects of the furan treatment regimens
To compare the relative hepatotoxicity of the treatment regimens, wild-type B6C3F1 female mice were treated with furan by gavage as follows: one single dose of 15 mg/kg bw, one dose of 15 mg/kg bw per week for 3 weeks, or five daily doses of 15 mg/kg bw per week for 6 weeks. The animals were sacrificed 24 h after the final dose. Blood was collected to determine the concentration of ALT, and the liver was prepared for histopathological analysis.
Furan was toxic to the liver as indicated by the elevated levels of ALT in all furan-exposed animals (Fig. 3). No significant difference was observed between the three different treatment regimens, suggesting that chronic treatment of furan does not alter the initial toxic response. The elevation was comparable to that reported by Fransson-Steen et al. for the daily administration of 15 mg/kg bw for 3 weeks (103 ± 34 units/L) [18].
Histopathological lesions were detected in left liver lobes in all furan-treated animals. The spectrum of microscopic lesions in liver included multifocal hepatocellular necrosis, oval cell hyperplasia, pigment-laden Kupffer cells, increased hepatocyte mitoses with occasional atypical mitosis, cytomegalic and karyomegalic hepatocytes, and reactive leukocyte infiltration in the areas of necrosis. Corn oil-treated livers did not show any abnormal features (Fig. 4A–C). Livers from animals treated with a single dose of furan had a few small foci of hepatocellular necrosis and reactive leukocyte infiltration (Fig. 4D). After the 3-week treatment with furan, the livers showed multi-focal areas of hepatocellular necrosis of increased magnitude admixed with reactive leukocytes, and an accentuated lobular pattern with mid-zonal hepatocytes that were moderately swollen with brightly eosinophilic cytoplasm (Fig. 4E); there were also minimal areas of sub-capsular hepatocellular necrosis. After the 6-week treatment, microscopic lesions in the liver were similar to those found after the treatment for 3 weeks, but most severe with areas of parenchyma collapse. This furan-treated group uniquely showed oval cell hyperplasia, and karyomegalic and cytomegalic hepatocytes. Within the areas of accentuated lobular patterns, there was single-cell necrosis/apoptosis in addition to clearly increased hepatocyte mitosis. A notable finding was the presence of occasional atypical mitotic figures including lag-type mitoses with nonattached condensed chromatin (Fig. 4F, lower inset), hollow metaphase mitoses, and multipolar mitoses, some of which were present in cytomegalic hepatocytes (Fig. 4F). All three furan-treated groups showed infiltrating reactive leukocytes in the areas of hepatocellular necrosis.
Figure 4.
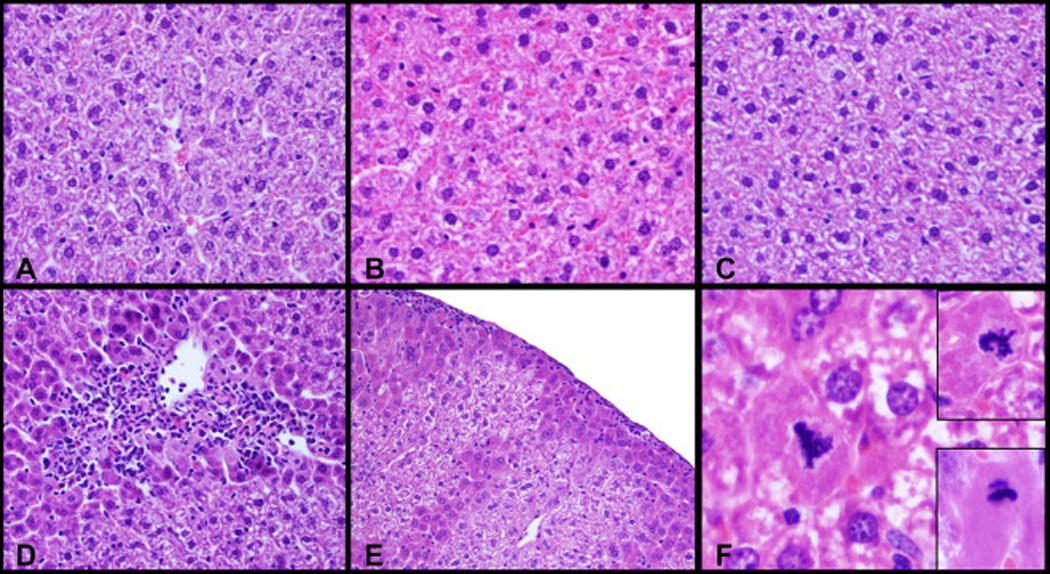
Photomicrographs of mouse left liver-lobe sections stained with haematoxylin and eosin (H&E). Female B6C3F1 mice were treated by gavage with 0 (A–C) or 15 mg/kg bw furan (D–F) in corn oil. The animals were sacrificed 24 h after receiving a single oral dose (A, D), three weekly doses (B, E) or five daily doses for 6 weeks (C, F). Furan treatment caused hepatocellular necrosis admixed with reactive leukocytes (panel D), an accentuated lobular pattern and minimal sub-capsular hepatocellular necrosis admixed with reactive leukocytes (panel E), and occasional atypical mitotic figures including multipolar mitoses in cytomegalic and normal hepatocytes (Panel F, and Panel F upper inset, respectively), and lag-type mitoses with nonattached condensed chromatin (Panel F, lower inset).
The increased hepatocyte proliferation observed by histopathological examination in the group treated for 6 weeks was further confirmed by Ki-67 immunohistochemistry in the liver. Ki-67 immunoreactivity was increased more than two-fold (p < 5 × 10−6) in the livers of mice receiving daily furan treatments during 6 weeks as compared with vehicle controls (Figs. 5 and 6). This change was statistically significant after multiple comparison adjustment. Treatments with a single dose or with three weekly doses did not produce a significant increase in Ki-67 staining.
Figure 5.
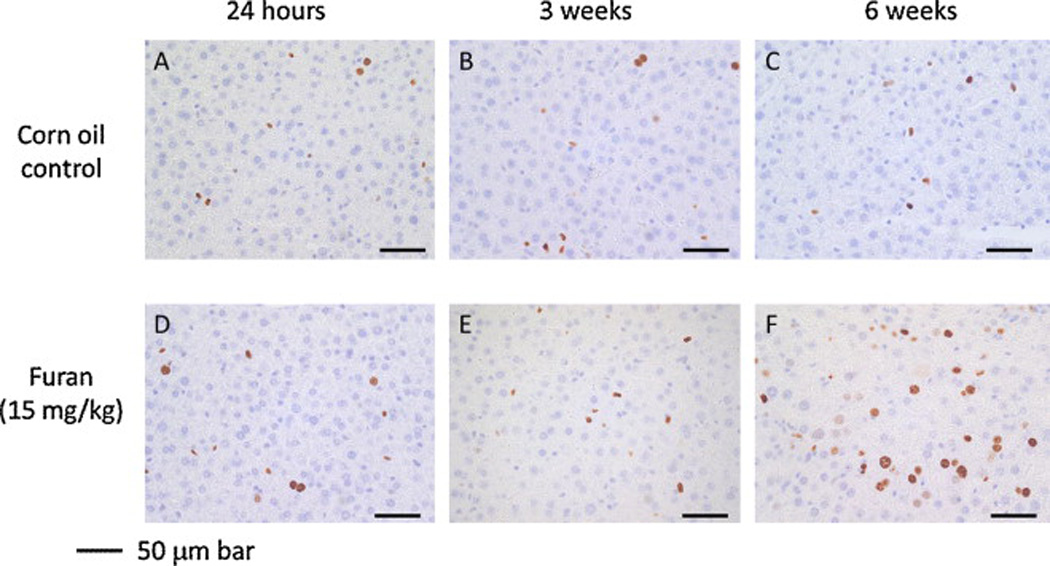
Representative pictures of liver Ki-67-staining following treatment Female B6C3F1 mice were treated by gavage with 0 (A–C) or 15 mg/kg bw furan (D–F) in corn oil by. The animals were sacrificed 24 h after receiving a single oral dose (A, D), three weekly doses (B, E) or five daily doses for 6 weeks (C, F). Livers were removed, fixed in formalin, and imbedded in paraffin. Tissue slices were probed for Ki-67 protein. Photographs were taken at 400× magnification with a Zeiss light microscope. Scale bars represent 50 µm.
Figure 6.
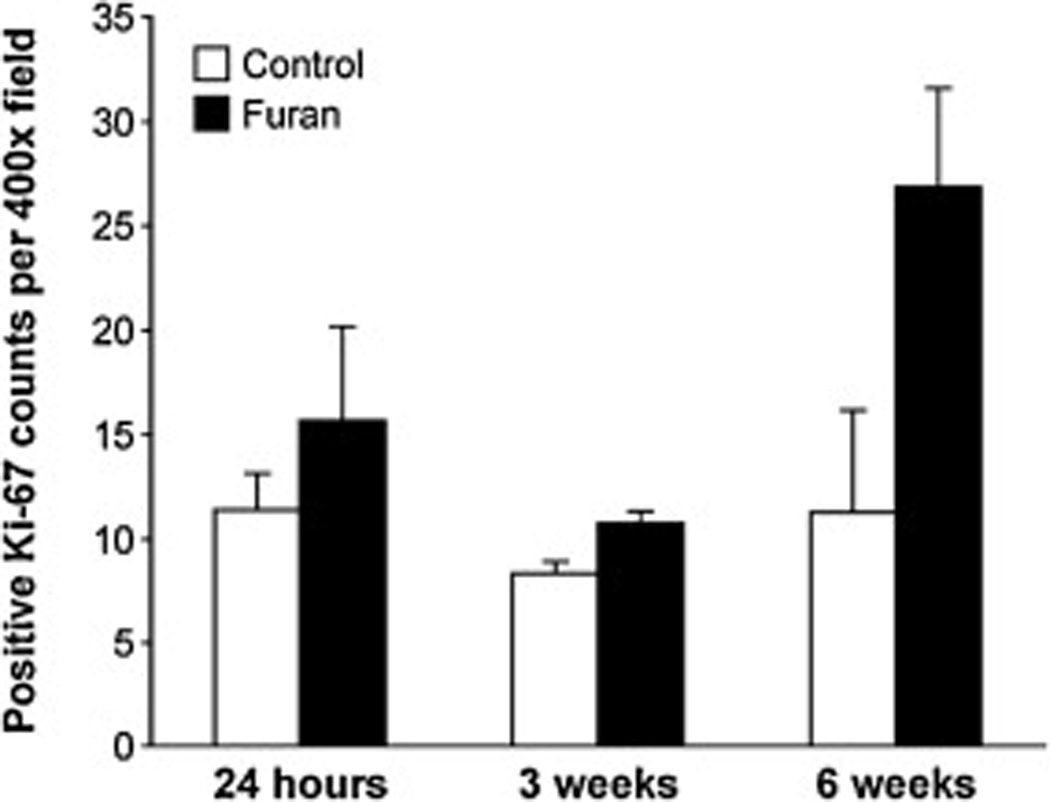
Liver Ki-67 staining in furan-treated mice and corn-oil controls Female B6C3F1 mice were treated by gavage with 0 or 15 mg/kg bw furan in corn oil. The animals were sacrificed 24 h after receiving a single oral dose, three weekly doses or five daily doses for 6 weeks. Livers were removed, fixed in formalin, and imbedded in paraffin. Tissue slices were probed for Ki-67, and photographs were taken at 400× magnification with a light microscope (Fig. 5). Twenty pictures per tissue were examined for positive Ki-67-staining. Data are presented as average number of positive Ki-67 stains per field ±S.D. The levels in furan-treated animals were significantly different from those in the corn oil-treated controls in all treatment groups (24 h, p < 0.02; 3 weeks, p < 2.6 × 10−11; 6 weeks, p < 5.0 × 10−6) as determined by the method of generalized estimating equations.
4. Discussion
The data presented in this report support the hypothesis that the mechanism of action of furan has a genotoxic component. Although furan did not increase the cII transgene mutation-frequency in Big Blue mice, furan-dependent changes in the mutation spectrum were observed. Furan induced small shifts in the cII mutation-spectrum in the 6-week study, but much larger changes in the 3-week study where the total dose of furan was considerably reduced. In both cases, there was a significant decrease in GC to AT transition mutations. In the 3-week study, the proportion of transversion mutations significantly increased, with the largest change occurring for GC to TA mutations. It is interesting to note that GC to TA transversions were a major mutation detected in H-ras in liver tumors from furan-treated mice [27,28].
Our histopathological findings support the hypothesis that the dosing regimen has an influence on the mutation spectrum due to differential toxicity. Microscopic lesions in the liver were more severe with increasing number of doses and longer duration of treatment, similar to the findings in previous reports [19,20]. In contrast, the 3 weekly doses induced a less toxic response. In addition, cell proliferation was greatly enhanced in animals receiving daily furan exposure during 6 weeks. Perhaps, compensatory hyper-proliferation following cytotoxicity led to a more rapid repopulation with undamaged rather than with damage-compromised cells, causing a dilution of damaged cells with non-mutated cells.
The BDA-dependent increase in AT to CG transversions in Big Blue mouse embryonic fibroblasts suggests that this metabolite is a mutagenic compound. In vitro, it reacts with 2′-deoxyadenosine, 2′-deoxycytidine and 2′-deoxyguanosine to form adducts [10,11]. The mutagenic potential of these adducts is unknown, but they have been detected in bacteria at mutagenic concentrations of BDA [15]. The observation that BDA primarily caused an increase in AT to CG transversions in the cII gene suggests that the 2′-deoxyadenosine adduct may have the highest mutagenic potential.
Since the changes in the BDA-associated mutation spectrum are different from those observed in livers of furan-treated mice, BDA may not be directly responsible for the in vivo genotoxic activity of furan. This hypothesis assumes that the cellular response to BDA-derived DNA damage is similar in mouse liver and mouse embryonic fibroblasts. It is also possible that BDA is not the ultimate DNA-reactive species formed in mouse liver, and that it is different in the two biological systems. Further studies characterizing the DNA damage responsible for the mutations observed in mouse liver and mouse embryonic fibroblasts are required.
Alternatively, the mutation spectrum seen after exposure to furan is consistent with an indirect genotoxic mechanism where the toxicity of this chemical generates inflammation-derived reactive species that react with DNA to form mutagenic adducts. The histopathological studies indicate that inflammation occurs during both treatment regimens and is associated with necrotic areas within the liver. These results are consistent with those in other reports [18,19,44]. Our findings are also consistent with previous reports indicating that several weeks of treatment with carcinogenic doses of furan up-regulated genes related to oxidative stress, inflammation and DNA-damage responses in mouse liver [20,45]. Comparable observations have been reported for furan-treated rats [22,46]. Therefore, it is possible that furan induces oxidative stress, which, in turn, results in DNA damage.
The furan-induced cII mutation spectrum is similar to that reported for models of oxidative and inflammatory stress. 8-Oxoguanine, a major DNA adduct resulting from oxidative stress, is generally thought to code for adenine, resulting in GC to TA transversions [47]. Ultraviolet A radiation, which induces significant levels of oxidative DNA damage, increased GC to TA transversions in the cII transgene isolated from Big Blue mouse embryonic epithelial cells [48]. Small tandem base deletions were also induced. Chronic infection with Helicobacter pylori induced gastric mutations in Big Blue transgenic mice, causing an increased frequency of GC to TA, AT to CG and AT to TA mutations in the cII transgene [49]. The mutational spectrum observed after treatment with furan is different from that reported for other non-genotoxic carcinogens: oxepam, phenobarbital and Wyeth 14,643. Oxepam and Wyeth 14,643 increased the percentage of frameshift mutations in the cII transgene, whereas phenobarbital also caused a significant decrease in GC to CG mutations [31]. A comparison between the in vivo cII mutation spectra suggests that the mode of action of furan may be different from that of these three compounds. Future studies will explore the ability of furan to induce oxidative and nitrosative damage to liver DNA.
Furan is, at best, a weak mutagen in the Big Blue system. Similarly, it did not significantly increase H-ras codon-61 mutations in mice [50]. Furan also did not induce a change in cII gene mutation frequency in Big Blue rats [51]. One disadvantage of the Big Blue mutagenesis assay is that it has limited sensitivity to clastogens [52]. Furan induced chromosomal aberrations but no sister chromatid exchange in bone marrow of mice following intraperitoneal administration [2]. The detection of DNA strand-breaks in splenocytes following oral exposure of mice to carcinogenic doses of furan required cell division [26]. We observed atypical mitotic figures with some degree of abnormal arrangement of chromosomes and spindle fibers, such as lag-type mitosis with non-attached condensed chromatin, multipolar mitoses with atypical configuration of the equatorial plate, and hollow metaphase mitoses, as described previously [53,54]. The presence of atypical mitotic figures in hepatocytes exposed to the 6-week treatment regimen (Fig. 4F) is suggestive of abnormal cell division, and is consistent with previous findings of furan-induced polyploidy and micronucleus formation [26]. These events may lead to mitotic chromosomal instability, and contribute to the tumorigenic activity of furan [55]. If these cells survive and/or divide further, transformation into neoplastic hepatocytes may eventually occur.
5. Conclusions
In summary, these experiments suggest that furan is a weak mutagen in mouse liver, inducing primarily GC to TA transversions in the cII transgene. The type of DNA damage causing these mutations requires further investigation. These studies also suggest that toxicity at carcinogenic doses masks the genotoxic effects of furan. However, it also is likely that toxicity is important for the carcinogenic effects of furan since it may be critical for inducing DNA damage in this particular animal model. These data are consistent with a mechanism of carcinogenesis in which chronic toxicity with subsequent inflammation and secondary cell proliferation triggers cancer development in furan-exposed rodents. In addition, exposure to furan resulted in aberrant mitosis, which also may contribute to the carcinogenic properties of this chemical.
Supplementary Material
Scheme 1.
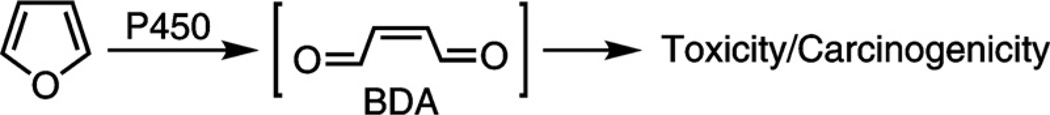
Metabolic activation of furan to the reactive metabolite, BDA.
Acknowledgments
We thank Leah Gates and Matilde Sullivan for their assistance with the animal studies, Dr. Ding Lu for the preparation of BDA, and Wieslawa Kosinska for her help with the cII mutagenesis assay. The sequencing analysis was performed by the BioMedical Genomics Center at the University of Minnesota. The Sequencher® software used for sequence alignments was provided by Minnesota Super-computing Institute for Advanced Computational Research at the University of Minnesota. We also thank Josh Parker and Paula Overn of the Masonic Cancer Center Comparative Pathology Shared Resource for their technical assistance.
Funding information
This work was supported by the National Institute of Environmental Health Sciences at the National Institutes of Health [R01 ES10577]. ANT was supported by a Diversity supplement to this grant. MH was supported by an American Recovery and Reinvestment Act supplement to this grant as well as by an Undergraduate Research Opportunity Program Grant from the University of Minnesota. The University of Minnesota Masonic Cancer Center Analytical Biochemistry and Biostatistics Shared Resources are supported in part by the National Cancer Institute at the National Institutes of Health [CCSG CA-77598]. The Biostatistics Shared Resource is also funded in part by the National Center for Research Resources of the National Institutes of Health to the University of Minnesota Clinical and Translational Science Institute.
Footnotes
Conflict of interest
No conflicts.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.mrgentox.2014.04.024.
Contributor Information
Ashley N. Terrell, Email: terre062@umn.edu.
Mailee Huynh, Email: huynh082@umn.edu.
Alex E. Grill, Email: gril0043@umn.edu.
Ramesh C. Kovi, Email: rckovi@umn.edu.
M. Gerard O’Sullivan, Email: gos@umn.edu.
Joseph B. Guttenplan, Email: Joseph.Guttenplan@nyumc.org.
Yen-Yi Ho, Email: yho@umn.edu.
Lisa A. Peterson, Email: peter431@umn.edu.
References
- 1.International Agency for Research on Cancer. Furan, Dry Cleaning, Some Chlorinated Solvents and Other Industrial Chemicals. IARC: Lyon, France; 1995. p. 393. [PMC free article] [PubMed] [Google Scholar]
- 2.National Toxicology Program. Toxicology and Carcinogenesis Studies of Furan in F344/N Rats and B6C3F1 Mice. Research Triangle Park, NC: US Department of Health and Human Services, Public Health Service, National Institutes of Health; 1993. [Google Scholar]
- 3.National Toxicology Program. 12th Report on Carcinogens. Washington DC: US Department of Health and Human Services; 2011. [Google Scholar]
- 4.Burka LT, Washburn KD, Irwin RD. Disposition of [14C]furan in the male F344 rat. J. Toxicol. Environ. Health. 1991;34:245–257. doi: 10.1080/15287399109531564. [DOI] [PubMed] [Google Scholar]
- 5.Parmar D, Burka LT. Studies on the interaction of furan with hepatic cytochrome P-450. J. Biochem. Toxicol. 1993;8:1–9. doi: 10.1002/jbt.2570080103. [DOI] [PubMed] [Google Scholar]
- 6.Carfagna MA, Held SD, Kedderis GL. Furan-induced cytolethality in isolated rat hepatocytes: correspondence with in vivo dosimetry. Toxicol. Appl. Pharmacol. 1993;123:265–273. doi: 10.1006/taap.1993.1245. [DOI] [PubMed] [Google Scholar]
- 7.Kedderis GL, Carfagna MA, Held SD, Batra R, Murphy JE, Gargas ML. Kinetic analysis of furan biotransformation by F-344 rats in vivo and in vitro. Toxicol. Appl. Pharmacol. 1993;123:274–282. doi: 10.1006/taap.1993.1246. [DOI] [PubMed] [Google Scholar]
- 8.Mugford CA, Carfagna MA, Kedderis GL. Furan-mediated uncoupling of hepatic oxidative phosphorylation in Fischer-344 rats: an early event in cell death. Toxicol. Appl. Pharmacol. 1997;144:1–11. doi: 10.1006/taap.1997.8121. [DOI] [PubMed] [Google Scholar]
- 9.Chen LJ, Hecht SS, Peterson LA. Characterization of amino acid and glutathione adducts of cis-2-butene-1,4-dial, a reactive metabolite of furan. Chem. Res. Toxicol. 1997;10:866–874. doi: 10.1021/tx9700174. [DOI] [PubMed] [Google Scholar]
- 10.Byrns MC, Predecki DP, Peterson LA. Characterization of nucleoside adducts of cis-2-butene-1,4-dial, a reactive metabolite of furan. Chem. Res. Toxicol. 2002;15:373–379. doi: 10.1021/tx0101402. [DOI] [PubMed] [Google Scholar]
- 11.Byrns MC, Vu CC, Peterson LA. The formation of substituted 1,N6-etheno-2′-deoxyadenosine and 1,N2-etheno-2′-deoxyguanosine adducts by cis-2-butene-1,4-dial, a reactive metabolite of furan. Chem. Res. Toxicol. 2004;17:1607–1613. doi: 10.1021/tx049866z. [DOI] [PubMed] [Google Scholar]
- 12.Peterson LA, Cummings ME, Vu CC, Matter BA. Glutathione trapping to measure microsomal oxidation of furan to cis-2-butene-1,4-dial. Drug Metab. Dispos. 2005;33:1453–1458. doi: 10.1124/dmd.105.004432. [DOI] [PubMed] [Google Scholar]
- 13.Kellert M, Brink A, Richter I, Schlatter J, Lutz WK. Tests for genotoxicity and mutagenicity of furan and its metabolite cis-2-butene-1,4-dial in L5178Y tk+/− mouse lymphoma cells. Mutat. Res. 2008;657:127–132. doi: 10.1016/j.mrgentox.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Marinari UM, Ferro M, Sciaba L, Finollo R, Bassi AM, Brambilla G. DNA-damaging activity of biotic and xenobiotic aldehydes in chinese hamster ovary cells. Cell Biochem. Funct. 1984;2:243–248. doi: 10.1002/cbf.290020411. [DOI] [PubMed] [Google Scholar]
- 15.Byrns MC, Vu CC, Neidigh JW, Abad JL, Jones RA, Peterson LA. Detection of DNA adducts derived from the reactive metabolite of furan, cis-2-butene-1,4-dial. Chem. Res. Toxicol. 2006;19:414–420. doi: 10.1021/tx050302k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson LA, Naruko KC, Predecki D. A reactive metabolite of furan, cis-2-butene-1,4-dial, is mutagenic in the Ames assay. Chem. Res. Toxicol. 2000;13:531–534. doi: 10.1021/tx000065f. [DOI] [PubMed] [Google Scholar]
- 17.Wilson DM, Goldsworthy TL, Popp JA, Butterworth BE. Evaluation of genotoxicity, pathological lesions, and cell proliferation in livers of rats and mice treated with furan. Environ. Mol. Mutagen. 1992;19:209–222. doi: 10.1002/em.2850190305. [DOI] [PubMed] [Google Scholar]
- 18.Fransson-Steen R, Goldsworthy TL, Kedderis GL, Maronpot RR. Furan-induced liver cell proliferation and apoptosis in female B6C3F1 mice. Toxicology. 1997;118:195–204. doi: 10.1016/s0300-483x(97)03618-4. [DOI] [PubMed] [Google Scholar]
- 19.Moser GJ, Foley J, Burnett M, Goldsworthy TL, Maronpot R. Furan-induced dose-response relationships for liver cytotoxicity, cell proliferation, and tumorigenicity (furan-induced liver tumorigenicity) Exp. Toxicol. Pathol. 2009;61:101–111. doi: 10.1016/j.etp.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Cordelli E, Leopardi P, Villani P, Marcon F, Macri C, Caiola S, Sinis-calchi E, Conti L, Eleuteri P, Malchiodi-Albedi F, Crebelli R. Toxic and genotoxic effects of oral administration of furan in mouse liver. Mutagenesis. 2010;25:305–314. doi: 10.1093/mutage/geq007. [DOI] [PubMed] [Google Scholar]
- 21.Hickling KC, Hitchcock JM, Chipman JK, Hammond TG, Evans JG. Induction and progression of cholangiofibrosis in rat liver injured by oral administration of furan. Toxicol. Pathol. 2010;38:213–229. doi: 10.1177/0192623309357945. [DOI] [PubMed] [Google Scholar]
- 22.Hickling KC, Hitchcock JM, Oreffo V, Mally A, Hammond TG, Evans JG, Chipman JK. Evidence of oxidative stress and associated DNA damage, increased proliferative drive, and altered gene expression in rat liver produced by the cholangiocarcinogenic agent furan. Toxicol. Pathol. 2010;38:230–243. doi: 10.1177/0192623309357946. [DOI] [PubMed] [Google Scholar]
- 23.Mortelmans K, Haworth S, Lawlor T, Speck W, Tainer B, Zeiger E. Salmonella mutagenicity tests. II. Results from the testing of 270 chemicals. Environ. Mutagen. 1986;7(suppl. l):1–119. [PubMed] [Google Scholar]
- 24.Glatt H, Schneider H, Liu Y. V79-hCYP2E1-hSULT1A1, a cell line for the sensitive detection of genotoxic effects induced by carbohydrate pyrolysis products and other food-borne chemicals. Mutat. Res. 2005;580:41–52. doi: 10.1016/j.mrgentox.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Durling LJ, Svensson K, Abramsson-Zetterberg L. Furan is not genotoxic in the micronucleus assay in vivo or in vitro. Toxicol. Lett. 2007;169:43–50. doi: 10.1016/j.toxlet.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 26.Leopardi P, Cordelli E, Villani P, Cremona TP, Conti L, De Luca G, Crebelli R. Assessment of in vivo genotoxicity of the rodent carcinogen furan: evaluation of DNA damage and induction of micronuclei in mouse splenocytes. Mutagenesis. 2010;25:57–62. doi: 10.1093/mutage/gep043. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds SH, Stowers SJ, Patterson RM, Maronpot RR, Aaronson SA, Anderson MW. Activated oncogenes in B6C3F1 mouse liver tumors: implications for risk assessment. Science. 1987;237:1309–1316. doi: 10.1126/science.3629242. [DOI] [PubMed] [Google Scholar]
- 28.Johansson E, Reynolds S, Anderson M, Maronpot R. Frequency of Ha-ras-1 gene mutations inversely correlated with furan dose in mouse liver tumors. Mol. Carcinog. 1997;18:199–205. doi: 10.1002/(sici)1098-2744(199704)18:4<199::aid-mc3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 29.Heddle JA, Dean S, Nohmi T, Boerrigter M, Casciano D, Douglas GR, Glickman BW, Gorelick NJ, Mirsalis JC, Martus HJ, Skopek TR, Thybaud V, Tindall KR, Yajima N. In vivo transgenic mutation assays. Environ. Mol. Mutagen. 2000;35:253–259. doi: 10.1002/(sici)1098-2280(2000)35:3<253::aid-em11>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 30.Dean SW, Brooks TM, Burlinson B, Mirsalis J, Myhr B, Recio L, Thybaud V. Transgenic mouse mutation assay systems can play an important role in regulatory mutagenicity testing in vivo for the detection of site-of-contact mutagens. Mutagenesis. 1999;14:141–151. doi: 10.1093/mutage/14.1.141. [DOI] [PubMed] [Google Scholar]
- 31.Singh VK, Ganesh L, Cunningham ML, Shane BS. Comparison of the mutant frequencies and mutation spectra of three non-genotoxic carcinogens, oxazepam, phenobarbital, and Wyeth 14,643, at the lambda cII locus in Big Blue transgenic mice. Biochem. Pharmacol. 2001;62:685–692. doi: 10.1016/s0006-2952(01)00722-5. [DOI] [PubMed] [Google Scholar]
- 32.Hohn B. In vitro packaging of lambda and cosmid DNA. Methods Enzymol. 1979;68:299–309. doi: 10.1016/0076-6879(79)68021-7. [DOI] [PubMed] [Google Scholar]
- 33.Butterworth BE, Sprankle CS, Goldsworthy SM, Wilson DM, Goldsworthy TL. Expression of myc, fos, and Ha-ras in the livers of furan-treated F344 rats and B6C3F1 mice. Mol. Carcinog. 1994;9:24–32. doi: 10.1002/mc.2940090106. [DOI] [PubMed] [Google Scholar]
- 34.Haseman JK, Hailey JR, Morris RW. Spontaneous neoplasm incidences in Fischer 344 rats and B6C3F1 mice in two-year carcinogenicity studies: a National Toxicology Program update. Toxicol. Pathol. 1998;26:428–441. doi: 10.1177/019262339802600318. [DOI] [PubMed] [Google Scholar]
- 35.Shane BS, Smith-Dunn DL, de Boer JG, Glickman BW, Cunningham ML. Mutant frequencies and mutation spectra of dimethylnitrosamine (DMN) at the lacI and cII loci in the livers of Big Blue transgenic mice. Mutat. Res. 2000;452:197–210. doi: 10.1016/s0027-5107(00)00081-6. [DOI] [PubMed] [Google Scholar]
- 36.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sobol RW, Watson DE, Nakamura J, Yakes FM, Hou E, Horton JK, Ladapo J, Van Houten B, Swenberg JA, Tindall KR, Samson LD, Wilson SH. Mutations associated with base excision repair deficiency and methylation-induced genotoxic stress. Proc. Natl. Acad. Sci. U.S.A. 2002;99:6860–6865. doi: 10.1073/pnas.092662499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies R, Gant TW, Smith LL, Styles JA. Tamoxifen induces G:C->T:A mutations in the cII gene in the liver of lambda/lacI transgenic rats but not at 5′-CpG-3′ dinucleotide sequences as found in the lacI transgene. Carcinogenesis. 1999;20:1351–1356. doi: 10.1093/carcin/20.7.1351. [DOI] [PubMed] [Google Scholar]
- 39.Adams WT, Skopek TR. Statistical test for the comparison of samples from mutational spectra. J. Mol. Biol. 1987;194:391–396. doi: 10.1016/0022-2836(87)90669-3. [DOI] [PubMed] [Google Scholar]
- 40.Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, Fazzari M, Jin B, Brown AM, Lipkin M, Edelmann W. Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14937–14942. doi: 10.1073/pnas.232579299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunson DB, Tindall KR. Bayesian analysis of mutational spectra. Genetics. 2000;156:1411–1418. doi: 10.1093/genetics/156.3.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeger SL, Liang KY, Albert PS. Models for longitudinal data: a generalized estimating equation approach. Biometrics. 1988;44:1049–1060. [PubMed] [Google Scholar]
- 43.Sandercock LE, Hahn JN, Li L, Luchman HA, Giesbrecht JL, Peterson LA, Jirik FR. Mgmt deficiency alters the in vivo mutational spectrum of tissues exposed to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Carcinogenesis. 2008;29:866–874. doi: 10.1093/carcin/bgn030. [DOI] [PubMed] [Google Scholar]
- 44.Gill S, Kavanagh M, Barker M, Weld M, Vavasour E, Hou Y, Cooke GM. Sub-chronic oral toxicity study of furan in B6C3F1 mice. Toxicol. Pathol. 2011;39:787–794. doi: 10.1177/0192623311412980. [DOI] [PubMed] [Google Scholar]
- 45.Jackson AF, Williams A, Recio L, Waters MD, Lambert IB, Yauk CL. Case study on the utility of hepatic global gene expression profiling in the risk assessment of the carcinogen furan. Toxicol. Appl. Pharmacol. 2014;274:63–77. doi: 10.1016/j.taap.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 46.Ding W, Petibone DM, Latendresse JR, Pearce MG, Muskhelishvili L, White GA, Chang CW, Mittelstaedt RA, Shaddock JG, McDaniel LP, Doerge DR, Morris SM, Bishop ME, Manjanatha MG, Aidoo A, Heflich RH. In vivo genotoxicity of furan in F344 rats at cancer bioassay doses. Toxicol. Appl. Pharmacol. 2012;261:164–171. doi: 10.1016/j.taap.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 47.Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 48.Besaratinia A, Synold TW, Xi B, Pfeifer GP. G-to-T transversions and small tandem base deletions are the hallmark of mutations induced by ultraviolet a radiation in mammalian cells. Biochemistry. 2004;43:8169–8177. doi: 10.1021/bi049761v. [DOI] [PubMed] [Google Scholar]
- 49.Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124:1408–1419. doi: 10.1016/s0016-5085(03)00266-x. [DOI] [PubMed] [Google Scholar]
- 50.Banda M, Recio L, Parsons BL. ACB-PCR measurement of spontaneous and furan-induced H-ras codon 61 CAA to CTA and CAA to AAA mutation in B6C3F1 mouse liver. Environ. Mol. Mutagen. 2013;54:659–667. doi: 10.1002/em.21808. [DOI] [PubMed] [Google Scholar]
- 51.McDaniel LP, Ding W, Dobrovolsky VN, Shaddock JG, Jr, Mittelstaedt RA, Doerge DR, Heflich RH. Genotoxicity of furan in Big Blue rats. Mutat. Res. 2012;742:72–78. doi: 10.1016/j.mrgentox.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 52.Lambert IB, Singer TM, Boucher SE, Douglas GR. Detailed review of transgenic rodent mutation assays. Mutat. Res. 2005;590:1–280. doi: 10.1016/j.mrrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 53.Scarpelli DG, Von Haam E. A study of mitosis in cervical epithelium during experimental inflammation and carcinogenesis. Cancer Res. 1957;17:880–884. [PubMed] [Google Scholar]
- 54.Van Leeuwen AM, Pieters WJ, Hollema H, Burger MP. Atypical mitotic figures and the mitotic index in cervical intraepithelial neoplasia. Virchows Arch. 1995;427:139–144. doi: 10.1007/BF00196518. [DOI] [PubMed] [Google Scholar]
- 55.Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat. Rev. Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.