

Abstract
While the SCN controls the circadian clock, further evidence suggests the existence of a food-entrainable oscillator (FEO) that links behavior to changes in food availability such as during restricted feeding (RF). We found that the activity of AgRP/NPY neurons changed rhythmically during RF suggesting that these neurons are a component of the FEO. We next ablated AgRP/NPY neurons in neonates with diphtheria toxin resulting in the loss of ∼50% of AgRP/NPY neurons. Body weight and food intake were unchanged in adult animals after neonatal ablation, as were the responses to leptin treatment, leptin withdrawal, food deprivation and ghrelin treatment. However, ablated animals showed 30% mortality within 4 days of RF. Moreover, the recovery of body weight and food intake in surviving animals lagged behind controls with an absence of food anticipatory activity even after three days. These findings identify AgRP/NPY neurons as a key cellular component of the food-entrained oscillator.
Keywords: AgRP, Hypothalamus, Food anticipatory activity (FAA), Food entrainable oscillator (FEO)
1. Introduction
Molecular clocks play a key role in coordinating physiology and behavior with environmental cues [1]. The circadian oscillator in the suprachiasmatic nucleus (SCN) is regulated by environmental light/dark cues conveyed from the retina and controls the sleep wake cycle and other behaviors in mammals [2,3]. At the molecular level, this circadian clock(s) relies on daily oscillations in the transcription and translation of set of evolutionarily conserved clock genes [4]. In the SCN, circadian oscillations in clock gene expression change cellular activity within this structure to control circadian rhythms [5,6]. Circadian rhythms in clock gene expression are also observed in many brain regions outside the SCN [7] as well as in peripheral, non-neuronal tissues [8,9].
While a light entrained oscillator regulates sleep wake cycle and other processes, this is not the only biological clock. When food availability is restricted to a single period during a fixed time of the day, over the course of a few days animals adapt to this schedule and increase their feeding during the window of food availability, even if it is in the light phase, a time when animals do not normally eat. . In addition, in this restricted feeding paradigm animals show a marked increase in locomotor activity shortly before the onset of the time when food is made available. This increased activity is referred to as food anticipatory activity, FAA [10,11]. FAA is also associated with increase in body temperature, adrenal corticosterone secretion and gastrointestinal motility. This biological rhythm, timed to a window of food availability is unaffected by SCN ablation, confirming the existence of a food-entrainable oscillator (FEO) that is separate from the SCN [12,13]. Moreover previous reports have demonstrated that restricted feeding can entrain and shift the circadian rhythms in peripheral tissues as well as other brain areas outside of the SCN even when oscillation in the SCN remains phase-locked to the light/dark cycle [14–17]. These findings show that the timing of food availability can cause a phase uncoupling among multiple circadian oscillators outside the SCN and can even dominate the circadian oscillator. However, while the demonstration that FAA and other physiologic responses develop after restricted feeding, the anatomic sites and neuronal populations that comprise the food entrained oscillator have not been elucidated.
Recently, we employed a new profiling technology to identify cell types in brain that are activated in response to an acute or chronic stimulus [18]. We used this method to identify neural populations in the hypothalamus that are activated when the food availability of animals is restricted to a 3 h window during the light phase. In this previous study, we found that dynorphin neurons in the DMH play a role in the response to restricted feeding. We also found that AgRP/NPY neurons in the arcuate nucleus are activated when animals are subjected to a restricted feeding paradigm. AgRP neurons in the arcuate nucleus of the hypothalamus co-express NPY [19] and regulate food intake and metabolism. Optogenetic activation of these neurons promote food intake [20,21]. These neurons also express the ghrelin receptor [22] and are responsive to plasma ghrelin which has been shown to rise prior to meal times and has been hypothesized to promote feeding during RF [23–25].
We thus set out to evaluate the possibility that AGRP neurons also play a role in the response to restricted feeding and the development of FAA. In this report, we confirm a key role for AgRP/NPY neurons in the response to restricted feeding suggesting that it is a key neural component of the food entrainable oscillator.
2. Methods and materials
2.1. Animal treatment
All experiments were carried out in accordance to regulations of the Institutional Animal Care and Use Committee at the Rockefeller University. All transgenic mice used in this study were previously published. The following line was used to express Cre recombinase in AgRP cells: AgRP-IRES-Cre (Jackson Labs Stock 012899, Agrptm1(cre)Lowl/J). The following reporter line was used, where a lox-STOP-lox sequence prevents expression of the reporter in the absence of Cre recombinase: ROSA-loxSTOPlox-tdTomato (Jackson Labs Stock 007909, B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J). AgRP DTR animals were generated by crossing heterozygous AgRP-IRES-Cre animals with animals homozygous for ROSA-loxSTOPlox-iDTR (Jackson Labs Stock 007900, C57BL/6-Gt(ROSA)26Sortm1(HBEGF)Awai/J). Animals heterozygous for AgRP-IRES-Cre and ROSA-loxSTOPlox-iDTR were termed AgRP DTR and animals heterozygous for only ROSA-loxSTOPlox-iDTR were termed DTR. Unless otherwise stated, mice were group housed under a standard 12 h light/dark cycle (lights on from 8:00 A.M. to 8:00 P.M.) and fed regular chow (LabDiet 5053). To ablate AgRP neurons, postnatal day 3 (P3) litters were injected once with diptheria toxin [50 ng/g, subcutaneous (s.c), Sigma–Aldrich D0564]. For fasting experiments, animals were transferred to new cages and food deprived for at least 18 h before they were either perfused for immunohistochemistry or re-fed. For ghrelin experiments, animals were given ghrelin (66 μg, intraperitoneal, i.p, Tocris Bioscience #1465) and subsequent food intake measured. Food was removed from cages for animals used for immunohistochemistry. For scheduled feeding, mice were allowed access to food between ZT4 and ZT7 or between ZT15 and ZT18 each day for 14 days and body weight and food intake were measured each day.
2.2. Leptin treatment
Miniosmotic pumps (Alzet, 2001) with mean pump rate of 0.98 μl/h were filled with leptin (Amylin Pharmaceutical) to achieve dose of 2.5 μg/h for 8 days. Pumps were incubated in sterile PBS at 37 °C for at least 6 h before subcutaneous (s.c) implantation into animals. Animals were anesthetized with isoflurane when pumps were implanted or removed.
2.3. Locomotion analysis
10–12 weeks old male AgRP DTR and DTR mice were acclimatized to the experimental chambers for at least 24 h prior to data collection using the Oxymax Lab Animal Monitoring System (Columbus Instruments). Only ambulatory beam breaks in the x and y axis were included in the analysis.
2.4. Immunohistochemistry
Mice were first anesthetized with isoflurane followed by transcardial perfusion with PBS and then 10% formalin. Brains were dissected, incubated in 10% formalin overnight at 4 °C and 40 μm sections were prepared using a vibratome. Free floating sections were blocked for 1 h at room temperature in blocking buffer (PBS, 0.1% Triton-X, 2% goat serum, 3% BSA) and then incubated overnight at 4 °C with primary antibodies. The next day, sections were washed in washing buffer (PBS, 0.1% Triton-X) 3 times for 20 min, incubated with dye-conjugated secondary antibodies at 1:1000 for 1 h at room temperature, washed in washing buffer and then mounted. Primary antibodies used were rabbit anti-NPY (1:2000, Bachem T4070) and rabbit anti-cfos (Santa Cruz, sc52).
2.5. Microscopy and quantification
Stained brain sections were imaged using Zeiss LSM 510 laser scanning confocal microscope. All parameters were kept constant for quantification purposes. Fluorescence intensity and cell counts were carried out using ImageJ.
2.6. Statistical analysis
Data present as mean ± standard error of the mean (SEM). Student's t test were used to determine significance of the difference of the mean between AgRP DTR and control animals. p < 0.05 represented statistical significance.
3. Results
3.1. AgRP neurons are activated during restricted feeding (RF)
Although the role of AgRP neurons in metabolic homeostasis is well studied, their role in circadian-regulated feeding behavior has not. We first used cfos expression to test whether these neurons are activated by restricted feeding. We mated AgRP-Cre mice to a lox-STOP-lox tdTomato reporter line to generate AgRP tdTomato animals that express the fluorescent reporter tdTomato only in AgRP cells. These animals were then subjected to a RF paradigm in which animals were only allowed access to food during a 3 h window between ZT 4 and ZT 7. As mentioned, mice normally eat between ZT 12–24. After 10 days of RF, animals were perfused at different times during the day and the expression of the immediate early gene, cfos was probed using immunohistochemistry. cfos expression in AgRP neurons was low 2 h before the start of the scheduled feeding window but increased thereafter peaking at ZT 4 just prior to food being made available (Figure 1A–F, M). cfos expression in these neurons then fell during the feeding window, and remained low 2 h after the feeding window ended (Figure 1G–M). However, if food was withheld during ZT 4 and ZT 7 in fully trained animals, the number of AgRP neurons that expressed cfos continued to rise during the feeding window and was still increasing 2 h after the feeding window i.e.; at ZT 9 (Figure 1M–S). These observations suggest that the activity of AgRP neurons is synchronized with food availability and that they might play a role in the behavioral response to scheduled feeding.
Figure 1.
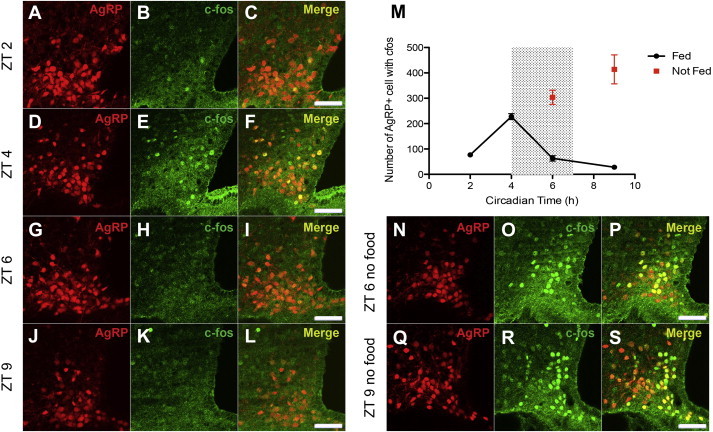
Expression of c-fos in AgRP neurons during restricted feeding. (A–C) 2 h before meal time, few AgRP neurons expressed cfos (77 ± 2.646). (D–F) At the scheduled start of meal time, the number of AgRP neurons that expressed c-fos peaked (227.7 ± 11.62). (G–L) The number of AgRP neurons that expressed c-fos continued to decrease 2 h into the meal window and 2 h after the end of meal (63 ± 10.60 and 28 ± 4.726 respectively). (M) Quantification of colocalization of AgRP and cfos over time with (black) or without (red) access to food. (N–S) When no food was presented, the number of c-fos positive AgRP neurons continued to increase 2 h into the feeding window and 2 h after the scheduled end of meal (304 ± 28 and 414 ± 57 respectively). n = 3 for each time point. Scale bar = 50 μm.
3.2. Ablation of AgRP neurons
To study the functional role AgRP neurons in scheduled feeding, we ablated these neurons by generating mice that expressed the human diptheria toxin receptor only in AgRP neurons by mating AgRP-Cre animals with a lox-STOP-lox iDTR line. We then injected DTX (50 ng/g) subcutaneously into neonatal animals. We used neonatal animals because prior reports showed that injection into adult animals resulted in profound hypophagia and death [26,27]. In contrast, these same reports showed that neonatal animals develop normally and fail to show any phenotypic effects after neonatal AgRP ablation suggesting that compensatory neural mechanisms enable the neonatal animals to maintain normal food intake when fed ad libitum. Consistent with previous reports, there was a substantial decrease in the number of AgRP neurons after administration of diptheria toxin (DTX) to neonatal animals [26,27] (Figure 2A–L). Subcutaneous injection of DTX at postnatal day 3 led to loss of more than 50% of AgRP neurons on postnatal day 7 with a similarly marked decrease in NPY immunoreactivity in AgRP DTR animals compared to controls (Figure 2A–C). NPY is co-expressed by the vast majority of AgRP neurons [19].
Figure 2.
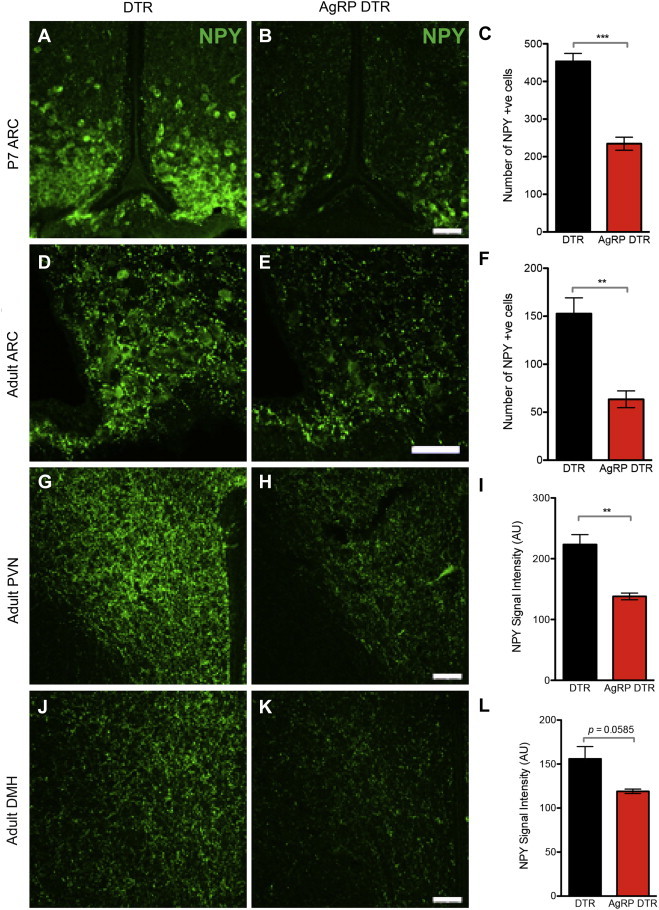
Ablation of AgRP neurons. (A–C) NPY immunostaining in the arcuate nucleus (ARC) at postnatal day 7 in control vs. AgRP DTR animals (DTR 453 ± 21.13 vs AgRP DTR 234.5 ± 17.33, p < 0.001). (D–E) Loss of NPY immunostaining in the ARC of 8-week-old AgRP DTR animals (DTR 152.8 ± 16.49 vs AgRP DTR 63.33 ± 8.743, p < 0.01). Decreased NPY signal intensity in the paraventricular nucleus (PVN) in adult AgRP DTR animals (DTR 223.3 ± 16.27 vs AgRP DTR 138 ± 5.508, p < 0.01). (J–L) Decreased NPY signal intensity in the dorsal medial hypothalamic nucleus (DMH) in adult AgRP DTR animals (DTR 156 ± 13.87 vs AgRP DTR 119 ± 2.517, p = 0.0585). n ≥ 3 in each group. Scale bar = 50 μm.
The aforementioned prior reports did not evaluate the effect of neonatal AgRP neural ablation on the number of AgRP neurons in adults. We found that animals receiving DTX at 3 days of age still showed decreased NPY immunoreactivity in 8-week-old animals (Figure 2D–F). The number of NPY fibers was similar in neonatal and adult animals after neonatal AgRP ablation suggesting that very little regeneration of AgRP neurons took place after DTX treatment. We also found a marked decrease in NPY projections in the paraventricular hypothalamic nucleus (PVN) and the dorsal medial hypothalamic nucleus (DMH) (Figure 2G–L), sites to which AGRP neurons are known to project. Similarly we did not observe a significant change in the number of AgRP projections in adult vs. neonatal animals after neonatal ablation.
3.3. Phenotypic assessment of AgRP DTR animals
Previous studies of neonatal animals after AgRP ablation have shown that the food intake is normal, in contrast to AgRP ablation of adult animals which leads to extreme anorexia and death [26]. Consistent with these reports, we found that neonatal ablation of AgRP neurons did not alter viability. Body weight (Figure 3A) and food intake (Figure 3B) of both male and female neonatal ablated animals were comparable to their littermate controls. Since AgRP neurons express leptin receptor and are known leptin targets [28], we next assayed the leptin responsiveness of AgRP DTR animals after treatment with high dose leptin (2.5 μg/h) via osmotic pumps for 8 days. The extent of weight loss and decrease of food intake following high dose leptin was similar in AgRP DTR animals vs. littermate controls (Figure 3C–D). We also tested whether the response to hypoleptinemia was altered by measuring food intake after removing the leptin pumps from AgRP ablated and control animals. We found that after withdrawal of high dose leptin treatment, the increase food intake and weight was similar in animals with neonatal AgRP ablation vs. controls (Figure 3C–D).
Figure 3.
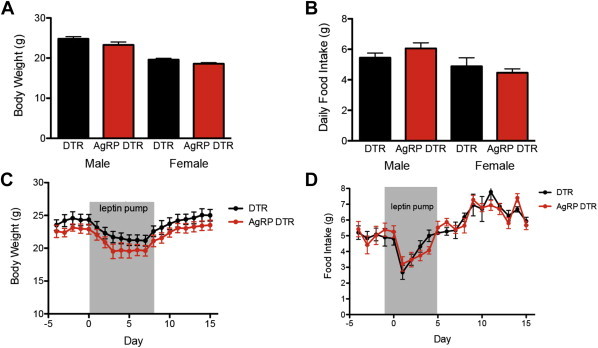
Metabolic phenotype after loss of AgRP neurons. (A) Body weight of 8 week old male and female animals (Male: DTR 24.82 ± 0.5274 vs AgRP DTR 23.3 ± 0.701, Female: DTR 19.62 ± 0.3135 vs AgRP DTR 18.58 ± 0.2763). (B) Daily food intake of 8 week old male and female animals (Male: DTR 5.45 ± 0.2982 vs AgRP DTR 6.057 ± 0.3677, Female: DTR 4.883 ± 0.5588 vs AgRP DTR 4.463 ± 0.2528). (C, D) Body weight and food intake in response to high dose leptin and subsequent leptin withdrawal. n ≥ 7 in each group.
AgRP neurons become activated when animals are food restricted (Figure 4A–F) so we next assayed the response of AgRP DTR animals to a 24 h fast. We first assayed cfos expression in these neurons after food restriction and found that there were significantly fewer cfos positive cells in the arcuate nucleus of AgRP DTR animals compared to controls (Figure 4G–I). Note the reduction in the number of cells expressing cfos was consistent with the loss of AGRP neurons suggesting that the cfos response in surviving cells was unchanged. Despite of the loss of AgRP neurons and the similarly low number of cfos positive cells in AgRP DTR animals, the food intake of these animals after an 18 h fast was unaltered compared to controls (Figure 4J).
Figure 4.
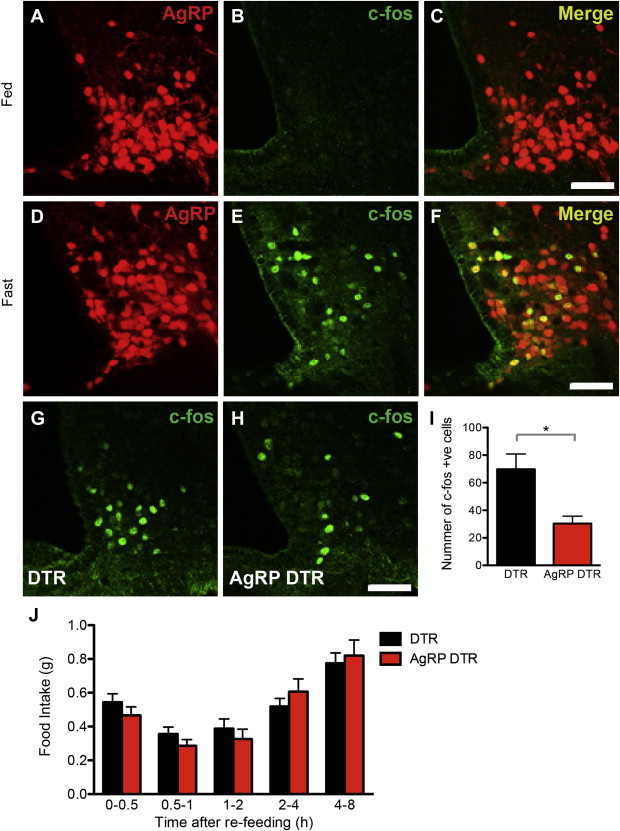
Response of AgRP DTR animals to overnight fast. (A–F) Overnight deprivation of food induced expression of cfos was mainly localized to AgRP neurons. (G–I) cfos immunostaining in the arcuate nucleus after overnight fast in control vs. AgRP DTR animals (DTR 67.75 ± 11.12 vs AgRP 30.33 ± 5.364, p < 0.05). n = 4 in each group (J) Food intake of DTR and AgRP DTR animals after overnight fast. n ≥ 10 in each group. Scale bar = 50 μm.
Because AgRP neurons also express the ghrelin receptor (Ghsr), we also assayed the response of AgRP DTR animals to ghrelin. We found that most of the cfos immunoreactivity in the arcuate nucleus colocalized with AgRP and NPY (Figure 5A–F). Similar to the case for food restriction, ghrelin injection during the light phase resulted in much a smaller number of cfos positive cells in the arcuate of AgRP DTR animals (Figure 5G–I). However, despite the decreased number of cfos positive cells, the food intake of AGRP DTR animals after ghrelin treatment was indistinguishable from that of control animals (Figure 5J). Taken together, these results show that animals with AgRP ablation do not show alterations of baseline food intake and weight and that their responses to leptin treatment, leptin withdrawal, fasting and ghrelin treatment are all normal.
Figure 5.
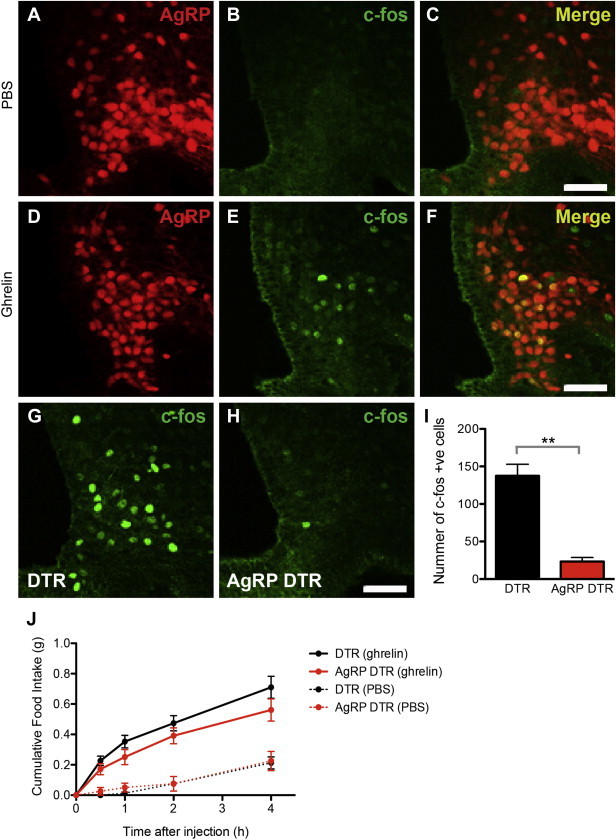
Response of AgRP DTR animals to ghrelin. (A–F) cfos expression in the arcuate nucleus was mainly colocalized to AgRp neurons after ghrelin injection. (G–I) cfos immunostaining in the arcuate nucleus after ghrelin injection in DTR vs. AgRP DTR animals (DTR 137.5 ± 15.39 vs AgRP DTR 23.33 ± 5.365, p < 0.01). n = 4 in each group (J) Food intake of DTR and AgRP DTR animals after either PBS or ghrelin injection. n ≥ 8 in each group. Scale bar = 50 μm.
3.4. Response of AgRP DTR animals to RF in the light phase
We next compared the response to restricted feeding of AgRP DTR animals that received DTX at three days of age to their littermate controls (also injected with DTX). 12-week-old animals were maintained on a 12 h light dark cycle since weaning at which time, food availability was changed to a period between ZT 4 and ZT 7. This was continued for 14 days. Consistent with previous reports, control animals showed a transient reduction in food intake but quickly acclimated to this change steadily increasing their food intake over the course of the experiment with no mortality. In contrast, we noted increased mortality among the AgRP DTR mice with 10% mortality at day 2 and 30% mortality at day 4 (Figure 6A). Moreover, the AgRP DTR animals that survived the first 4 days of RF and remained viable through the end of the experiment failed to acclimate normally to the restricted feeding protocol. Wild type mice that are given food only between ZT 4–7 consume less food over the first three days as they acclimate to the change in food availability. However, by day 4 normal mice consume an equivalent amount of food in the restricted window as they consumed previously during the entire dark phase. In contrast, AgRP DTR animals consumed less food and weighed less than control mice from day 4 to day 9 with food intake only reaching the weight of control mice after day 10 (Figure 6B–C).
Figure 6.
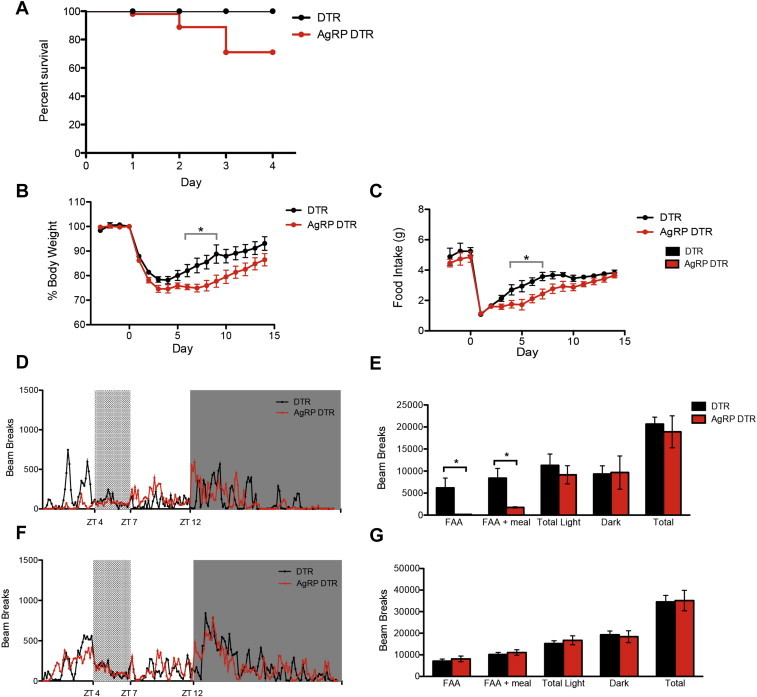
Response of AgRP DTR animals to restricted feeding during the light phase. (A) Survival plot of DTR and AgRP animals on restricted feeding. n ≥ 10 for each group (B, C) Percent body weight and daily food intake of animals on restricted feeding. Body weight of AgRP DTR animals was significantly lower from day 6 to day 9 (p < 0.05) and food intake was also significantly lower from day 4 to day 7 (p < 0.05). n ≥ 6 in each group (D) 24 h plot of activity of DTR and AgRP DTR animals after 3 days on restricted feeding protocol. Error bars were excluded for clarity of plot. (E) Bin plots of activity counts after 3 days on restricted feeding protocol. AgRP DTR animals showed little or no feeding anticipatory activity (FAA) (DTR 6193 ± 2207 vs AgRP DTR 182.5 ± 61.65, p < 0.05) (F) 24 h plot of activity of DTR and AgRP DTR animals after 10 days on restricted feeding protocol. Error bars were excluded for clarity of plot. (G) Bin plots of activity counts after 10 days on restricted feeding protocol. n = 4 for each group.
Animals that have become acclimated to the restricted feeding protocol exhibit markedly increased locomotor activity known as feeding anticipatory activity (FAA) beginning at ZT 2, two hours prior to the time when food is made available. Thus, animals acclimated to the scheduled feeding paradigm exhibit increased locomotion, (FAA) in the two hours prior to the presentation of food. We next monitored the 24-h activity of AgRP DTR animals after 3 days of RF, by which time wild type animals show robust FAA [10]. Consistent with the reduced food consumption at this time, AgRP DTR animals did not show an increase in locomotion in the two hour window prior to the scheduled meal three days after food availability was shifted to the light phase (Figure 6D–E). This defect in locomotion before meal time was not due to a general deficit in locomotion since total activity counts for AgRP DTR animals was similar to controls. However after 10 days of restricted feeding the FAA of AgRP DTR animals was indistinguishable from controls. Taken together, these data demonstrate that AgRP neurons play an important role in the ability of animals to sense and adapt to a temporal change in food availability normally. The delayed acclimation to this change leads some animals to die of starvation while those animals that survive still show a significant delay in the recovery of food intake and the onset of FAA.
3.5. Response of AgRP DTR animals to RF in the dark phase
As a further control, we monitored survival, food intake, weight and FAA in AgRP DTR mice when food availability was limited to three hours during the dark phase which, as mentioned, is when wild type mice, which are nocturnal, consume nearly all of their food. AgRP DTR animals given food only during a 3 h window in the dark phase between ZT 15 and ZT 18 showed an equivalent increase of food intake and body weight to controls throughout the experiment (Figure 7A–B). After 3 days of RF in the dark phase, 24 h activity analyses further revealed that AgRP DTR animals showed increased locomotion prior to the start of meal time, although the magnitude of FAA was significantly lower than controls (Figure 7C–D). Thus the deficit in adapting to RF was more profound when food was only available during the light phase, a time when mice generally move less and eat less.
Figure 7.
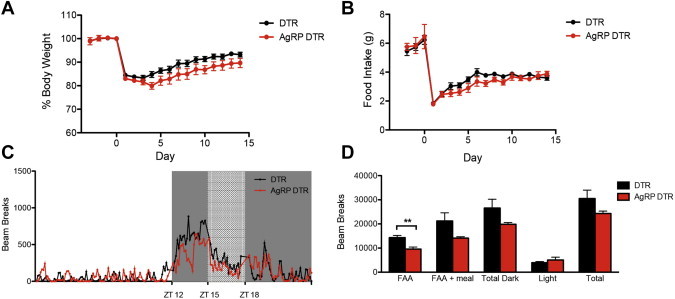
Response of AgRP DTR animals to restricted feeding during the dark phase. (A, B) Percent body weight and daily food intake of animals on restricted feeding. AgRP DTR animals weight and food consumption was comparable to DTR animals. n ≥ 7 in each group. (C) 24 h plot of activity of DTR and AgRP DTR animals after 3 days on restricted feeding protocol. Error bars were excluded for clarity of plot. (D) Bin plots of activity counts after 3 days on restricted feeding protocol. AgRP DTR exhibited feeding anticipatory activity (FAA) though to a significantly lower extent than DTR animals (DTR 14310 ± 942 vs AgRP DTR 9579 ± 831.1, p < 0.01). n = 4 in each group.
4. Discussion
AgRP neurons which are localized in the arcuate nucleus are both necessary and sufficient for feeding in adult animals [26,27]. However animals in which AgRP neurons are ablated as neonates survive suggesting that the increased plasticity of the central nervous system in younger animals allows such animals to survive after the loss of these neurons. Here we show that the ablation of AgRP neurons in neonatal animals results in significant abnormalities in the response to a temporal alteration of food availability. These abnormalities include increased mortality in animals subjected to a protocol that elicits FAA with a significant delay in the recovery of food intake and body weight and an absence of FAA even in those animals that survive. These abnormalities are not observed when food is limited to an equivalent window in the dark phase suggesting that AgRP neurons are essential for the ability of animals to adapt to a change in food availability at circadian times when food is uncoupled from the circadian oscillator (i.e.; moved to the light phase). In contrast, the loss of AgRP neurons has no effect on baseline food intake, body weight, or the response to food restriction, leptin and ghrelin treatment or leptin withdrawal. In aggregate, these findings suggest that AgRP neurons are a key component of a food entrained oscillator that allows animals to predict in time when nutrient will be available and adjust their behavior accordingly. Moreover the observation that AgRP neural ablation in neonatal animals affects only the response to restricted feeding suggests that a key function of these neurons is to control this set of responses.
In this study, we used a Cre-lox system to express the human diptheria toxin receptor only in AgRP neurons and were able ablate 50% of these neurons. This extent of ablation is consistent with a previous report using the same Cre-lox strategy [27]. We do note that in a separate report using a knockin strategy as reported by Luquet et al. [26] there was more than 90% ablation of AgRP neurons. It is unclear why these strategies result in differing degrees of ablation. Interestingly, neonatal ablation of AgRP neurons using the knockin method (with near complete ablation) resulted in animals becoming obese after 12 weeks of age with a metabolic shift towards lipid oxidation which is a somewhat paradoxical in light of the known effect of AgRP neurons to increase feeding [29]. The animals with ∼90% ablation exhibited decreased food intake after a 24 h fast and ghrelin challenge in contrast to the failure of a more limited ablation to have an effect as shown in our studies. Our data show that a loss of only 50% of AgRP is not sufficient to impair responses to food deprivation and ghrelin injection while the hyperphagia that develops after glucoprivation is not impaired even after the loss of the vast majority of AgRP neurons [30]. Though the extent of AgRP ablation observed here is incomplete, the key finding is that the response to RF in animals with AgRP ablation was still impaired even when 50% of AgRP neurons were intact, suggesting that the response to RF is quite sensitive to perturbations of AgRP neurons. The nutrient signal responsible for linking AgRP neuronal activity to circadian in time is not known though recent studies have shown that hypothalamic autophagy [31] and mitochondria dynamics [32] regulate AgRP neuronal function in response to changes in energy availability.
The SCN, which responds to light, is believed to be the primary circadian oscillator, but the biological rhythms that develop in response to changes in food availability are known to persist even in the absence of the SCN. These data have thus suggested that there is a food entrainable oscillator that is separate and independent of the light entrained oscillator [12,13]. Previous work to identify the anatomical sites of the food entrainable oscillator have focused on structures in the posterior to the SCN that regulate feeding behavior such as the dorsal medial hypothalamus (DMH) as well as the arcuate nucleus.
The possible role of the DMH was previously assayed by analyzing the periodicity of the expression of the canonical clock genes, Period 1 (mPer1) and Period 2 (mPer2), both of which were expressed rhythmically in the DMH during RF [33]. However lesioning studies of the DMH reported disparate results. Thus while Landry et al. [34] showed that radio-frequency induced lesions of the DMH did not impair FAA, Gooley et al. [35] reported that ibotenic acid induced lesions of the DMH attenuated FAA. The different finding of these studies may be attributable to a variety of factors including lesion type and the behavioral assays that were used.
In addition to the DMH, rhythmic expression of clock genes were also reported in other brain regions, including the arcuate nucleus, after a change in feeding schedule though the identity of the specific population was not determined [36,37]. The clock genes mPer1 and mPer2 are also expressed in the arcuate nucleus with RF [33] while endogenous mPer2 is expressed rhythmically in the arcuate nucleus in slice preparations of a mPer2 luciferase knock-in mouse [38]. These data show that AgRP neurons show rhythmic behaviors and are consistent with our conclusion that AgRP neurons are a neural component of the food entrainable oscillator. While Mistlberger and Antle [39] showed that monosodium glutamate (MSG) induced lesions of the arcuate nucleus enhanced FAA in rats during RF, the possible role AgRP/NPY neurons was not assessed and moreover this finding is the opposite of what we observe after AgRP ablation which reduces FAA. However, it must be noted that lesions of the arcuate using MSG not only results in the loss of AgRP/NPY neurons but also adjacent cell populations such as POMC neurons. Thus the increase in FAA after non-specific arcuate lesions is likely to be a result the net effect of damage to several different cell types and the identity of the putative cell type(s) that normally suppresses FAA is not known.
Arcuate neurons that co-express the neuropeptides AgRP and NPY [19] also express the leptin receptor and are inhibited by leptin [28]. NPY is a potent orexigenic neuropeptide and promotes feeding in sated or food-deprived animals when administered centrally [40] while AgRP is an antagonist of the melanocortin-4 receptor (MC4R), which when activated suppresses feeding [41]. While the effect of AgRP/NPY neurons in metabolic homeostasis is well-characterized, their involvement in the temporal control of food intake has been less clear at least prior to the current study. Consistent with our data, studies of leptin deficient ob/ob mice and Zucker rats, which exhibit hyperphagia as well as increased AgRP/NPY activity, showed increased FAA after a change in food schedule and this effect can be suppressed by recombinant leptin treatment [42,43]. In addition, AgRP DTR animals exhibited a defect in adapting to RF during the light phase despite partial loss of AgRP neurons, suggesting that a full ‘complement’ of AgRP neurons is essential for the normal response of animals to this restricted feeding paradigm. It has been recently shown that AgRP neurons are heterogynous with respect to their projections in the central nervous system [44] and it is possible that different AgRP populations suffer different degrees of cell loss after diptheria toxin treatment. It is also possible that the impairment to RF observed here could be due to developmental effects of neonatal AgRP neuronal ablation since development of downstream dopamine neurons in the ventral tegmental area (VTA) is altered after AgRP neuron ablation [45]. Further studies will be necessary to understand whether distinct AgRP subpopulations are responsible FAA as well as to elucidate the cellular mechanisms that couple food availability to the activity of these neurons. Since mPer1 and mPer2 are also expressed rhythmically in the arcuate nucleus during RF [33], it is possible that arcuate AgRP neurons express canonical clock genes in response to food availability. Intriguingly, mice lacking known canonical clock components in all tissue exhibited normal FAA and response to RF [46], suggesting that oscillators that link nutrient availability to behavior (i.e; FAA) may be different from the known clock genes that regulate circadian rhythm.
It has been hypothesized that the gut hormone ghrelin is a peripheral signal that targets hypothalamic nuclei to increase locomotor activity in anticipation to a scheduled meal. Ghrelin acts through the growth hormone secretagogue receptor (GHSR) which is widely expressed throughout the hypothalamus, including in AgRP neurons in the arcuate nucleus [22,47,48]. Interestingly, animals under RF exhibited rhythmic patterns of ghrelin secretion with plasma levels peaking at 2 h prior to which when food was available [49]. Consistent with our data, GHSR knockout mice also exhibited attenuated FAA [50]. The fact that FAA was not abolished but only impaired in GHSR knockout mice suggests that there must be other undiscovered peripheral cues that regulate meal pattern behavior. Intriguingly, ghrelin knockout mice displayed normal FAA [51], suggesting that another unknown ligand of GHSR could induce FAA and/or compensate for the absence of ghrelin. Since GHSR is expressed widely in the hypothalamus, of which AgRP neurons are a small population, it is thus not surprising that FAA can still be observed in those AgRP ablated animals that survive albeit with a significantly delayed onset. Furthermore, shRNA knockout of GHSR in the dorsomedial hypothalamic nucleus and the ventromedial hypothalamic nucleus of rats did not abolish FAA with some anticipatory activity still persisting [52]. Taken together, these data suggest a distributed network of brain regions that regulate meal pattern behavior that includes AgRP neurons in the arcuate nucleus Thus, disrupting one node of this network only attenuates but does not completely abolish FAA.
It is well documented that circadian rhythm of the SCN is primarily determined by light/dark cues while food availability can entrain peripheral tissues [14–17]. It is intriguing to note that light availability can also be used as a cue for animals to begin food seeking behavior since animals have to be awake to consume food. Restricted feeding during the light phase, when mice are usually at rest and do not feed, uncouples oscillations in peripheral tissues from the central pacemaker to coordinate body physiology in response to food intake. At the same time, cues from the peripheral must influence non-SCN brain regions to change behavior from rest to food seeking. Our data suggest that AgRP neurons in the arcuate nucleus are a key site where peripheral and possibly other cues are detected independent of visual cues (i.e.; light) that allow an animal to adapt to a new meal time during a period when mice are normally at rest and do not consume food.
5. Conclusion
Here we show that AgRP neurons are a critical component of the food entrained oscillator and play an important role to control meal pattern. These findings provide a starting point for future studies to identify the cellular mechanisms that link changes in food availability to the activity of these neurons as part of a learned behavioral change that allows animals to adapt to changing environmental conditions.
Acknowledgments
We thank the JPB Foundation and the Rockefeller Foundation for supporting this research. Z.A.K. also acknowledges support from the New York Stem Cell Foundation, the Klingenstein Fund, Sloan Foundation, the McKnight Foundation, the Rita Allen Foundation, and the Brain and Behavior Research Foundation. The funding sources were not involved in the research or manuscript preparation.
Contributor Information
Keith Tan, Email: ktan@rockefeller.edu.
Jeffrey M. Friedman, Email: friedj@rockefeller.edu.
Conflict of interest
There are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
References
- 1.Rosenwasser A.M., Boulos Z., Terman M. Physiology & Behavior. 1981;27:33. doi: 10.1016/0031-9384(81)90296-1. [DOI] [PubMed] [Google Scholar]
- 2.Lowrey P.L., Takahashi J.S. Annual Review of Genomics and Human Genetics. 2004;5:407. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hastings M.H., Reddy A.B., Maywood E.S. Nature Reviews Neuroscience. 2003;4:649. doi: 10.1038/nrn1177. [DOI] [PubMed] [Google Scholar]
- 4.Reppert S.M., Weaver D.R. Nature. 2002;418:935. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 5.Fuller P.M., Lu J., Saper C.B. Science. 2008;320:1074. doi: 10.1126/science.1153277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gavrila A.M., Robinson B., Hoy J., Stewart J., Bhargava A., Amir S. Neuroscience. 2008;154:409. doi: 10.1016/j.neuroscience.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 7.Guilding C., Piggins H.D. European Journal of Neuroscience. 2007;25:3195. doi: 10.1111/j.1460-9568.2007.05581.x. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki S., Numano R., Abe M., Hida A., Takahashi R., Ueda M. Science. 2000;288:682. doi: 10.1126/science.288.5466.682. [DOI] [PubMed] [Google Scholar]
- 9.Balsalobre A., Damiola F., Schibler U. Cell. 1998;93:929. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 10.Mistlberger R.E. Neuroscience & Biobehavioral Reviews. 1994;18:171. doi: 10.1016/0149-7634(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 11.Stephan F.K., Biol J. Rhythms. 2002;17:284. doi: 10.1177/074873040201700402. [DOI] [PubMed] [Google Scholar]
- 12.Stephan F.K., Swann J.M., Sisk C.L. Behavioral and Neural Biology. 1979;25:346. doi: 10.1016/s0163-1047(79)90415-1. [DOI] [PubMed] [Google Scholar]
- 13.Krieger D.T., Hauser H., Krey L.C. Science. 1977;197:398. doi: 10.1126/science.877566. [DOI] [PubMed] [Google Scholar]
- 14.Damiola F., Le Minh N., Preitner N., Kornmann B., Fleury-Olela F., Schibler U. Genes & Development. 2000;14:2950. doi: 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hara R., Wan K., Wakamatsu H., Aida R., Moriya T., Akiyama M. Genes to Cells. 2001;6:269. doi: 10.1046/j.1365-2443.2001.00419.x. [DOI] [PubMed] [Google Scholar]
- 16.Stokkan K.A., Yamazaki S., Tei H., Sakaki Y., Menaker M. Science. 2001;291:490. doi: 10.1126/science.291.5503.490. [DOI] [PubMed] [Google Scholar]
- 17.Wakamatsu H., Yoshinobu Y., Aida R., Moriya T., Akiyama M., Shibata S. European Journal of Neuroscience. 2001;13:1190. doi: 10.1046/j.0953-816x.2001.01483.x. [DOI] [PubMed] [Google Scholar]
- 18.Knight Z.A., Tan K., Birsoy K., Schmidt S., Garrison J.L., Wysocki R.W. Cell. 2012;151:1126. doi: 10.1016/j.cell.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn T.M., Breininger J.F., Baskin D.G., Schwartz M.W. Nature Neuroscience. 1998;1:271. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 20.Atasoy D., Betley J.N., Su H.H., Sternson S.M. Nature. 2012;488:172. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aponte Y., Atasoy D., Sternson S.M. Nature Neuroscience. 2011;14:351. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willesen M.G., Kristensen P., Rømer J. Neuroendocrinology. 1999;70:306. doi: 10.1159/000054491. [DOI] [PubMed] [Google Scholar]
- 23.Mistlberger R.E. Physiology & Behavior. 2011;104:535. doi: 10.1016/j.physbeh.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 24.LeSauter J., Hoque N., Weintraub M., Pfaff D.W., Silver R. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13582. doi: 10.1073/pnas.0906426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhagen L.A.W., Egecioglu E., Luijendijk M.C.M., Hillebrand J.J.G., Adan R.A.H., Dickson S.L. European Neuropsychopharmacology. 2011;21:384. doi: 10.1016/j.euroneuro.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Luquet S., Perez F.A., Hnasko T.S., Palmiter R.D. Science. 2005;310:683. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 27.Gropp E., Shanabrough M., Borok E., Xu A.W., Janoschek R., Buch T. Nature Neuroscience. 2005;8:1289. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- 28.Flier J.S. Cell. 2004;116:337. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- 29.Joly-Amado A., Denis R.G.P., Castel J., Lacombe A., Cansell C., Rouch C. EMBO Journal. 2012;31:4276. doi: 10.1038/emboj.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luquet S., Phillips C.T., Palmiter R.D. Peptides. 2007;28:214. doi: 10.1016/j.peptides.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 31.Kaushik S., Rodriguez-Navarro J.A., Arias E., Kiffin R., Sahu S., Schwartz G.J. Cell Metabolism. 2011;14:173. doi: 10.1016/j.cmet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dietrich M.O., Liu Z.-W., Horvath T.L. Cell. 2013;155:188. doi: 10.1016/j.cell.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mieda M., Williams S.C., Richardson J.A., Tanaka K., Yanagisawa M. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12150. doi: 10.1073/pnas.0604189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landry G.J., Simon M.M., Webb I.C., Mistlberger R.E. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2006;290:R1527. doi: 10.1152/ajpregu.00874.2005. [DOI] [PubMed] [Google Scholar]
- 35.Gooley J.J., Schomer A., Saper C.B. Nature Neuroscience. 2006;9:398. doi: 10.1038/nn1651. [DOI] [PubMed] [Google Scholar]
- 36.Verwey M., Khoja Z., Stewart J., Amir S. Neuroscience Letters. 2008;440:54. doi: 10.1016/j.neulet.2008.05.043. [DOI] [PubMed] [Google Scholar]
- 37.Moriya T., Aida R., Kudo T., Akiyama M., Doi M., Hayasaka N. European Journal of Neuroscience. 2009;29:1447. doi: 10.1111/j.1460-9568.2009.06697.x. [DOI] [PubMed] [Google Scholar]
- 38.Guilding C., Hughes A.T.L., Brown T.M., Namvar S., Piggins H.D. Molecular Brain. 2009;2:28. doi: 10.1186/1756-6606-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mistlberger R.E., Antle M.C. Brain Research. 1999;842:73. doi: 10.1016/s0006-8993(99)01836-3. [DOI] [PubMed] [Google Scholar]
- 40.Billington C.J., Levine A.S. Current Opinion in Neurobiology. 1992;2:847. doi: 10.1016/0959-4388(92)90144-a. [DOI] [PubMed] [Google Scholar]
- 41.Ollmann M.M., Wilson B.D., Yang Y.K., Kerns J.A., Chen Y., Gantz I. Science. 1997;278:135. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 42.Ribeiro A.C., Ceccarini G., Dupré C., Friedman J.M., Pfaff D.W., Mark A.L. PLoS ONE. 2011;6:e23364. doi: 10.1371/journal.pone.0023364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mistlberger R.E., Marchant E.G. Physiology & Behavior. 1999;66:329. doi: 10.1016/s0031-9384(98)00311-4. [DOI] [PubMed] [Google Scholar]
- 44.Betley J.N., Cao Z.F.H., Ritola K.D., Sternson S.M. Cell. 2013;155:1337. doi: 10.1016/j.cell.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dietrich M.O., Bober J., Ferreira J.G., Tellez L.A., Mineur Y.S., Souza D.O. Nature Neuroscience. 2012;15:1108. doi: 10.1038/nn.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Storch K.-F., Weitz C.J. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6808. doi: 10.1073/pnas.0902063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guan X.M., Yu H., Palyha O.C., McKee K.K., Feighner S.D., Sirinathsinghji D.J. Brain Research – Molecular Brain Research. 1997;48:23. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- 48.Zigman J.M., Jones J.E., Lee C.E., Saper C.B., Elmquist J.K. Journal of Comparative Neurology. 2006;494:528. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drazen D.L., Vahl T.P., D'Alessio D.A., Seeley R.J., Woods S.C. Endocrinology. 2006;147:23. doi: 10.1210/en.2005-0973. [DOI] [PubMed] [Google Scholar]
- 50.Blum I.D., Patterson Z., Khazall R., Lamont E.W., Sleeman M.W., Horvath T.L. Neuroscience. 2009;164:351. doi: 10.1016/j.neuroscience.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szentirmai E., Kapás L., Sun Y., Smith R.G., Krueger J.M. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2010;298:R467. doi: 10.1152/ajpregu.00557.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Merkestein M., van Gestel M.A., van der Zwaal E.M., Brans M.A., Luijendijk M.C., van Rozen A.J. International Journal of Obesity (London) 2014;38:610. doi: 10.1038/ijo.2013.131. [DOI] [PubMed] [Google Scholar]