

Abstract
Understanding how muscle contraction orchestrates insulin-independent muscle glucose transport may enable development of hyperglycemia-treating drugs. The prevailing concept implicates Ca2+ as a key feed forward regulator of glucose transport with secondary fine-tuning by metabolic feedback signals through proteins such as AMPK. Here, we demonstrate in incubated mouse muscle that Ca2+ release is neither sufficient nor strictly necessary to increase glucose transport. Rather, the glucose transport response is associated with metabolic feedback signals through AMPK, and mechanical stress-activated signals. Furthermore, artificial stimulation of AMPK combined with passive stretch of muscle is additive and sufficient to elicit the full contraction glucose transport response. These results suggest that ATP-turnover and mechanical stress feedback are sufficient to fully increase glucose transport during muscle contraction, and call for a major reconsideration of the established Ca2+ centric paradigm.
Keywords: Exercise, Skeletal muscle, Ca2+, AMPK, Stretch
1. Introduction
Muscle contraction during exercise is a more potent physiological stimulus of skeletal muscle glucose uptake than even maximal insulin [1] and a highly effective prophylactic against the hyperglycemia associated with the development of type 2 diabetes mellitus [2]. Despite these drug-target-wise attractive features, the molecular signalling mechanisms by which muscle contraction augments glucose transport are, compared to insulin, relatively understudied and remain poorly defined (for review see Ref. [3]).
The signals underlying increased glucose transport with contraction have long been speculated to include a feedforward signalling component, activated directly by depolarization-induced SR Ca2+ release through the ryanodine receptors. In addition, feedback signals are activated secondarily in response to Ca2+-activated contraction to further fine-tune the glucose transport response, including probably activation of the metabolic stress responsive kinase 5′ AMP-activated protein kinase (AMPK) by ATP-turnover and mechanical stress-activated signals [4,5]. The belief that Ca2+ by itself is adequate to stimulate glucose transport is based to a major extent on seminal work by John Holloszy and co-workers where 3–4 mM caffeine was shown to increase SR Ca2+ release and glucose transport in incubated rat muscles without changes in muscle tension, ATP or Creatine phosphate [6]. In rats, caffeine-stimulation was sufficient to double glucose transport rate in slow-twitch oxidative soleus muscle and triple it in fast-twitch glycolytic epitrochlearis [7,8] without activating AMPK [7]. This suggested that Ca2+ per se could stimulate a substantial increase in muscle glucose transport. However, our group and others subsequently found 3–4 mM caffeine increased AMPK activation and nucleotide-turnover in muscles from mice and rats [9–11], presumably due to the considerable energy-demand posed by sarco/endoplasmatic reticulum Ca2+ ATPase (SERCA)-dependent Ca2+ reuptake [12]. Furthermore, caffeine-stimulated glucose transport was potently inhibited in muscles from muscle-specific dominant-negative kinase-dead α2 AMPK mice [10,13], suggesting that the caffeine-response largely depends on ATP turnover-mediated activation of AMPK rather than on Ca2+ as such.
To clarify the relative sufficiency and necessity of SR Ca2+ vs. feedback signals to contraction-stimulated glucose transport, we presently combined contractile myosin blockers, AMPK transgenic mice, Ca2+ ATPase inhibitors and electrical stimulated contraction, in incubated mouse muscles, a classical model system allowing cell culture-like manipulations and full environmental control of fully differentiated, contraction-competent, striated muscle.
2. Materials and methods
2.1. Antibodies, reagents and immunoblotting
All antibodies and reagents used were commercially available. Details on antibodies used and immunoblotting specifics are included in Supplemental Experimental Procedures.
2.2. Ex vivo muscle incubation
Soleus and EDL muscles from anaesthetized female C57BL/6 wildtype and muscle-specific KD AMPK overexpressing [4] mice were incubated in continuously gassed (95% O2/5% CO2) modified Krebs–Ringer–Henseleit-buffer at 30 °C. After 10–15 min rest, buffer containing inhibitors was added for 1 h, followed by stimulation with CPA, insulin, electrical stimulation, AICAR or passive stretch as described in the figure legends and in the Supplemental Experimental Procedures.
2.3. Cytosolic Ca2+ measurements
Changes in Ca2+ concentration were measured as Fluo-3 fluorescence in confluent 3 h serum-starved L6 myotubes. See Supplemental Experimental Procedures for details.
2.4. AMPK activity
Immunoprecipitated AMPK trimer activity was measured in vitro using 32P-labelled ATP incorporation into a substrate peptide. See Supplemental Experimental Procedures for details.
2.5. Statistics
The data were analysed using T-test or one-way, two-way or two-way repeated measures ANOVA with Tukey's post hoc test. The significance level was set at p < 0.05.
3. Results
3.1. Myosin ATPase blockade with BTS and blebbistatin blocks contraction but not SR Ca2+ release
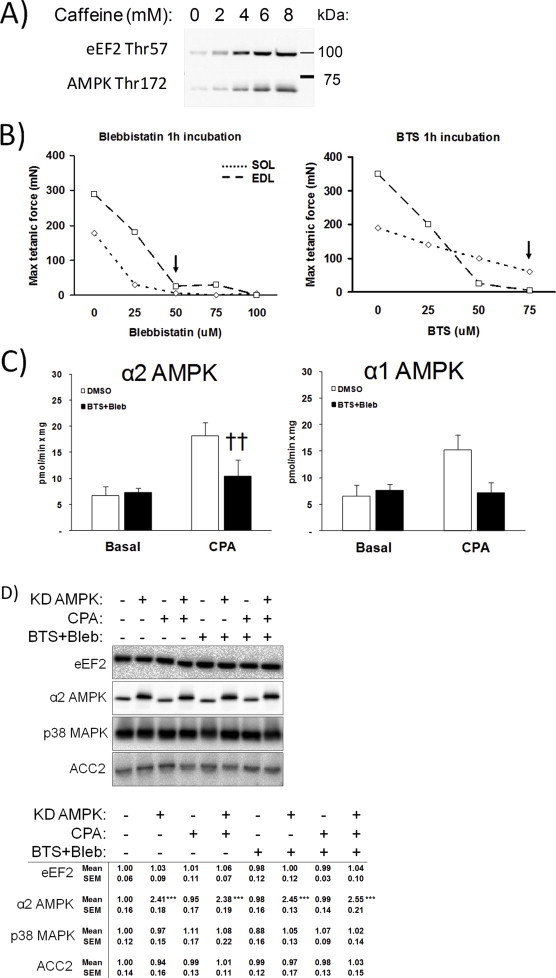
Ex vivo incubated mouse slow-twitch oxidative soleus and fast-twitch glycolytic extensor digitorum longus (EDL) muscles were stimulated with the SERCA-inhibitor cyclopiazonic acid (CPA) at increasing doses to inhibit Ca2+ reuptake, thus allowing a sustained Ca2+ leak from the sarco/endoplasmatic reticulum (SR). In soleus muscle, this caused a dose-dependent increase in the Ca2+-regulated Thr57 phosphorylation of eEF2 by eEF2 kinase, a readout of SR Ca2+ release [14], in addition to increased phosphorylation of AMPK Thr172 (Figure 1A). This pattern is reminiscent of the increase in eEF2 and AMPK phosphorylation seen in soleus stimulated with 4–6 mM caffeine (Figure S1A). Glucose transport was elevated significantly above baseline using 50 μM CPA (Figure 1B). No effect of CPA was observed in mouse EDL (Figure 1A), probably due to the known differences in Ca2+ handling proteins between type II fibres compared to type I fibres [15]. Pretreatment with the SR Ca2+ channel blocker dantrolene prevented both the CPA-stimulated eEF2 and AMPK phosphorylations (Figure 1C) and directly measured Ca2+ release in L6 myotubes (Figure 1D), showing their dependence on SR Ca2+ release. The fast and slow contractile myosin heavy chain type II ATPase inhibitors, BTS and blebbistatin (Bleb) did not affect Ca2+ release (Figure 1D) consistent with previous reports [16–18].
Figure 1.
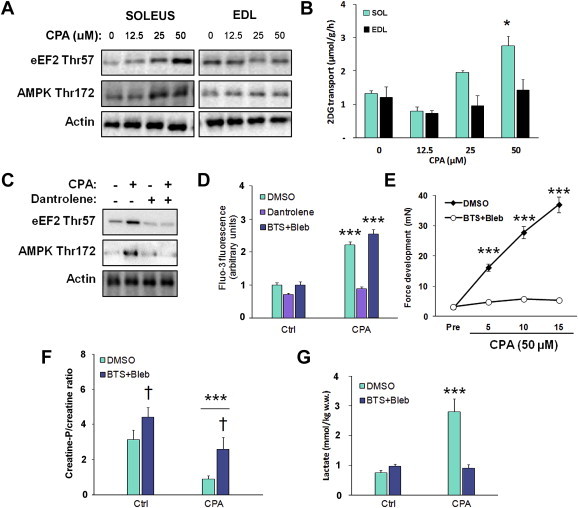
Optimization of the cyclopiazonic acid (CPA)-induced tonic contraction-model. A) Signalling blots from cyclopiazonic acid (CPA, 15′ stimulation) dose–response experiment in mouse soleus and EDL muscles (n = 2) and B) Dose–response of cyclopiazonic acid (CPA)-stimulated glucose transport (15 min), n = 2/muscle, *p < 0.05 using Tukey's post hoc test. C) Prevention of the response to 50 μM CPA in soleus muscle by pretreatment with the sarcoplasmatic reticulum Ca2+ release blocker dantrolene (60 μM, 1 h), n = 4. D) Fluo-3 Ca2+ fluorescence +/− CPA (50 μM, 1′) in L6 myotubes pretreated with DMSO, dantrolene (60 μM, 20′) or myosin ATPase inhibitors BTS (75 μM, 20′) and blebbistatin (50 μM, 20′), n = 2. E) CPA-stimulated force-development timecourse in soleus muscles treated +/− myosin ATPase blockers, n = 6–8. F) Creatine-phosphate/creatine ratio in SOL muscles stimulated with CPA (50 μM, 15 min) in the presence or absence of BTS (50 μM)+Bleb (75 μM), n = 6–8, *** ANOVA main-effect of CPA, †p < 0.05 ANOVA main-effect of BTS + Bleb. G) CPA-stimulated lactate production +/− myosin ATPase blockers, n = 6–8. Data are mean ± S.E.M.
The CPA-induced rise in cytosolic Ca2+ leak elicited a sustained submaximal muscle contraction (Figure 1E). Inhibition of contractile function by BTS + Bleb blocked tension development (Figure 1E + dose–response curves in Figure S1B), caused a general increase in the creatine phosphate/creatine (CrP/Cr)-ratio (Figure 1F) and markedly reduced CPA-induced metabolic stress measured as lactate concentration (Figure 1G).
3.2. Myosin ATPase blockade does not affect insulin or AICAR-stimulated signalling or glucose transport
We were initially concerned that Bleb in particular might have off-target effects since MyoII has been implicated in insulin-stimulated GLUT4 translocation and glucose transport in adipocytes [19]. However, neither insulin nor AICAR-stimulated signalling (Figure 2A+C) or glucose transport (Figure 2B+D) were inhibited by BTS + Bleb, suggesting that the effects of myosin ATPase inhibition are specific to contraction.
Figure 2.
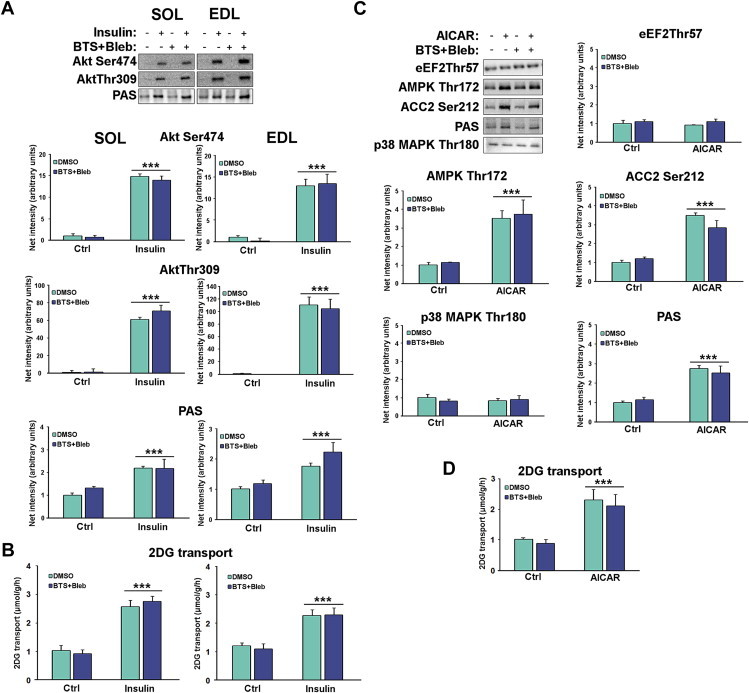
Neither insulin nor AICAR-stimulated signalling or glucose transport are affected by myosin ATPase blockers. A) Representative western blots and quantifications of control and insulin-stimulated (60 nM, 20 min) phosphorylations in SOL and EDL and B) corresponding 2DG transport in SOL and EDL. n = 9, ***p < 0.001 ANOVA main-effect of insulin. C) Representative western blots and quantifications of control vs. AICAR-stimulated (2 mM, 40 min) phosphorylations and D) corresponding 2DG transport (right) in EDL, n = 9, ***p < 0.001 ANOVA main-effect of AICAR. Data are mean ± S.E.M.
3.3. Reduced CPA-stimulated glucose transport with BTS + blebbistatin correlates with lower AMPK and mechanical stress-, but not SR Ca2+ dependent, signalling
Myosin ATPase inhibition in soleus muscle potently dampened the CPA-induced phosphorylation of AMPK and p38 MAPK, (Figure 2A) activated by ATP-turnover [4] and mechanical stress [20], respectively. SR Ca2+ dependent phosphorylation of eEF2 was unaffected (Figure 3A). BTS + Bleb significantly inhibited CPA-stimulated α2 AMPK activity and had a qualitatively similar effect on α1 AMPK activity (Figure S1C). Myosin ATPase blockade potently reduced CPA-stimulated glucose transport (Figure 3).
Figure 3.
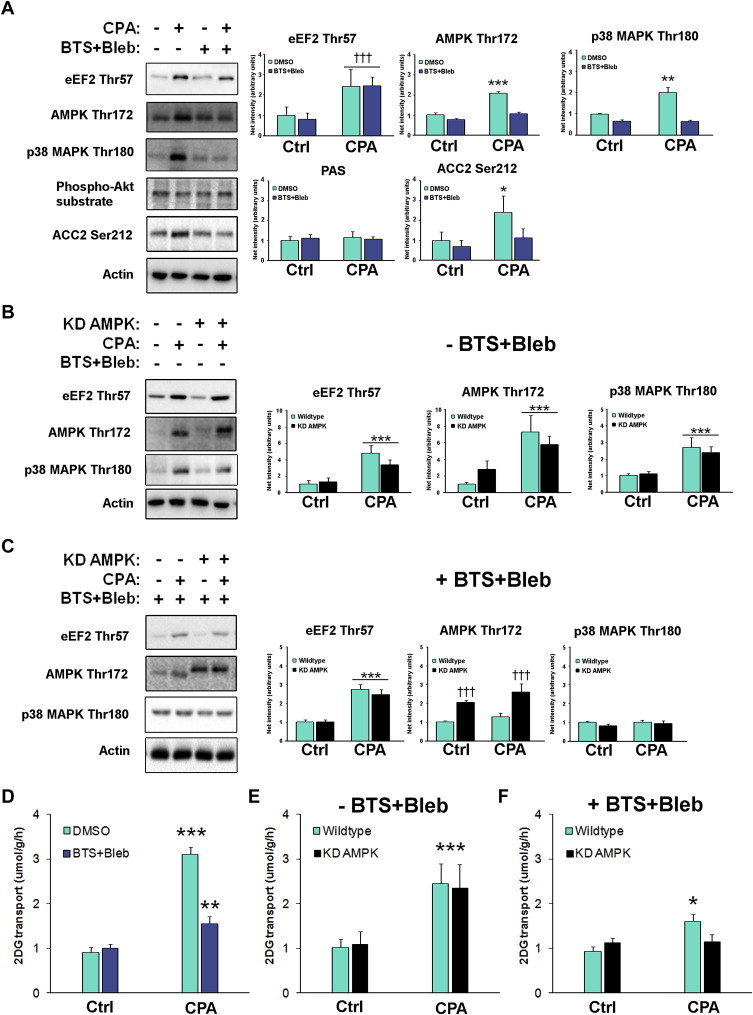
Cyclopiazonic acid (CPA)-induced glucose transport depends on AMPK and likely mechanical stress but not SR Ca2+. A) Quantifications of immunoblots from CPA-stimulated SOL muscles +/− BTS + Bleb. Quantified protein phosphorylation are indicated above the graphs throughout, n = 6, †††p < 0.001 ANOVA main-effect of CPA, */**/***p < 0.05/0.01/0.001 Tukey's post hoc test effect of CPA. B) Quantifications of immunoblots from CPA-stimulated wildtype and kinase-dead (KD) AMPK overexpressing SOL muscles, n = 6, ***p < 0.001 ANOVA main-effect of CPA. C) Quantifications of immunoblots from CPA-stimulated wildtype and KD AMPK overexpressing SOL muscles in the presence of BTS + Bleb. n = 6, p < 0.001 ANOVA main-effect of CPA, ††† ANOVA genotype main-effect. CPA-stimulated 2-deoxyglucose (2DG) transport (bottom graph) in mouse soleus D) +/− myosin ATP blockers E) in wildtype and KD AMPK mice F) combining myosin ATPase blockers with KD AMPK overexpression, n = 6. */**/***p < 0.05/0.01/0.001 CPA-effect using Tukey's post hoc test. Data are mean ± S.E.M.
Next, we asked if the residual CPA-stimulated glucose transport-response in BTS + Bleb-treated soleus muscles required AMPK signalling. As expected, our signalling readouts including the phosphorylation of AMPK by upstream kinases on Thr172 were not affected by muscle-specific overexpression of kinase-dead α2 (KD) AMPK (Figure 3B). CPA-stimulated glucose transport was unaffected by muscle-specific overexpression of KD AMPK (Figure 3E).
Similar to the findings with BTS + Bleb in Figure 3A, combining BTS + Bleb with KD AMPK inhibition only reduced AMPK Thr172 and p38 MAPK Thr180/Tyr182 phosphorylation, but did not affect eEF2 Thr57 phosphorylation (Figure 3C). Interestingly, despite the normal SR Ca2+ dependent signalling, the residual glucose transport-response in the presence of BTS + Bleb was completely abolished in the presence of KD AMPK (Figure 3F). No effect of CPA or BTS + Bleb was found for total protein expression (Figure S1D). Overall, these data show that CPA-induced contraction-stimulated glucose transport can be completely prevented despite normal SR Ca2+ release. This strongly implies that SR Ca2+ in itself is insufficient to stimulate glucose transport. Rather, this appears to require input from AMPK and likely mechanical stress-signalling.
3.4. Contraction-stimulated glucose transport is related to metabolic and mechanical stress but not Ca2+ transients
A sustained increase in cytosolic Ca2+ by CPA is quite dissimilar from the transient Ca2+ spikes elicited by physiological muscle excitation–contraction coupling. Therefore, we also investigated the effect of contractile inhibition on the glucose transport-response elicited by intermittent electrical field stimulation of muscle contraction. Given that two previous reports saw widely different effect of BTS on the muscle glucose transport-response in rodent fast-twitch muscles (∼50% reduction vs. no effect) [21,22], these previously published stimulation protocols were included in our experimental setup and directly compared to a third less intense protocol (representative force-traces shown in Figure 4A). With these protocols, muscles were stimulated for 0.1%, 0.3% and 0.7% of the total time (% net stimulation time (NST)) and a clear intensity-dependent effect on fatigue rates measured for the initial 3 min was observable (Figure 4B). The tension development during electrical stimulation was all but prevented by the addition of myosin ATPase inhibitors (Figure 4B). Inhibition of cross-bridge cycling markedly reduced metabolic stress measured as CrP/Cr-ratio (Figure 4C) and muscle lactate (Figure 4D).
Figure 4.
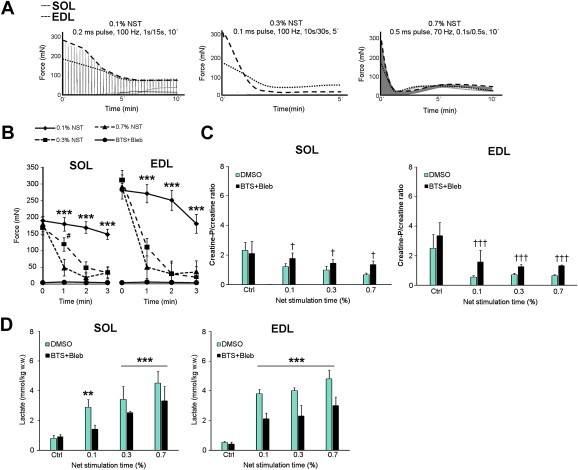
Metabolic responses to electrically stimulated contraction protocols representing different intensities. A) Representative force curves from low (0.1% net stimulation time (NST)), intermediate (0.3% NST) and high (0.7% NST) intensity electrical stimulation regimens in mouse soleus (SOL) and extensor digitorum longus (EDL) muscles B) quantification of the peak force production for the first 3 min with 0.1, 0.3 and 0.7% NST electrical stimulation regimens and myosin ATPase blocker treated muscles, ***p < 0.001 0.1% NST vs. BTS + Bleb, #p < 0.05 0.1% NST vs. 0.3% NST using Tukey's post hoc test. C) Creatine-phosphate/creatine ratio in SOL and EDL muscles under the conditions described above, †/†††p < 0.05/0.001 ANOVA BTS + Bleb main effect. D) Lactate production in SOL and EDL, **/***p < /0.01/0.001 contraction-effect vs. ctrl using Tukey's post hoc test. n = 6–8. Data are mean ± S.E.M.
The effect of myosin ATPase inhibition on the electrically stimulated glucose transport-response was highly dependent on the stimulation protocol, ranging from no difference in glucose transport-stimulation when applying the most intense 0.7% NST protocol in EDL to a complete prevention of glucose transport using the mild 0.1% NST protocol in both soleus and EDL muscles (Figure 5A+B). Consistent with our CPA-experiments, the electrically stimulated increases in eEF2 Thr57 phosphorylation were unaffected by myosin ATPase inhibitors whereas the increases in AMPK Thr172 and p38 MAPK Thr180/Tyr182 phosphorylation were markedly blunted by inhibition of contraction by the myosin inhibitors (Figure 5C). No effect of ex vivo contraction or BTS + Bleb was observed for total protein expression (Figure S2A).
Figure 5.
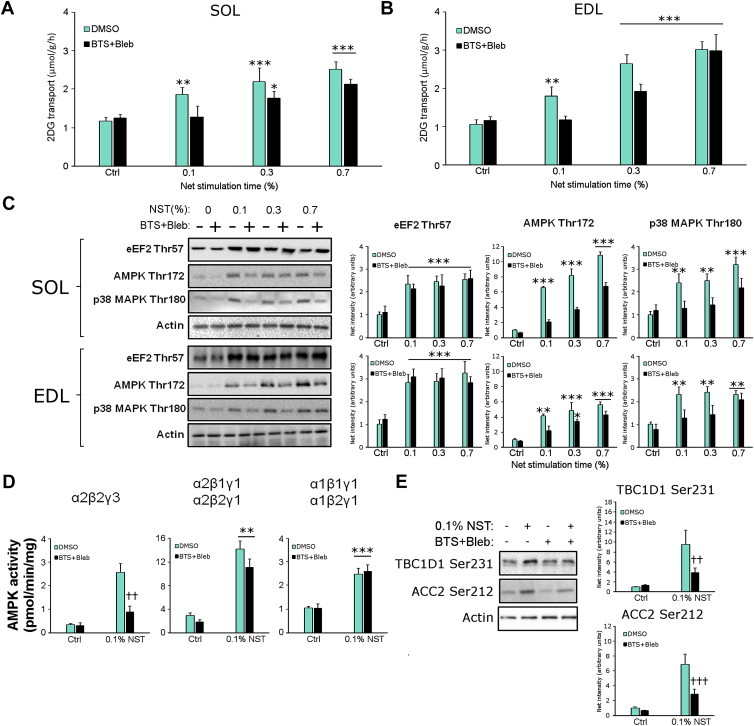
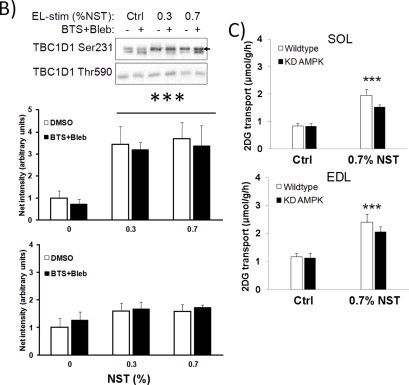
Low-intensity electrically-induced contraction-stimulated glucose transport but not Ca2+ release is abolished by myosin ATPase blockade. 2-deoxyglucose (2DG) transport in A) soleus (SOL) and B) extensor digitorum longus (EDL). C) Representative western blots and quantifications in SOL (top) and EDL (bottom). D) AMPK heterotrimer activities in EDL and E) Representative western blots and quantifications of known AMPK substrates TBC1D1 Ser231 and ACC2 Ser212 in EDL. */**/***p < 0.05/0.01/0.001 contraction-effect vs. ctrl or in (B) 0.1% NST vs. 0.3% NST using Tukey's post hoc test, #p < 0.05 contraction-effect 0.3% NST vs. 0.7% NST, ††p < 0.01 ANOVA contraction × inhibitor interaction. n = 6–8. Data are mean ± S.E.M.
AMPK kinase activity measurements on subsets of AMPK trimer complexes in vitro revealed that the residual increase in AMPK Thr172 phosphorylation with myosin ATPase inhibitors reflected a normal increase of γ1 AMPK associated trimer activity but a very potent inhibition of the α2β2γ3 AMPK trimer (Figure 5D). The decreased α2β2γ3 AMPK activation was associated with decreased phosphorylation of TBC1D1 Ser231 and ACCbeta Ser212 (Figure 5E), previously suggested to be α2β2γ3 AMPK substrates in exercising humans [23,24]. Using more intense 0.3% and 0.7% NST stimulation, no discernible difference in TBC1D1 Ser231 phosphorylation was observed between control and BTS + Bleb-treated muscles (Figure S2B).
Since the residual CPA-stimulated glucose transport-response in the presence of BTS + Bleb is AMPK-dependent, this was also tested for electrical stimulation. Given that BTS + Bleb-treated muscles only increase glucose transport in response to intense stimulation, we asked if the residual glucose transport-response to the most intense 0.7% NST stimulation was reduced in KD AMPK muscles. However, no significant effect of KD AMPK expression was seen on the residual increase in glucose transport stimulated with the 0.7% protocol (Figure S2C). Still, similar to CPA-stimulated glucose transport, the experiments using low intensity 0.1% NST electrical stimulation define conditions where contraction-stimulated glucose transport is abolished (Figure 5A+B) and associated with decreased α2β2γ3 AMPK (Figure 5D) and mechanical stress-signalling but not increased SR Ca2+ on its own (Figure 5C).
3.5. AMPK and mechanical stress additively increase glucose transport through SR Ca2+ independent mechanisms
Stimulation of AMPK activity and application of mechanical stress (i.e. passive stretch) have both been shown to stimulate glucose transport in incubated rodent muscles without affecting indices of SR Ca2+ release. However, the interaction between these stimuli is not known, nor is it clear how much of the total contraction-stimulated glucose transport-response is rallied by combined stimulation with AMPK and mechanical stress. To test this, a series of additivity-experiments were performed combining AICAR and passive stretch.
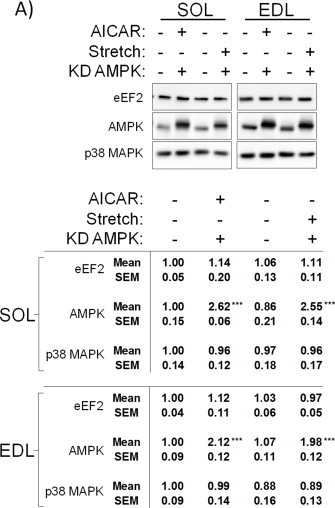
We first performed AICAR and passive stretch additivity experiments in soleus and EDL muscles from wildtype and KD AMPK expressing mice. AMPK and p38 MAPK phosphorylations were increased by AICAR and passive stretch, respectively, whereas neither stimulus altered eEF2 phosphorylation (Figure 6A+B). AMPK-dependent phosphorylation of ACC2 was blocked in KD AMPK expressing muscles (Figure 6A+B). No effect of AICAR or stretch were observed on total protein expression (Figure S3).
Figure 6.
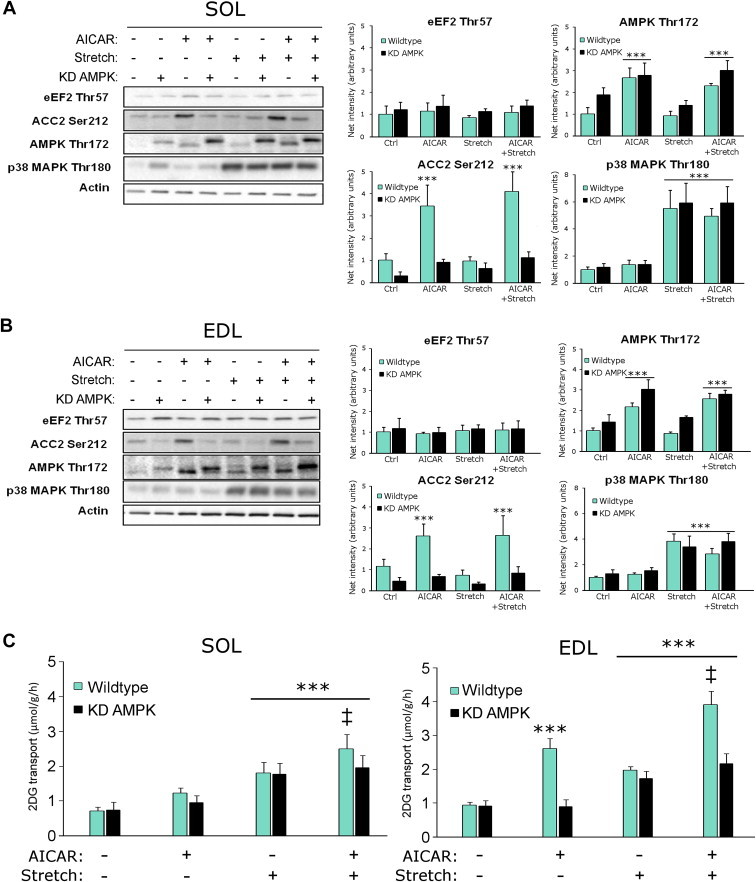
Combined AMPK activation and mechanical stress-stimulation mobilize glucose transport by parallel mechanisms without increasing Ca2+ release. A) Representative western blots and quantifications from wildtype and kinase-dead (KD) AMPK expressing soleus and B) extensor digitorum longus (EDL) stimulated with AICAR (2 mM, 40′), passive stretch (50 mN, 15′) or the two combined, ***p < 0.001 vs. control using Tukey's post hoc test. C) 2-deoxyglucose (2DG) transport in wildtype and KD AMPK SOL and EDL, stimulated with AICAR (2 mM for 40′) or stretch (50 mN, 15′) or the two stimuli combined, ***p < 0.001 increase above control using Tukey's post hoc test. ‡p < 0.05 AICAR vs. AICAR + stretch. n = 6. Data are mean ± S.E.M.
Glucose transport was increased by passive stretch in both muscles (Figure 6C+D) whereas AICAR only increased glucose transport significantly in EDL muscle (Figure 6D). In both soleus and EDL, combined AICAR + stretch additively stimulated glucose transport compared to AICAR or stretch alone (Figure 6C+D). Overexpression of KD AMPK abolished AICAR but not stretch-stimulated effects on glucose transport (Figure 6C+D), confirming that stretch-stimulated glucose transport is AMPK-independent [20]. Overall, these experiments support a model where AMPK activation and stretch independently and additively increase glucose transport.
3.6. Combined AICAR + stretch fully recruit the contraction, but not insulin, stimulated glucose transport response
To directly compare the AICAR + stretch-stimulated glucose transport-response to contraction and insulin-stimulated glucose transport, AICAR + stretch-stimulation was further combined with either insulin or intermediate intensity 0.3% NST electrically stimulated contraction in the absence or presence of the phosphatidylinositol-3-kinase (PI3K) inhibitor wortmannin.
Signalling-wise, Akt Ser474 was only increased by insulin and eEF2 Thr57 phosphorylation was only increased by contraction (Figure 7A+B). AMPK and p38 MAPK phosphorylation responses were generally as expected, apart from some unexpected interactions with wortmannin, including potentiation of contraction-stimulated AMPK activation by wortmannin and a strong inhibitory effect of wortmannin on p38 MAPK phosphorylation by stretch (Figure 7A+B).
Figure 7.
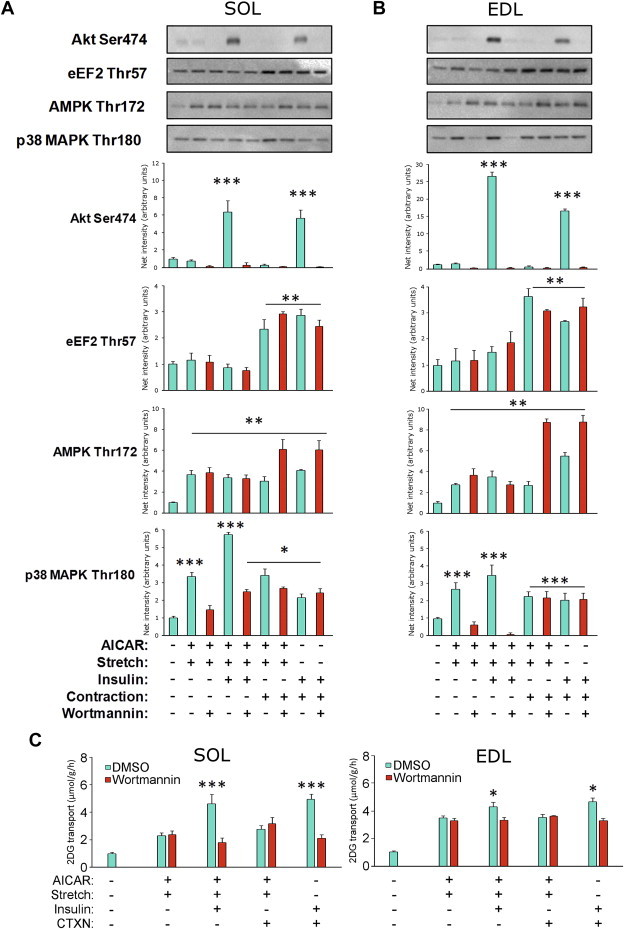
AICAR and stretch recruit the full contraction, but not insulin-stimulated glucose transport response. Representative western blots and quantifications from A) soleus (SOL) and B) extensor digitorum longus (EDL) muscles in which AICAR + cyclic stretch stimulation was combined with either insulin (60 nM, 20′) or contraction (CTXN, 0.3% NST protocol) in the presence or absence of the PI3K inhibitor wortmannin (wmn, 500 nM, 1 h). Phosphorylations measured are indicated above the individual graphs. C) 2DG transport measurements in SOL (left) and EDL (right) under the same conditions. */**/***p < 0.05/0.01/0.001 increase above control using Tukey's post hoc test, n = 4–6. Data are mean ± S.E.M.
Similar to the contraction-response, the glucose transport-response to AICAR + stretch was insensitive to wortmannin in both soleus (Figure 7C left) and EDL (Figure 7C right) muscles. Furthermore, AICAR + stretch-stimulated glucose transport was fully additive to insulin- but not contraction-stimulated glucose transport. Lastly, the glucose transport-response to AICAR + stretch was comparable in magnitude to the contraction-stimulated glucose transport-response in both muscles (Figure 7C). These results show that AICAR + stretch can fully mobilize the full contraction-stimulated glucose transport-response without increasing SR Ca2+ dependent signalling. Similar to contraction, this response is additive to insulin-stimulation and PI3K-independent.
3.7. p38 MAPK is not required for stretch-stimulated glucose transport
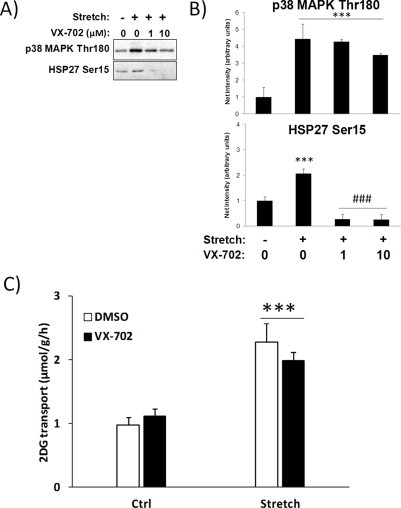
Stretch-stimulated glucose transport has previously been suggested to be mediated by p38 MAPK [20]. Complete blockade of p38 MAPK-dependent signalling to HSP27 using the novel p38 MAPK inhibitor indicated the effectiveness of the blocker VX-702 but it did not reduce stretch-stimulated glucose transport (Figure S4), indicating that p38 MAPK may not be essential to stretch-stimulated glucose transport.
4. Discussion
The premise that SR Ca2+ release per se increases glucose transport in muscle has remained largely undisputed and continues to be reiterated and built upon in the scientific literature. However, here we demonstrated that 1) SR Ca2+ release is unable to increase glucose transport in the absence of muscle contraction-related feedback signals and 2) that the full contraction-response in glucose transport can be obtained without increased SR Ca2+ dependent signalling. Apart from disputing what has become a text-book paradigm, this study is to our knowledge the first study to completely block and thus account for 100% of the glucose transport-response during contraction induced by either CPA or electrical stimulation and to reconstitute 100% of the contraction-response by combining two of its proposed components, AMPK activation and mechanical stress.
Muscle contraction is a complex stimulus that activates a myriad of independent signalling pathways that may regulate a given endpoint. Over the years, this has made it extremely challenging to convincingly assign a given signalling protein to glucose transport-regulation by contraction. Our current work shows that the parallel and independent inputs from AMPK and mechanical stress are likely both required for glucose transport-stimulation and sufficient to recruit the full contraction-sensitive glucose transport response. The signalling mechanisms used by mechanical stress to increase glucose transport are presently unknown. Any protein partly required for contraction-stimulated glucose transport independently of AMPK is a candidate for regulating stretch-stimulated glucose transport, including Rac1 [25], reactive oxygen species [20,26] and the AMPK-related kinase SNARK [27].
A number of studies in isolated rodent muscles have proposed that various Ca2+-regulated proteins are necessary for the full contraction-stimulated glucose transport-response, including PKC isoforms [28–30], Ca2+/calmodulin-dependent kinase (CaMK)II [31], NSPL1 [32] and CaMKK [33,34]. From our study, SR-Ca2+ release is clearly neither sufficient nor required to elicit the full contraction-like glucose transport-response. Worth noting, however, AICAR stimulation takes 40 min to maximally stimulate glucose transport while ex vivo contraction-stimulated glucose transport peaks within 5–10 min. Therefore, other signals, including perhaps Ca2+, may accelerate the completion of the glucose transport-response during contraction. Furthermore, our data do not rule out a contribution from other Ca2+ sources, most notably extracellular Ca2+ entry through voltage and/or stretch-sensitive plasma membrane Ca2+ channels. Silencing of the transient receptor potential (TRP) 3 Ca2+ channel in mouse flexor digitorum brevis single muscle fibres reduced insulin-stimulated glucose transport [35], suggesting a role of extracellular Ca2+ in insulin-stimulated GLUT4 vesicle membrane fusion. Interestingly, pharmacological or stretch-stimulated TRP3-mediated Ca2+ entry resulted in increased activation of CaMKII, Rac1 and ROS production in mouse heart [36], all of which have previously been linked to contraction-stimulated glucose transport [25,26,37,38]. However, pharmacological activator and inhibitor-studies do not support an essential role of TRP channels during skeletal muscle contraction [39]. For the voltage-gated L-type Ca2+ channels, inhibitor-specificity may limit conclusions regarding their requirement for muscle glucose transport [40]. Based on these studies, a contribution from other surface Ca2+ channels and non SR Ca2+ dependent activation of for instance CaMKII cannot be excluded at present and should be further investigated.
The present studies rely heavily on the specificity of BTS and Blebbistatin. BTS was first characterized in 2001 as a specific inhibitor of the fast-twitch (Myosin heavy chain (MHC) type II), but not slow-twitch muscle/cardiac MyoII (MHC type I) isoform [16]. Remarkably, concentrations as high as 100 μM BTS did not affect the activity of any of the enzymes tested including kinesin, pyruvate kinase, lactate dehydrogenase, non-muscle human platelet MyoIIa nor did it affect electrically stimulated Ca2+ transients. A few years later, Bleb was found to specifically inhibit non-muscle MyoIIa and b, in addition to skeletal muscle fast-twitch and porcine slow-twitch heart MyoII without effects on other myosin classes [17,41]. Also, Bleb inhibited the contractility of mouse cardiomyocytes without affecting action potentials or Ca2+ entry [18]. Still, we feared that Bleb might have off-target effects since MyoII has been implicated in insulin-stimulated GLUT4 translocation and glucose uptake in adipocytes [19,42–44]. However, no effect of BTS + Bleb treatment was observed on insulin or AICAR-stimulated signalling or glucose transport. Moreover, the decline in glucose transport-response with BTS + Bleb was absent in EDL at the highest intensity 0.7% NST contraction-intensity, despite a sustained effect on tension development. Therefore, an acute effect of MyoII inhibition on glucose transport or on GLUT4 independently of cross-bridge cycling is unlikely. Our data also indicate that MyoII isoforms are dispensable to both insulin and AICAR-stimulated glucose transport in mouse skeletal muscle.
To compare different muscle types, our study encompassed both the relatively slow-twitch oxidative soleus and fast-twitch glycolytic EDL muscle. With contraction, both muscles displayed qualitatively similar increments in glucose transport with increasing contraction-intensity, demonstrating that the dependence on tension development to stimulate glucose transport holds true for both slow twitch and fast twitch fibre types. This agrees with earlier conclusions in incubated rat muscles, where glucose transport correlated with force-production and metabolic stress rather than contraction-frequency [45]. The response to CPA differed markedly between the two muscle types, with a lack of CPA-stimulated eEF2 phosphorylation in EDL. A more sensitive tension-development in slow vs. fast-twitch muscle in response to Ca2+ has been described in rodents previously [46]. From a molecular viewpoint, this difference may relate to the relatively more extensive SR in fast-twitch fibres, the exclusive expression of the cytosolic Ca2+ buffer protein, parvalbumin and a less Ca2+ sensitive troponin C-isoform, in addition to higher expression of Ca2+ handling proteins such as DHPRα1s, RyR1 and SERCA1 (For references see e.g. [15]). At higher rates of Ca2+ release, specifically when combining CPA with caffeine, our group has previously reported increased tension-development, eEF2 and AMPK phosphorylation in rat EDL [14]. However, we have repeatedly observed that glucose transport is reduced, not increased, by CPA + caffeine-treatment in mouse EDL (unpublished observation), perhaps associated with triggering of cell necrosis or apoptosis by elevated Ca2+. Therefore, the CPA-studies were limited to soleus muscle. Nonetheless, our electrical stimulation-data allowed us to demonstrate a similar uncoupling of SR Ca2+ activated signalling and glucose transport in both soleus and EDL muscles. Overall, these data indicate that both slow-twitch oxidative and fast-twitch glycolytic muscles depend mainly on signals secondary to cross-bridge cycling to elicit contraction-stimulated glucose transport.
Unexpectedly, the inhibitory effect of BTS + Bleb on contraction-stimulated glucose transport varied from complete inhibition at the lowest intensity 0.1% NST electrical stimulation protocol to no effect in EDL during stimulation with the highest intensity 0.7% NST protocol. This observation highlights several points. Firstly, different electrical stimulation protocols provide different answers, implying that ideally several different electrical stimulation protocols should be evaluated for a given intervention. Secondly, the less potent BTS + Bleb inhibition at higher intensities could indicate either that glucose transport at higher intensities relies on distinct mechanisms not related to tension-development as proposed previously [22], or that electrical stimulation-related artefacts such as ROS-production by electrolysis [47] increasingly mobilize compensatory mechanisms. Whether such compensatory mechanisms during intense electrical field stimulation are physiologically meaningful or rather electrical stimulation artefacts is presently unknown.
Based on studies using pharmacological activators, AMPK is necessary (see [48] for references) and likely sufficient [49] to stimulate glucose transport in skeletal muscle, in particular those dominated by type II fibres [8,50]. Thus, AMPK activation can clearly increase glucose transport, with the downstream mechanisms proposed to involve TBC1D1 [51], NO synthases [52,53], PIKfyve [54], NSPL1 [32,55] and/or the RalA GAP protein GARNL1 [56], with RalA previously being linked to GLUT4 translocation in muscle downstream of Rac1 [57]. However, the contribution of AMPK during contraction-stimulated glucose transport remains somewhat controversial. Thus, investigations by various groups in the same model as presently used, the kinase-dead α2 AMPK overexpressing mouse, have found partly decreased, normal or increased contraction or exercise-stimulated glucose uptake in this model compared to wildtype (see [48] for references). Adding to the uncertainty, double knockout of both regulatory AMPK β-subunits abolished AMPK-activity and potently reduced contraction- and treadmill running stimulated glucose transport [58] whereas double knockout of the catalytic AMPK α-subunits did not affect contraction-stimulated glucose transport [59]. In the present study, passive stretch and AICAR additively increased glucose transport, and the residual ∼1/3rd of CPA-stimulated glucose transport that was not blocked by BTS + Bleb, was abolished in the KD AMPK mice. Thus, while AMPK activation can clearly blend with other signals to elicit the full contraction-stimulated glucose transport-response, the contribution from AMPK during contraction may be hard to isolate in various KO models due to compensation and redundancy of other glucose transport-stimulating signals during contraction.
In conclusion, combined AMPK activation and mechanical stress can fully account for the contraction-stimulated glucose transport-response ex vivo, strongly suggesting that AMPK and stretch-activated signalling, but not SR Ca2+ per se, regulates glucose transport during skeletal muscle contraction.
Acknowledgements
Funding was from the Weimann Foundation (TEJ), the Danish Independent Research Council/Medicine and the Danish Independent Research Council/Natural Sciences (TEJ, YA, LSH, EAR) and the University of Copenhagen Excellence Program 2016 and UNIK Research Program, the Novo Nordisk Foundation and the Lundbeck Foundation (EAR).
Betina Bolmgren, Jesper Birk and Nicoline Resen Andersen are thanked for skilled technical assistance. Peter Schjerling is thanked for genotyping the kinase-dead AMPK mice. TEJ, AJR and EAR designed the experiments. TEJ, LSH, AJR, YA, AM, SJM, collected and analysed data. TEJ wrote the paper with input from all co-authors.
Conflict of interest
The authors declare no conflict of interest.
Appendix A. Supplementary material
The following are the Supplementary material related to this article:
References
- 1.James D.E., Kraegen E.W., Chisholm D.J. Muscle glucose metabolism in exercising rats: comparison with insulin stimulation. American Journal of Physiology. 1985;248:E575–E580. doi: 10.1152/ajpendo.1985.248.5.E575. [DOI] [PubMed] [Google Scholar]
- 2.Goodyear L.J., Kahn B.B. Exercise, glucose transport, and insulin sensitivity. Annual Review of Medicine. 1998;49:235–261. doi: 10.1146/annurev.med.49.1.235. [DOI] [PubMed] [Google Scholar]
- 3.Richter E.A., Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiological Reviews. 2013;93:993–1017. doi: 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- 4.Mu J., Brozinick J.T., Jr., Valladares O., Bucan M., Birnbaum M.J. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Molecular Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 5.Ihlemann J., Ploug T., Hellsten Y., Galbo H. Effect of tension on contraction-induced glucose transport in rat skeletal muscle. American Journal of Physiology. 1999;277:E208–E214. doi: 10.1152/ajpendo.1999.277.2.E208. [DOI] [PubMed] [Google Scholar]
- 6.Youn J.H., Gulve E.A., Holloszy J.O. Calcium stimulates glucose transport in skeletal muscle by a pathway independent of contraction. American Journal of Physiology. 1991;260:C555–C561. doi: 10.1152/ajpcell.1991.260.3.C555. [DOI] [PubMed] [Google Scholar]
- 7.Wright D.C., Hucker K.A., Holloszy J.O., Han D.H. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes. 2004;53:330–335. doi: 10.2337/diabetes.53.2.330. [DOI] [PubMed] [Google Scholar]
- 8.Wright D.C., Geiger P.C., Holloszy J.O., Han D.H. Contraction- and hypoxia-stimulated glucose transport is mediated by a Ca2+-dependent mechanism in slow-twitch rat soleus muscle. American Journal of Physiology. Endocrinology and Metabolism. 2005;288:E1062–E1066. doi: 10.1152/ajpendo.00561.2004. [DOI] [PubMed] [Google Scholar]
- 9.Raney M.A., Turcotte L.P. Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. Journal of Applied Physiology. 2008;104:1366–1373. doi: 10.1152/japplphysiol.01282.2007. [DOI] [PubMed] [Google Scholar]
- 10.Jensen T.E., Rose A.J., Hellsten Y., Wojtaszewski J.F., Richter E.A. Caffeine-induced Ca(2+) release increases AMPK-dependent glucose uptake in rodent soleus muscle. American Journal of Physiology. Endocrinology and Metabolism. 2007;293:E286–E292. doi: 10.1152/ajpendo.00693.2006. [DOI] [PubMed] [Google Scholar]
- 11.Egawa T., Hamada T., Ma X., Karaike K., Kameda N., Masuda S. Caffeine activates preferentially alpha1-isoform of 5′AMP-activated protein kinase in rat skeletal muscle. Acta Physiologica (Oxford) 2011;201:227–238. doi: 10.1111/j.1748-1716.2010.02169.x. [DOI] [PubMed] [Google Scholar]
- 12.Smith I.C., Bombardier E., Vigna C., Tupling A.R. ATP consumption by sarcoplasmic reticulum Ca(2)(+) pumps accounts for 40–50% of resting metabolic rate in mouse fast and slow twitch skeletal muscle. PLoS ONE. 2013;8:e68924. doi: 10.1371/journal.pone.0068924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abbott M.J., Bogachus L.D., Turcotte L.P. AMPKalpha2 deficiency uncovers time dependency in the regulation of contraction-induced palmitate and glucose uptake in mouse muscle. Journal of Applied Physiology. 2011;111:125–134. doi: 10.1152/japplphysiol.00807.2010. [DOI] [PubMed] [Google Scholar]
- 14.Rose A.J., Alsted T.J., Jensen T.E., Kobbero J.B., Maarbjerg S.J., Jensen J. A Ca(2+)-calmodulin-eEF2K-eEF2 signalling cascade, but not AMPK, contributes to the suppression of skeletal muscle protein synthesis during contractions. The Journal of Physiology. 2009;587:1547–1563. doi: 10.1113/jphysiol.2008.167528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baylor S.M., Hollingworth S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. The Journal of Physiology. 2003;551:125–138. doi: 10.1113/jphysiol.2003.041608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung A., Dantzig J.A., Hollingworth S., Baylor S.M., Goldman Y.E., Mitchison T.J. A small-molecule inhibitor of skeletal muscle myosin II. Nature Cell Biology. 2002;4:83–88. doi: 10.1038/ncb734. [DOI] [PubMed] [Google Scholar]
- 17.Straight A.F., Cheung A., Limouze J., Chen I., Westwood N.J., Sellers J.R. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- 18.Dou Y., Arlock P., Arner A. Blebbistatin specifically inhibits actin-myosin interaction in mouse cardiac muscle. American Journal of Physiology. Cell Physiology. 2007;293:C1148–C1153. doi: 10.1152/ajpcell.00551.2006. [DOI] [PubMed] [Google Scholar]
- 19.Steimle P.A., Fulcher F.K., Patel Y.M. A novel role for myosin II in insulin-stimulated glucose uptake in 3T3-L1 adipocytes. Biochemical and Biophysical Research Communications. 2005;331:1560–1565. doi: 10.1016/j.bbrc.2005.04.082. [DOI] [PubMed] [Google Scholar]
- 20.Chambers M.A., Moylan J.S., Smith J.D., Goodyear L.J., Reid M.B. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. The Journal of Physiology. 2009;587:3363–3373. doi: 10.1113/jphysiol.2008.165639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blair D.R., Funai K., Schweitzer G.G., Cartee G.D. A myosin II ATPase inhibitor reduces force production, glucose transport, and phosphorylation of AMPK and TBC1D1 in electrically stimulated rat skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2009;296:E993–E1002. doi: 10.1152/ajpendo.91003.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandstrom M.E., Zhang S.J., Westerblad H., Katz A. Mechanical load plays little role in contraction-mediated glucose transport in mouse skeletal muscle. The Journal of Physiology. 2007;579:527–534. doi: 10.1113/jphysiol.2006.123372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birk J.B., Wojtaszewski J.F. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. The Journal of Physiology. 2006;577:1021–1032. doi: 10.1113/jphysiol.2006.120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frosig C., Pehmoller C., Birk J.B., Richter E.A., Wojtaszewski J.F. Exercise-induced TBC1D1 Ser237 phosphorylation and 14-3-3 protein binding capacity in human skeletal muscle. The Journal of Physiology. 2010;588:4539–4548. doi: 10.1113/jphysiol.2010.194811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sylow L., Jensen T.E., Kleinert M., Mouatt J.R., Maarbjerg S.J., Jeppesen J. Rac1 is a novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Diabetes. 2013;62:1139–1151. doi: 10.2337/db12-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandstrom M.E., Zhang S.J., Bruton J., Silva J.P., Reid M.B., Westerblad H. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. The Journal of Physiology. 2006;575:251–262. doi: 10.1113/jphysiol.2006.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koh H.J., Toyoda T., Fujii N., jung M.M., Rathod A., Middelbeek R.J. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15541–15546. doi: 10.1073/pnas.1008131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen T.E., Maarbjerg S.J., Rose A.J., Leitges M., Richter E.A. Knockout of the predominant conventional PKC isoform, PKCalpha, in mouse skeletal muscle does not affect contraction-stimulated glucose uptake. American Journal of Physiology. Endocrinology and Metabolism. 2009;297:E340–E348. doi: 10.1152/ajpendo.90610.2008. [DOI] [PubMed] [Google Scholar]
- 29.Wojtaszewski J.F., Laustsen J.L., Derave W., Richter E.A. Hypoxia and contractions do not utilize the same signaling mechanism in stimulating skeletal muscle glucose transport. Biochimica et Biophysica Acta. 1998;1380:396–404. doi: 10.1016/s0304-4165(98)00011-7. [DOI] [PubMed] [Google Scholar]
- 30.Li Q., Zhu X., Ishikura S., Zhang D., Gao J., Sun Y. Ca2+ signals promote GLUT4 exocytosis and reduce its endocytosis in muscle cells. American Journal of Physiology. Endocrinology and Metabolism. 2014;307:E209–E224. doi: 10.1152/ajpendo.00045.2014. [DOI] [PubMed] [Google Scholar]
- 31.Witczak C.A., Jessen N., Warro D.M., Toyoda T., Fujii N., Anderson M.E. CaMKII regulates contraction- but not insulin-induced glucose uptake in mouse skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2010;298:E1150–E1160. doi: 10.1152/ajpendo.00659.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ikemoto T., Hosoya T., Takata K., Aoyama H., Hiramatsu T., Onoe H. Functional role of neuroendocrine-specific protein-like 1 in membrane translocation of GLUT4. Diabetes. 2009;58:2802–2812. doi: 10.2337/db09-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jensen T.E., Rose A.J., Jorgensen S.B., Brandt N., Schjerling P., Wojtaszewski J.F. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. American Journal of Physiology. Endocrinology and Metabolism. 2007;292:E1308–E1317. doi: 10.1152/ajpendo.00456.2006. [DOI] [PubMed] [Google Scholar]
- 34.Abbott M.J., Edelman A.M., Turcotte L.P. CaMKK is an upstream signal of AMP-activated protein kinase in regulation of substrate metabolism in contracting skeletal muscle. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2009;297:R1724–R1732. doi: 10.1152/ajpregu.00179.2009. [DOI] [PubMed] [Google Scholar]
- 35.Lanner J.T., Bruton J.D., Assefaw-Redda Y., Andronache Z., Zhang S.J., Severa D. Knockdown of TRPC3 with siRNA coupled to carbon nanotubes results in decreased insulin-mediated glucose uptake in adult skeletal muscle cells. FASEB Journal. 2009;23:1728–1738. doi: 10.1096/fj.08-116814. [DOI] [PubMed] [Google Scholar]
- 36.Kitajima N., Watanabe K., Morimoto S., Sato Y., Kiyonaka S., Hoshijima M. TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochemical and Biophysical Research Communications. 2011;409:108–113. doi: 10.1016/j.bbrc.2011.04.124. [DOI] [PubMed] [Google Scholar]
- 37.Witczak C.A., Jessen N., Warro D.M., Toyoda T., Fujii N., Anderson M.E. CaMKII regulates contraction-induced, but not insulin-induced, glucose uptake in mouse skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2010;298:E1150–E1160. doi: 10.1152/ajpendo.00659.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merry T.L., Steinberg G.R., Lynch G.S., McConell G.K. Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. American Journal of Physiology. Endocrinology and Metabolism. 2010;298:E577–E585. doi: 10.1152/ajpendo.00239.2009. [DOI] [PubMed] [Google Scholar]
- 39.Lanner J.T., Katz A., Tavi P., Sandstrom M.E., Zhang S.J., Wretman C. The role of Ca2+ influx for insulin-mediated glucose uptake in skeletal muscle. Diabetes. 2006;55:2077–2083. doi: 10.2337/db05-1613. [DOI] [PubMed] [Google Scholar]
- 40.Cartee G.D., Briggs-Tung C., Holloszy J.O. Diverse effects of calcium channel blockers on skeletal muscle glucose transport. American Journal of Physiology. 1992;263:R70–R75. doi: 10.1152/ajpregu.1992.263.1.R70. [DOI] [PubMed] [Google Scholar]
- 41.Limouze J., Straight A.F., Mitchison T., Sellers J.R. Specificity of blebbistatin, an inhibitor of myosin II. Journal of Muscle Research and Cell Motility. 2004;25:337–341. doi: 10.1007/s10974-004-6060-7. [DOI] [PubMed] [Google Scholar]
- 42.Choi Y.O., Ryu H.J., Kim H.R., Song Y.S., Kim C., Lee W. Implication of phosphorylation of the myosin II regulatory light chain in insulin-stimulated GLUT4 translocation in 3T3-F442A adipocytes. Experimental & Molecular Medicine. 2006;38:180–189. doi: 10.1038/emm.2006.22. [DOI] [PubMed] [Google Scholar]
- 43.Fulcher F.K., Smith B.T., Russ M., Patel Y.M. Dual role for myosin II in GLUT4-mediated glucose uptake in 3T3-L1 adipocytes. Experimental Cell Research. 2008;314:3264–3274. doi: 10.1016/j.yexcr.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung le T.K., Hosaka T., Harada N., Jambaldorj B., Fukunaga K., Nishiwaki Y. Myosin IIA participates in docking of Glut4 storage vesicles with the plasma membrane in 3T3-L1 adipocytes. Biochemical and Biophysical Research Communications. 2010;391:995–999. doi: 10.1016/j.bbrc.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 45.Ihlemann J., Ploug T., Hellsten Y., Galbo H. Effect of stimulation frequency on contraction-induced glucose transport in rat skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2000;279:E862–E867. doi: 10.1152/ajpendo.2000.279.4.E862. [DOI] [PubMed] [Google Scholar]
- 46.Stephenson D.G., Williams D.A. Calcium-activated force responses in fast- and slow-twitch skinned muscle fibres of the rat at different temperatures. The Journal of Physiology. 1981;317:281–302. doi: 10.1113/jphysiol.1981.sp013825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Derave W., Straumann N., Olek R.A., Hespel P. Electrolysis stimulates creatine transport and transporter cell surface expression in incubated mouse skeletal muscle: potential role of ROS. American Journal of Physiology. Endocrinology and Metabolism. 2006;291:E1250–E1257. doi: 10.1152/ajpendo.00060.2006. [DOI] [PubMed] [Google Scholar]
- 48.Jensen T.E., Richter E.A. Regulation of glucose and glycogen metabolism during and after exercise. The Journal of Physiology. 2012;590:1069–1076. doi: 10.1113/jphysiol.2011.224972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai Y.C., Kviklyte S., Vertommen D., Lantier L., Foretz M., Viollet B. A small-molecule benzimidazole derivative that potently activates AMPK to increase glucose transport in skeletal muscle: comparison with effects of contraction and other AMPK activators. Biochemical Journal. 2014;460:363–375. doi: 10.1042/BJ20131673. [DOI] [PubMed] [Google Scholar]
- 50.Jørgensen S.B., Viollet B., Andreelli F., Frosig C., Birk J.B., Schjerling P. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. The Journal of Biological Chemistry. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 51.An D., Toyoda T., Taylor E.B., Yu H., Fujii N., Hirshman M.F. TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes. 2010;59:1358–1365. doi: 10.2337/db09-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee-Young R.S., Griffee S.R., Lynes S.E., Bracy D.P., Ayala J.E., McGuinness O.P. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. The Journal of Biological Chemistry. 2009;284:23925–23934. doi: 10.1074/jbc.M109.021048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas M.M., Wang D.C., D'Souza D.M., Krause M.P., Layne A.S., Criswell D.S. Muscle-specific AMPK beta1beta2-null mice display a myopathy due to loss of capillary density in nonpostural muscles. FASEB Journal. 2014;28:2098–2107. doi: 10.1096/fj.13-238972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y., Lai Y.C., Hill E.V., Tyteca D., Carpentier S., Ingvaldsen A. Phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) is an AMPK target participating in contraction-stimulated glucose uptake in skeletal muscle. Biochemical Journal. 2013;455:196–206. doi: 10.1042/BJ20130644. [DOI] [PubMed] [Google Scholar]
- 55.Ikemoto T., Suzuki M., Onoe H. Involvement of a phosphorylation-mediated pathway to regulate the function of NSPL1 in exercise. Journal of Veterinary Medical Science. 2011;73:733–738. doi: 10.1292/jvms.10-0543. [DOI] [PubMed] [Google Scholar]
- 56.Chen Q., Quan C., Xie B., Chen L., Zhou S., Toth R. GARNL1, a major RalGAP alpha subunit in skeletal muscle, regulates insulin-stimulated RalA activation and GLUT4 trafficking via interaction with 14-3-3 proteins. Cell Signal. 2014;26:1636–1648. doi: 10.1016/j.cellsig.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 57.Nozaki S., Ueda S., Takenaka N., Kataoka T., Satoh T. Role of RalA downstream of Rac1 in insulin-dependent glucose uptake in muscle cells. Cell Signal. 2012;24:2111–2117. doi: 10.1016/j.cellsig.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 58.O'Neill H.M., Maarbjerg S.J., Crane J.D., Jeppesen J., Jorgensen S.B., Schertzer J.D. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lantier L., Fentz J., Mounier R., Leclerc J., Treebak J.T., Pehmoller C. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB Journal. 2014;28:3211–3224. doi: 10.1096/fj.14-250449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.