

Abstract
Kinase Suppressor of Ras (KSR) is a molecular scaffold that interacts with the core kinase components of the ERK cascade, Raf, MEK, ERK to provide spatial and temporal regulation of Ras-dependent ERK cascade signaling. Interruption of this mechanism can have a high influence in inhibiting the downstream signaling of the mutated tyrosine kinase receptor kinase upon ligand binding. Still none of the studies targeted to prevent the binding of Raf, MEK binding on kinase suppressor of RAS. In that perspective the cysteine rich C1 domain of scaffold proteins kinase suppressor of Ras-1 was targeted rather than its ATP binding site with small ligand molecules like flavones and anthocyanidins and analyzed through insilico docking studies. The binding energy evaluation shows the importance of hydroxyl groups at various positions on the flavone and anthocyanidin nucleus. Over all binding interaction shows these ligands occupied the potential sites of cysteine rich C1 domain of scaffold protein KSR.
Keywords: Kinase suppressor of Ras, MAPK signaling, flavones, anthocyanidins, in silico docking, AutoDock 4.2.6, Neoplasia
Background
Mitogen activated protein kinase (MAPK) pathway refers to a module of three kinases which are activated by sequentially phosphorylating each other in response to a diverse range of stimuli, such as cytokines, growth factors, neurotransmitters, cellular stress and cell adherence [1]. Accordingly, the pathway plays a pivotal role in many key cellular processes, ranging from growth control in all its variations, cell differentiation and survival to cellular adaptation to chemical and physical stress. Augmentation in activity or mutations in receptor tyrosine kinases, that leads to neoplasia [2, 3]. Receptor linked tyrosine kinases such as the epidermal growth factor receptor (EGFR) are activated by extracellular ligands. Binding of epidermal growth factor (EGF) to the EGFR activates the tyrosine kinase activity of the cytoplasmic domain of the receptor. The EGFR becomes phosphorylated on tyrosine residues. Docking proteins such as GRB2 contains an SH2 domain that binds to the phosphotyrosine residues of the activated receptor [4]. GRB2 binds to the guanine nucleotide exchange factor SOS by way of the two SH3 domains of GRB2. When the GRB2-SOS complex docks to phosphorylated EGFR, SOS becomes activated. Activated SOS then promotes the removal of GDP from a member of the Ras subfamily (most notably H-Ras or K-Ras). Ras can then bind GTP and become active [5]
In between all these signaling process there is a role of scaffolding proteins for the activation of Raf [6, 7]. Scaffolds are defined as proteins with several domains that bind two or more components of a signaling pathway simultaneously. They bring signaling partners in close proximity to each other, link them in a multi-enzyme complex and facilitate their functional interaction. Within this complex, the kinases are shielded from the deactivating phosphatases, and interference with other signaling cascades is minimized [8, 9].
Kinase Suppressor of Ras (KSR1) is a molecular scaffold that interacts with the core kinase components of the ERK cascade, Raf, MEK, and ERK and provides spatial and temporal regulation of Ras-dependent ERK cascade signaling. CK2 is a component of the KSR1 scaffold complex that contributes to Raf kinase activation [10]. Raf-1 is a ceramide-activated kinase and that its C1 domain is involved in the ceramide-mediated response, Whereas KSR1 and its C1 domain is not getting activated in the same manner [11]. Unlike Raf-1, however, the kinase domain of KSR1 appears to be non-functional, suggesting that KSR-1 does not promote Ras signaling by phosphorylating target molecules [12, 13, 14]. So instead of targeting kinase domain, ligands directed at the cysteine rich C1 domain would have better functional activity in preventing Ras signaling upon inducing conformational changes along the scaffold protein preventing Raf-1, ERK binding to their site on KSR1. The crystal structure of cysteine rich C1 domain of kinase suppressor of Ras is shown in the (Figure 1). The conserved KSR1 domains include a 40 residue region unique to KSR1 proteins (CA1), a proline-rich region (CA2), a cysteine rich C1 domain (CA3), a serine/threonine-rich region (CA4), and a putative kinase domain (CA5). Similar to the domain organization of Raf-1, the smaller conserved domains of KSR1 are found in the N-terminal region, while the kinase-like domain occupies the C-terminal half of the protein. Unlike Raf 1, however, the kinase domain of KSR1 appears to be non functional, suggesting that KSR1 does not promote Ras signaling by phosphorylating target molecules. C1 domains are defined as regions of approximately 50 amino acid residues that contain the motif HX10-12CX2CX11-19CX2CX4 HX2-4CX5-9C [15]. C1A and C1B are the two repeat C1 domains located within the same protein. C1 domains were initially identified as the phorbol ester and 1, 2-dialyglycerol binding moieties of the protein kinase C (PKC) family of serine/ threonine kinases [16]. Ligands antagonizing the KSR1 scaffold activity and thereby interrupting the MAPK signaling pathway are not available yet. But researches were started focusing on to analyse its potential in binding with small molecules like flavonoids and other phytoconstituents. Present study focuses on binding affinity of some selected anthocyanidins and flavones on the basis of existing reviews [17]. Rare flavonoids like 2'-Hydroxygenistein was found to occupy the Raf binding site of KSR [18].
Figure 1.
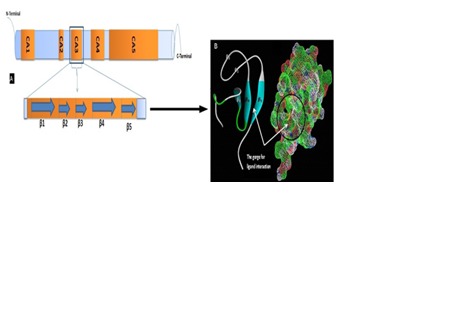
A) The schematic diagram shows the domains of knase suppressor of Ras with their terminals. The cysteine rich C1 domain − CA3 is emphasized to show their protein structure makup. The marking ( ) shown in the image indicates the respective beta sheets; B) Secondary structure showing the beta sheets (β1, β2, β3, β4, β5) containing amino acid residue at the gorge. The mesh diagram with encircled areas shows the gorge of KSR C1 domain. (Images have been visualized using Accelrys discovery studio 4.0 and Pymol viewer). The schematic diagram is created based on the information as per the literature.
In silico studies like protein-ligand docking has been carried out in order to find out a lead molecule for antagonizing the effect of KSR1. In this study latest version of Auto Dock 4.2.6 was utilized. AutoDock 4.2.6 features improved input checking and an output format suitable for automated analysis. Multiple search methods can be used in a single 4.2.6 job. Auto Dock 4.2.6 is an advanced docking platform which utilizes monte carlo simulated annealing and Lamarckian genetic algorithm (LGA) to create a set of possible conformations. LGA is used as a global optimizer and energy minimization as a local search method. For the evaluation of possible orientations, AMBER force field model in conjunction with free energy scoring function is used. Coordinate files preparation, atomic affinities (AutoGrid) calculation was made. Semi empirical free energy force field is used to evaluate conformations during docking. The Ligand and protein stay in an unbound conformation. Then binding is evaluated in two steps by force field. Force field evaluates intramolecular energetics during the translation from their unbounded states to the conformation of both ligand and protein into the form of bound state [19, 20]
Methodology
The crystal structure of the scaffold protein cysteine rich C1 domain of kinase suppressor of Ras was downloaded from RCSB protein data bank bearing the PDB code – 1KBE. Molecular docking was performed with MGL (Molecular Graphics Laboratory) tools–latest version of AutoDock 4.2.6 (release date: 2014-08-01), ligands were designed using Chem sketch [21]. Energy minimization was done with Chem Office package- Chem 3D ultra [22]. Interactive Graphic visualizers like Accelrys Discovery studio visualizer 4.0 [23], PyMol visualizer [24] was used.
Enzyme preparation and Ligand preparation:
Investigational ligands have designed and the ligands were optimized for energy minimization using MM2 force field [25]. The optimized ligands were shown in Figure 2.The ligands were selected according to the Lipinski's rule of five [26]. According to the drug likeness properties, the ligands showed zero violation of the Lipinski rule of five. Macromolecule has to be prepared, prior to docking process. Preparation involves removal of water molecule and any unwanted hetero atoms, because these will interfere in docking process. After refining enzyme macromolecule is saved as .pdb execution file.
Figure 2.
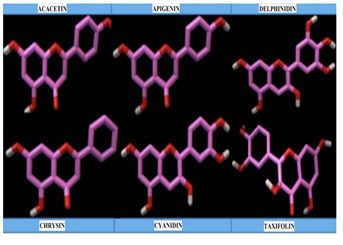
Energy minimized 3D-ligands used for docking study visualized in accelrys discovery studio 4.0
Validation of molecular docking:
To know the accuracy of molecular docking, the co-crystallized ligand was retrieved and again re-docked on to the binding site of ksr domain. If the RMSD value of the docked ligand is found to be less than 2.0Å, then the docking methodology is reliable [27]. Still current investigation is a prediction studies, we have retrieved the ligand (2-hydroxygenistein) from crystal structure as reported from the literature quoted in background section of this article from our previous findings and validated their docking efficiency.
Docking methodology:
Molecular docking was performed in making enzyme molecule rigid and ligand to get flexible, in this way different conformation arises during each run and the best conformer fits with lowest binding energy (kcal/mol). Auto Dock 4.2.6was used to automatically dock the ligands to the enzyme. In the latest version of Auto Dock 4.2.6 under windows platform, cygwin interface is not needed to perform the docking study. Rigid docking was performed using latest version of AutoDock 4.2 (4.2.6). The enzyme molecule is loaded and stored as ksr.pdb after assigning hydrogen bonds and kollman charges. The investigation ligand was loaded and their torsions along with rotatable bonds were assigned and the file is saved as ligand.pdbqt. Grid menu is toggled, after loading enzyme.pdbqt the map files were selected directly with setting up grid points with 106 × 124 × 74 dimensions for the searching of ligand within the active site of the enzyme molecule. This way the grid parameter files are created with setting up of map files directly. Followed by, setting up of docking parameter files with search parameter as genetic algorithm and docking parameter utilizing Lamarckian genetic algorithm. The Lamarckian genetic algorithm (LGA) was applied to deal with the protein– inhibitor interactions. These so-called state variables are the inhibitors genotype, and the resulting atomic coordinates together with the interaction and the intra-molecular energies are the inhibitors phenotype. The environmental adaptation of the phenotype is reverse transcribed into its genotype and become heritable traits. Each docking cycle or generation, consists of a regimen of fitness evaluation, crossover, mutation, and selection. Following up of grid parameter files (gpf) and docking log files (dlg), the command prompt is toggled and commands are typed in step wise for autogrid and autodock execution. The docked structures of the inhibitors are generated after a maximum number of evaluations [28].
Results & Discussion
Validation of molecular docking:
The actual (native) conformation of co-crystallized ligand (2'- hydroxygenistein) and re-docked conformation of the same ligand (2'-hydroxygenistein) is shown in Figure 3. The RMSD of all atoms between these two conformations is 1.7430Å. From that validation, investigating ligands are concluded to run for docking calculations.
Figure 3.
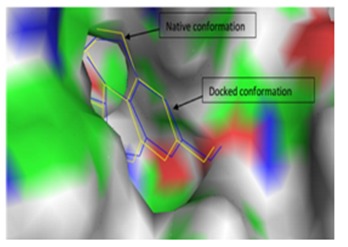
Validation of molecular docking. Surface image showing the native conformation and docked conformation of co-crystallized ligand (Image generated using Pymol Version 1.1)
Binding affinity and dissociation constant:
AutoDock 4.2 uses a semi-empirical free energy force field to evaluate conformations during docking simulations. The force field was parameterized using a large number of protein inhibitor complexes. The force field evaluates binding in two steps. The ligand and protein start in an unbound conformation. In the first step, the intramolecular energetics is estimated for the transition from these unbound states to the conformation of the ligand and protein in the bound state. The second step then evaluates the intermolecular energetic of combining the ligand and protein in their bound conformation. Table 1 list out the lowest binding energy of ligands at the gorge of the C1 domain of scaffold protein (KSR1). Among the investigated ligands cyanidin, Chrysin and Apigenin showed free energy of binding (ΔG) values -6.73, -6.56, -6.08 kcal/mol with a dissociation constant of 12.0uM, 15.66uM, 35.04uM respectively. Whereas ligands like delphinidin, acacetin and taxifolin possessed a considerable free energy of binding (ΔG) 5.74,-5.69, and -4.86 respectively.
Crystal structure of kinase suppressor of Ras-1 and its specificity :
The cysteine rich C1 domain is the investigational target site for the prediction on binding affinity character of flavonoids. KSR is an essential scaffolding protein to co-ordinate the assembly of Raf-MEK-ERK complexes in MAPK pathway of cellular proliferation [29]. The cysteine rich C1 domain containing amino acid chains extend from Gly 330 to Arg378. Aminoacids exposed at the active site gorge where Cys 346, Lys365, Gln344, Gln356, Trp341, Ile354, Phe355, Lys358, Lys365, Asn368 and Lys369, Lys365, Phe366. The crystal structure of C1 domain of KSR1 was found to contain cysteine residues at Cys377, Cys370, Cys366, Cys362, Cys 359, Cys349, and Cys 346.
Interaction analysis of flavones and anthocyanidins:
Occupancy of acacetin flexed their C7 hydroxyl groups towards Asn368 (2.04 Å) formed a hydrogen bonding but the opposite strand containing Thr338 (Figure 4 & Figure 5) doesn't involved in interaction even though oxygen atom of acacetin is in near vicinity. But in case of delphinidin, C7 hydroxyl group was replaced with a trihydroxyl phenyl group interacted at Thr338 (2.03 Å) through hydrogen bonding but that was absent in acacetin where it favored against Asn368. Where asC7 hydroxyl groups of taxifolin Hbond with the Thr338 (1.04 Å) at their catalytic site. Apigenin at its lowest binding energy (- 6.08kcal/mol) showed its interaction at Lys369 (2.03Å) where interaction was found to be absent through repeated docking studies and even in different poses generated during their run. This shows the Lys369 directed interaction of C7 hydroxyl groups of apigenin. Cyanidin with its free energy of binding (- 6.73kcal/mol) has a differential interaction at Phe355 and Val357 as a fragmentation type. On analyzing their hydrophobic interaction, they found to interact with Trp341 (5.22 Å) and Lys365 (3.15 Å). This makes their affinity stronger than other ligands under investigation. Bioflavonoids investigated in this study were found to interact with amino acids like ASN368, LYS365. Table 4 shows the amino acids involved in hydrogen bond interaction with the investigating ligands. Aminoacid residues at beta sheets β1, β4 was found to interact with the bioflavonoids and this gorge is the potential area of interaction.
Figure 4.
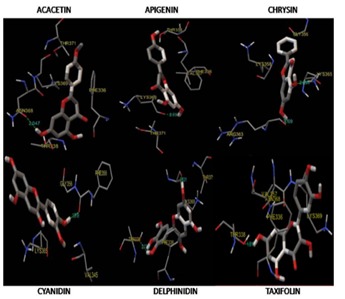
Image showing the binding interactions of the ligands used in the study along with their hydrogen bond length (Å). Images generated using MGLtools AutoDock 4.2.6 package (release date: 2014-08-01)
Figure 5.
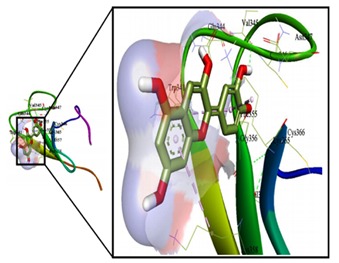
The gorge occupied area of cyanidin (left) has been emphasized (right), showing the non-bonding interactions (hydrophobic and hydrogen bond interactions) at the gorge of cystein rich C1domain of kinase suppressor of RAS. Image generated using acclerys discovery studio 4.0. Pale blue colour indicates the hydrophobic interaction and yellow colour indicates the hydrogen bonding.
Ligands structural specificity:
Experimental analysis on structure of KSR1 C1 domain showed that the lower two thirds of the protein surface are composed largely of positively charged residues. Located at the top of the C1 domain is a local hydrophobic region formed by residues in the hairpin structure; namely, L342, V345, M353, I354 and F355. These hydrophobic residues constitute the predicted ligand-binding pocket of the KSR1 C1 domain. The functional specificity and the structural features of the KSR1- C1 regulatory domains can provide valuable insight into their ligand-binding properties [30]. On analyzing ligand's structural specificity towards the hydrophobic site of ksr1, it explains the role of hydroxyl groups at C7 of flavone nucleus was found to interact through hydrogen bonding with most of the amino acids along the beta sheet ARG 363, LYS365, ASN368, and LYS369 (Figure 6). Some of the ligands showed fragmental binding (GLY356 and PHE355; LYS369; THR338) with the aminoacid residues. Lowest free energy of binding was shown by cyanidin which contains C7 hydroxyl group doesn't have any interaction at the gorge area. But instead the C4'substituted hydroxyl groups has its interaction through hydrogen bonding with Gly356 (1.919Å). Chrysin and taxifolin showed major interaction with ASN 368 and LYS 365. Over all from these binding affinity studies, the amino acids along the beta sheet (β4 and β5) has most of the interaction with the ligands and this might elicit conformational changes in the scaffold. This way these ligands can interrupt in Raf binding prior to ERK and Ras activation in Neoplasia through interaction at cysteine rich C1 domain of kinase suppressor of Ras-1.
Figure 6.
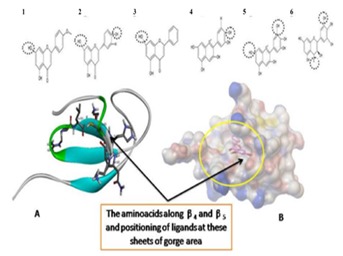
The ligands (1 to 6 as shown in table 1) above the crystal structure shows the hydroxyl groups (black dotted circles) involved in interaction through hydrogen bonding at the receptor site. A) Most of the interactions were with the amino acids along the beta sheet ARG 363, LYS365, ASN368, and LYS369 and B) Surface image with ligand (acacetin) positioning at its lowest binding energy (ΔG) at the gorge along the β4 and β5.
Conclusion
Computational designing and docking studies of potential bio flavonoids and anthocyanidins exhibited better binding affinity with kinase suppressor of Ras, the scaffold protein of MAPK signaling pathway. The binding energy evaluation shows the importance of hydroxyl groups at various positions on the flavone and anthocyanidin nucleus. Over all binding interaction shows these ligands occupied the essential area of Cysteine rich C1 domain of kinase suppressor of Ras-1. These ligands can be evaluated for in-vitro ksr-1 binding affinity studies and can be added as lead molecules in the development of drugs on targeting neoplasia through inhibition of MAPK signaling pathways.
Supplementary material
Footnotes
Citation:Karthik et al, Bioinformation 10(9): 580-585 (2014)
References
- 1.Chung KR, et al. MAP Kinase. 2013;2:17. [Google Scholar]
- 2.Cobb MH. Prog Biophys Mol Biol. 1999;71:479. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 3.Chang L, et al. Nature. 2001;410:37. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 4.Schulze WX, et al. Molecular systems biology. 2005;1:1. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zarich N, et al. Molecular Biology of the Cell. 2006;17:3591. doi: 10.1091/mbc.E05-12-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanoue T, Nishida E. Pharmacol Ther. 2002;93:193. doi: 10.1016/s0163-7258(02)00188-2. [DOI] [PubMed] [Google Scholar]
- 7.Pouyssegur J, et al. Eur J Biochem. 2003;270:3291. doi: 10.1046/j.1432-1033.2003.03707.x. [DOI] [PubMed] [Google Scholar]
- 8.Heinrich R, et al. Mol Cell. 2002;9:957. doi: 10.1016/s1097-2765(02)00528-2. [DOI] [PubMed] [Google Scholar]
- 9.Locasale JW, et al. Proc Natl Acad Sci. 2007;104:13307. doi: 10.1073/pnas.0706311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ritt DA, et al. Current Biology. 2007;17:179. doi: 10.1016/j.cub.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 11.Huwiler A, et al. Proc Natl Acad Sci USA. 1996;93:6959. [Google Scholar]
- 12.Therrien M, et al. Cell. 1995;83:879. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 13.Cacace JM, et al. Mol Cell Biol. 2000;20:5529. doi: 10.1128/mcb.20.15.5529-5539.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart S, et al. Mol Cell Biol. 1999;19:5523. doi: 10.1128/mcb.19.8.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Therrien M, et al. Cell. 1995;83:879. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 16.Driedger PE, Blumberg PM. Proc Natl Acad Sci. 1980;77:567. doi: 10.1073/pnas.77.1.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qingxin L, et al. Bio cell. 2011;35:71. [Google Scholar]
- 18.Dhananjayan K, Arunachalam S. J Pharm Res. 2014;8:423. [Google Scholar]
- 19. http://autodock.scripps.edu/faqshelp/tutorial.
- 20.Dhananjayan K, et al. Orient Pharm Exp Med. Doi: 10.1007/s13596-014-0160-8. [Google Scholar]
- 21.ACD/Structure Elucidator, version 12.01, Advanced Chemistry Development. Toronto, ON, Canada: Inc.; 2014. www.acdlabs.com. [Google Scholar]
- 22.Mills N, et al. J Am Chem Soc. 2006;128:13649. doi: 10.1021 /ja0697875. [Google Scholar]
- 23. http://accelrys.com/products/discovery-studio.
- 24.Daniel S, Bert LG. J Comput Aided Mol Des. 2010;24:417. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norman LA, et al. J Am Chem Soc. 1977;99:8127. [Google Scholar]
- 26.Lipinski CA, et al. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 27.Morris GM, et al. Journal of Computational Chemistry. 1998;19:1639. [Google Scholar]
- 28.Morris GM. J Comput Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennan DF, et al. Nature. 2011;472:366. doi: 10.1038/nature09860. [DOI] [PubMed] [Google Scholar]
- 30.Zhou M, et al. J Mol Biol. 2002;315:435. doi: 10.1006/jmbi.2001.5263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.