

Abstract
The mature heart valves are made up of highly organized extracellular matrix (ECM) and valve interstitial cells (VIC) surrounded by an endothelial cell layer. The ECM of the valves is stratified into elastin-, proteoglycan- and collagen-rich layers that confer distinct biomechanical properties to the leaflets and supporting structures. Signaling pathways have critical functions in primary valvulogenesis as well as maintenance of valve structure and function over time. Animal models provide powerful tools to study valve development and disease processes. Valve disease is a significant public health problem and increasing evidence implicates aberrant developmental mechanisms underlying pathogenesis. Further studies are necessary to determine regulatory pathway interactions underlying pathogenesis in order to generate new avenues for novel therapeutics.
Keywords: heart, cardiac development, animal models, valve disease
INTRODUCTION
Heart valves function to promote coordinated forward blood flow during the cardiac cycle. Valves are highly organized connective tissue structures populated with dynamic cell populations (1). Valvulogenesis occurs after the initial stages of cardiogenesis as a result of endocardial cushion formation and extensive remodeling of the extracellular matrix (ECM) (2, 3). The valve ECM is stratified, and the localized distribution of elastin, collagen, and proteoglycan underlies the biomechanical properties of the mature valve (4). Valve disease (stenosis or regurgitation) is a significant public health problem (5, 6). There are distinct types of valve malformations and disease that are characterized by ECM dysregulation, cellular disarray and often calcification. Any one of the four heart valves can be affected; however, the aortic valve is the most common site of disease (7). Aortic valve malformation, including bicuspid aortic valve (BAV), is present in 1–2% of the general population suggesting a developmental origin (8). Increasing evidence implicates aberrant developmental signaling pathways underlying valve disease pathogenesis (1–3). Faulty regulation of these interacting pathways results in maladaptive ECM remodeling, subtle malformation and ultimately disease.
VALVE ANATOMY AND STRUCTURE
Valve anatomy is complex (Figure 1). The mitral and tricuspid atrioventricular (AV) valves separate the atria from the ventricles, while the aortic and pulmonary semilunar (SL) valves separate the ventricles from the great arteries. AV valves have leaflets and SL valves have cusps. There is a specialized support structure specific to AV valves, while the distinct shape of SL valves creates a unique self-contained support structure within the arterial roots (9, 10). In contrast to the aorta, the aortic root is made up of the fibrous valve annulus region and the arterial tissue within the sinuses of Valsalva. The AV valves are characterized by large asymmetric leaflets hinged to ring shaped annuli on the secured end and tethered to the ventricles by an elaborate apparatus made up of the chordae tendineae and papillary muscles on the mobile end. The fibrous skeleton of the heart is continuous with the annulus fibrosa that constitutes the interconnected fibrous cartilage-like support apparatus of the tricuspid, mitral, and aortic valves. The annulus fibrosa is connected to the muscle of the heart in a manner that is analogous to the attachment of tendon to skeletal muscle (11, 12). The pulmonary valve is separated from the other valves by a muscular sleeve and has a poorly defined, less substantial annulus structure. The annuli of the AV valves are ring-shaped; however, the annulus of the aortic valve is crown-shaped resulting in the “semilunar” shape of the individual cusps (13, 14).
Figure 1.
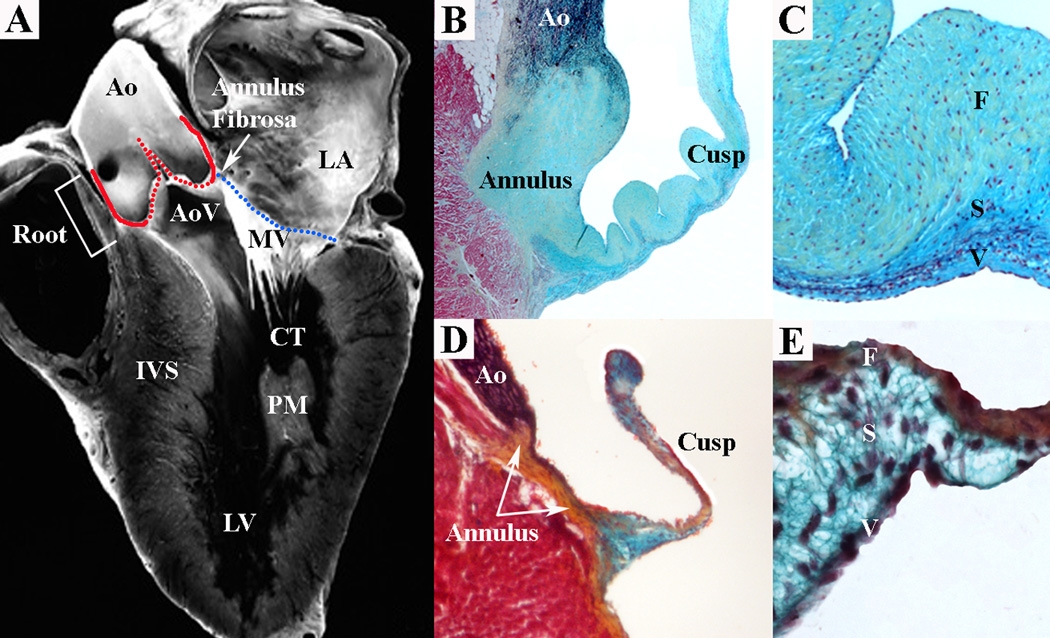
SL and AV valves with distinct structural and functional features are present in the human heart (A). The mitral valve (MV) is an AV valve and connects the left atrium (LA) to the left ventricle (LV). The MV consists of an annulus (A, blue line), leaflets and chordae tendineae (CT) that insert into papillary muscles (PM) in the myocardial wall. The aortic valve (AoV) is a SL valve and connects the LV to the aorta (Ao). The AoV consists of an annulus (A, red line) and cusps anchored within the aortic root (Root). Pentachrome staining shows valve ECM structure and composition in human (B,C) and mouse (D,E) aortic valves. At low magnification, SL valve tissue demonstrates cusp and annulus regions in human and mouse (B,D). At high magnification, aortic valve cusp architecture demonstrates similar ECM organization in human and mouse (C,E). The collagen-rich fibrosa layer (F) is oriented on the arterial aspect of the cusp, while the elastin-rich ventricularis layer (V) is oriented on the ventricular aspect of the cusp. The proteoglycan-rich spongiosa layer (S) interconnects the collagen and elastin fibers. IVS interventricular septum. (Panel A from reference (115), with permission.)
The mitral valve is composed of two leaflets, the anterior (or aortic) and posterior leaflets. The supporting tendinous cords (chordae tendineae) on the ventricular aspect of the valve leaflets insert into two well-defined papillary muscles that are continuous with the left ventricular myocardium. The posterior leaflet dominates the majority of the mitral valve annulus circumference, but the anterior leaflet is larger and makes up a greater area. Conversely, the tricuspid valve is composed of three leaflets, the anterior, posterior and septal leaflets, which attach to the ventricles via chordae tendineae to a large and variable number of seemingly unorganized papillary muscles within the trabecular right ventricle (9). The aortic valve is composed of three cusps, the left coronary, right coronary and non-coronary cusps, named for their relationship to the coronary arteries (15). The pulmonary valve is situated anterior and leftward relative to the aortic valve, and the mirror image “facing” cusps of the pulmonary valve are aligned in an orthogonal plane (13). Human valve thickness varies by valve and valve region, but in all valves is normally less than 1mm (16, 17). The AV valves are slightly thicker than the SL valves, and the left-sided valves are slightly thicker than the right-sided valves. The base and tip of the valves tend to be thicker, especially in the SL valves. The anterior leaflet of the mitral valve is in direct continuity with the aortic valve unlike the tricuspid valve, which is separated by muscular tissue from the pulmonary valve. Despite the common functional requirements of all heart valves, each valve is structurally different, and there is emerging molecular evidence that individual cusps and leaflets maintain distinct structural and biomechanical characteristics, potentially related to different intrinsic vulnerabilities to disease.
VALVE DEVELOPMENT
Valve morphogenesis
During embryonic development, the heart is the first organ to function, and it forms initially as a primitive tube composed of a myocardial cell layer surrounding an endocardial endothelial cell layer (3). The first indication of valve development during vertebrate embryogenesis is the formation of endocardial cushions in the outflow tract (OFT) and atrioventricular (AV) canal regions of the primitive heart tube (reviewed in (18)). Endocardial cushion formation is initiated when signaling factors emanating from the myocardium induce an epithelial to mesenchymal transition (EMT) of adjacent endocardial endothelial cells (19). This EMT generates mesenchymal progenitor cells that contribute to valvuloseptal structures and adult valve interstitial cells (20, 21). Initially the mesenchymal cells of the endocardial cushions are highly proliferative, and they are embedded in a loosely organized extracellular matrix (22). The endocardial cushion swellings in the OFT and AV canal function as valves to drive unidirectional blood flow in the primitive heart tube (23). Valve primordia corresponding to the distinct leaflets of the four valves arise with septation of the OFT and fusion of the AV canal cushions. Valve leaflet formation is characterized by thinning and elongation of the valve primordia, as well as remodeling of the ECM into layers rich in elastin (atrialis of AV valves/ventricularis of SL valves), fibrillar collagen (fibrosa), and proteoglycans (spongiosa) (18, 24). Likewise valve cell proliferation decreases during remodeling, and there is little to no proliferation of adult VICs (24, 25).
Embryonic origins of valve precursor cells
Heart valve cells come from multiple sources in the developing embryo. The endothelial cells that surround the valve leaflets form a continuous epithelial cell layer with the endocardium (2). In the OFT, both the endocardial and myocardial precursors arise from the secondary heart field (26). During the early stages of endocardial cushion formation, the mesenchymal cells of the AV and OFT cushions are derived from endothelial cells, as determined by Tie2-Cre;ROSA26R reporter lineage tracing in mice (27). In the mature AV valves, the VICs also are derived primarily, if not entirely, from Tie2-Cre expressing endothelial cells (20, 21). In mice, there is little if any contribution of VICs in the AV valves from epicardially-derived cells, as indicated by Wilms Tumor 1 (WT1)-Cre lineage analysis (28). However, chick-quail chimera studies in avian embryos have reported significant contributions of epicardium-derived cells in the developing AV valves (27, 29). In the developing OFT cushions, there are significant numbers of neural crest-derived cells, as demonstrated by Wnt1-Cre lineage studies in mice (27). In the mature SL valves, the cells of neural crest origin persist and are concentrated in individual cusps of the pulmonary and aortic valves (30)(Mead and Yutzey, unpublished). Overall, lineage tracing studies in mice demonstrate that the majority of VICs arise from endothelially-derived progenitors in the endocardial cushions. However, there is increasing evidence that specific subpopulations in individual valve leaflets arise from distinct embryonic sources. It is not known if these cells from diverse embryonic origins represent different subpopulations of VICs with specific contributions to mature valve structure and function.
Molecular regulation of valvulogenesis
Several developmentally important signaling pathways have critical functions in endocardial cushion induction and EMT. BMP2 signaling from the myocardium to the endocardium in the OFT and AV canal is required for the initial induction of EMT (31). Canonical Wnt signaling, as well as TGF-beta signaling, also are required for EMT and proliferation of mesenchymal endocardial cushion cells (19, 32, 33). Notch signaling in the endocardium regulates the repression of endothelial cell gene expression and also is required for EMT (34). The mesenchymal cells of the endocardial cushions are highly proliferative and express Twist1 and Msx transcription factors, characteristic of mesenchymal progenitor populations of many organ systems (18, 35). Twist1, along with Tbx20, promotes cell proliferation, migration and primitive ECM gene expression in the endocardial cushions and subsequently is down-regulated during valve remodeling (36, 37). Together, the valve progenitor cells of the endocardial cushions express many of the genes and cellular properties of mesenchymal cell types involved in development and regeneration, as well as tumor metastasis.
The generation of ECM compartments of the stratified valve leaflets is controlled by regulatory interactions shared with related connective tissue types (Figure 2) (12, 18). The transition from endocardial cushion to remodeling valve requires the transcription factor NFATc1, that promotes the expression of the ECM remodeling enzyme gene cathepsinK in valve endothelial cells, as well as osteoclasts in remodeling bone (38–40). Research over the past few years has demonstrated striking parallels in the interactions among signaling molecules, transcription factors, and structural proteins that control differentiation of connective tissues, such as cartilage, tendon, and bone, and also regulate compartmentalized ECM gene expression in the developing valves (12). For example, BMP2 signaling activates the Sox9 transcription factor and aggrecan gene expression in cartilage precursors as well as valve progenitors (41, 42). In addition, FGF4 signaling activates Scleraxis and tenascin in developing tendons as well as in the remodeling valves (41, 42). Wnt signaling, active in the developing valves and critical for early bone formation, promotes expression of genes characteristic of the collagen-rich fibrosa layer in cultured valve interstitial cells (43). The initiation and maintenance of valve leaflet stratification are likely affected by hemodynamics and biomechanical forces acting on the valves during the cardiac cycle (44). However, the molecular basis for the integration of valve function and cardiovascular physiology with ECM compartmentalization during development and later in life has not been elucidated.
Figure 2.

Regulatory interactions of signaling pathways and transcription factors in heart valve development. Signaling pathways including Notch, Transforming growth factor (TGF), Bone morphogenetic protein (BMP), and Wnt, with transcription factors including Twist1, Tbx20 and Msx1/2 are involved in endocardial cushion (EC) formation during early valvulogenesis. NFATc1 signaling contributes to elongation and remodeling of the EC. During valve maturation, BMP signaling induces cartilage-associated genes Sox9 and aggrecan. Fibroblast growth factor (FGF) signaling promotes expression of scleraxis and tenascin, which are characteristic of tendon cell lineages. These genes and pathways involved in valve development also are active in adult valve disease.
VALVE COMPOSITION AND FUNCTION
ECM composition and organization in the developing and mature valves
Normal valve function requires coordinated activity of complex structures. Gross and Kugel systematically described the histology of the human heart valves in 1931, and the proposed nomenclature for valve tissue organization is now established (45). The mature valve structure is made up of highly organized ECM that is compartmentalized into three layers, the fibrosa, spongiosa, and either the ventricularis of the SL valves or the atrialis of the AV valves (Figure 1). The fibrosa, which is situated on the ventricular aspect of AV valves and the arterial aspect of SL valves, is composed predominantly of fibrillar collagens (types I and III) that are circumferentially oriented and provide tensile stiffness (46–49). The atrialis layer of the AV valves and the ventricularis layer of the SL valves are composed primarily of radially-oriented filamentous elastic fibers that facilitate tissue motion (50, 51). Elastic fibers extend from the valve hinge to the closing or coapting edge and therefore do not run the entire length of a valve. The atrialis/ventricularis layer facilitates valve tissue movement by allowing extension and recoil of the valve during the cardiac cycle. The spongiosa makes up the middle area and is composed primarily of proteoglycans with interspersed collagen fibers. Proteoglycans are present throughout the valve thickness, but are the predominant matrix component of the middle layer and serve as an interface between the orthogonally arranged fibrosa and atrialis/ventricularis layers to provide tissue compressibility and integrity. The annulus, composed primarily of fibrous collagen, provides a buttress for dispersion of forces, and tethering of the cusp/leaflet free edges is required for tissue stabilization. In the AV valves, leaflets are connected to the ventricular myocardium by chordae tendineae, while in the SL valves, cusps are anchored directly to the arterial roots. There is redundant tissue at the tips of both AV and SL valves that provides functional valve closure or coaptation of the valve leaflets/cusps and ultimately competence when the valve is closed. There has to be a precise balance between stiffness and flexibility. Therefore the stoichiometry and distribution of ECM components is critical to proper valve function.
Mice lacking specific ECM proteins have developmental defects in valve formation and function (Table 1). In contrast to the highly structured stratified ECM layers of the mature valve, the ECM of the endocardial cushions is initially composed primarily of hyaluronan, and the mesenchymal cells generate a loosely organized collagen network permissive for cell migration (22). Mice lacking the hyaluronan synthetic enzyme Has2 exhibit loss of endocardial cushion swelling and lack of EMT (52). Loss of the proteoglycans perlecan or versican also leads to OFT cushion abnormalities and embryonic lethality (53, 54). Likewise loss of cartilage link protein1 (Crtl1), that interacts with hyaluronan and versican, also leads to valvuloseptal defects (55). Elastin gene expression is initiated in the remodeling valves during late embryonic and neonatal stages (24). Mice lacking elastin do not survive after birth due to vascular obstruction, and elastin heterozygous mice have aortic valve abnormalities in both structure and function in adulthood (56–58). Periostin regulates collagen fibrillogenesis in a variety of connective tissues, and loss of periostin in mice leads to abnormal valve morphogenesis and collagen organization (59, 60). Similarly, loss of the crosslinking collagens 5a1 and 11a1 leads to thickening of the SL and AV valves, with altered ratios of fibrillar collagens 1 and 3 indicative of remodeling defects (61). Together these studies demonstrate that the expression and organization of diverse ECM components is essential to the morphogenesis and structural integrity of the valves during development and consequently after birth.
Table 1.
Mouse mutations in ECM genes associated with heart valve abnormalities
| ECM | Genotype | Phenotype | Ref. |
|---|---|---|---|
| Proteoglycan-related | |||
| Hyaluronan | Has2−/− | Lethal E9.5a; Lack endocardial cushions | (52) |
| Versican | hdf | Lethal E10.5; Lack endocardial cushions; OFT defects | (53) |
| Perlecan | Perlecan−/− | Lethal E10-P0; OFT cushion defects; other heart defects | (104) |
| Cartilage Link Protein | Crtl1−/− | Lethal P0; valvuloseptal defects and other abnormalities | (55) |
| ADAMTS9 | Adamts9+/− | Aortic valve cusp and annulus malformations | (98) |
| Elastic fiber-related | |||
| Elastin | Eln−/− | P0 death from vascular obstruction | (56) |
| Eln+/− | Aortic valve cusp and annulus malformations | (58) | |
| Fibrillin-1 | Fbn1−/− | Lethality by P14 from vascular complications | (105) |
| Fbn1+/− | Mitral valve prolapse | (94) | |
| Fibulin-4 | Fibulin4-R/R | Adult thickened aortic valves and vascular defects | (106) |
| Fibrillar collagen-related | |||
| Periostin | Postn−/− | Spectrum of lethal and non-lethal valve defects | (59) |
| Collagen 1a1 | OIM | Thickening of adult semilunar valves | (Yutzey, unpublished) |
| Collagen 3a1 | Col3a1−/− | Aortic aneurysm; valves not examined | (95) |
| Collagen 11 | Col11a1−/− | P0 lethality; Thickened heart valves | (61, 107) |
Abbreviations: E embryonic day; P postnatal day; OFT outflow tract.
The ECM composition of the mature valves is dependent on the synthetic activity of the valve interstitial cells (VIC). During valve remodeling, the VICs express genes that encode fibrillar collagens, chondroitin sulfate proteoglycans, and elastin, associated with the stratified ECM of the valve leaflets (24, 25). The localized expression of specific ECM proteins characteristic of different connective tissue cell types suggests that there are different subpopulations of VICs in the stratified valves, but this has not yet been definitively demonstrated. Additional ECM remodeling enzymes such as matrix metalloproteases (MMPs), tissue inhibitors of matrix metalloproteases (TIMPS), and cathepsins also are expressed during valve maturation (17, 25). VICs from remodeling valves are highly synthetic, and cell proliferation is reduced relative to the endocardial cushion cells (21, 24). In normal adult valves, the VICs are largely quiescent with little or no cell proliferation and maintain baseline levels of ECM gene expression necessary for valve homeostasis (25).
Biomechanics and hemodynamics
Valve structure-function relationships provide important insight in understanding mechanisms of valve homeostasis as well as developmental and disease processes. The heart valves function essentially to maintain unobstructed unidirectional blood flow. The hemodynamics of the normal mature heart are well established (62). Blood flows from low pressure atria to higher pressure ventricles, which in turn supply the great arteries. The left side of the heart maintains significantly higher pressures than the right side. As a result, the impact of various physiologic forces depends on the position and hemodynamic environment of the valve. Valve composition and biomechanics reflect underlying hemodynamics. There are three basic loading states that affect valve tissue during the cardiac cycle: flexure, shear and tension. Flexure occurs when the valve is actively opening or closing, shear occurs when blood is passing through the open valve, and tension occurs when the valve is closed (4, 63). Shear, compressive, and longitudinal stresses contribute to valve deformation, or displacement of the valve tissue during the constant motion of the cardiac cycle (64). Valve tissue has exceptionally high strain because the tissue cycles to a completely unloaded state with each heart beat (49). These deformation forces result in a compensatory balance in cell matrix composition. For example, comparison of porcine aortic and pulmonary valves demonstrates that the left sided aortic valve is thicker, predominantly as a result of increased collagen expression and increased thickness of the fibrosa layer (Alfieri, Carruthers, Yutzey, and Sacks, unpublished data). The heart beats more than 100,000 times per day handling approximately 5 liters of blood per minute. Over the average lifetime, there are greater than 3 billion heart beats, or cardiac cycles. Valve failure may result from an underlying predisposing genotype and valve malformation that alters the response to physiologic stresses. The long held appreciation of age-related degeneration (“wear and tear”) and latent valve disease may in fact represent subtle defects in valve tissue maintenance as regulated by developmental pathways.
VALVE MALFORMATION AND DISEASE
Valve disease is a public health problem
Valve disease results in approximately 20,000 deaths annually (65). The prevalence of aortic valve disease is 2.5% in the United States, corrected for age (66). Aortic valve sclerosis, a marker of valve disease and cardiovascular risk, is present in more than 25% of the aged (67). The actual direct cost for valve disease in the United States alone has been estimated at 1 billion dollars per year (68). Taken together, the public health impact of valve disease and burden to society is underappreciated. Valve disease may manifest as stenosis, an obstruction to outflow, or regurgitation, a defective closure resulting in backward flow. Valve disease tends to progress. Ultimately, ventricular function can be compromised. Aortic valve stenosis is the most common form of valve disease and classically manifests as angina, syncope and heart failure. The diagnosis can be made clinically and confirmed by echocardiography, which quantifies the severity, and, over time, the progression of disease (62). The majority of valve disease at any age has an underlying valve malformation suggesting a genetic basis (8).
Congenital heart valve malformations occur in approximately 2% of live births, and the incidence is thought to be significantly higher since many cases remain subclinical and therefore unidentified. The two most common types of valve malformation are bicuspid aortic valve (BAV), an aortic valve with two rather than three cusps, and mitral valve prolapse (MVP), a mitral valve with redundant and billowing leaflets that prolapse into the left atrium. BAV has been estimated in up to 2% and MVP in up to 5% of the general population (5). In addition, valve defects occur in approximately 30% of cardiovascular malformations (CVM), including complex defects where valve disease is one component of the diagnosis, e.g. aortic valve stenosis is part of hypoplastic left heart syndrome and pulmonary valve stenosis is part of tetralogy of Fallot (69). There is considerable evidence that congenital valve malformations have a genetic basis and therefore represent abnormalities in development (69). BAV and MVP are common findings in patients with gene mutations that impact connective tissue homeostasis (Table 2). In nonsyndromic families, mutations in NOTCH1 have been identified in cases of BAV and calcific aortic valve disease (70). Family based linkage studies have identified disease loci on chromosomes 18q, 13q and 5q for BAV and 16p, 11p and 13q for MVP, however no genes have been identified (71–74). Importantly, these linkage studies represent a significant proportion of cases and therefore likely harbor the causes of malformation and disease. Pedigree analysis is consistent with complex inheritance, and in the context of reduced penetrance and variable expressivity, valve malformation may be the result of multiple predisposing genotypes. Taken together, valve malformation is a subtle and viable genetic defect that commonly manifests as significant disease later in life.
Table 2.
Human mutations in ECM genes associated with heart valve abnormalities
| Gene | Syndrome | Valve phenotype | Ref. |
|---|---|---|---|
| FIBRILLIN-1 (FBN1) | Marfan | Aortic root dilation, BAVa, MVP | (108) |
| ELASTIN (ELN) | Williams | SVAS, BAV, MVP | (109) |
| TGFβReceptor-1 (TGFBR1) | Loeys-Dietz | Aortic aneurism, MVP | (110) |
| COLLAGEN-1 (COL1A1) | Osteogenesis Imperfecta | Aortic valve prolapse, MVP | (96) |
| COLLAGEN-3 (COL3A1) | Ehlers-Danlos | Aortic root dilation, BAV, MVP | (111) |
| NOTCH-1 | BAV, CAVD, CVM | (70) | |
| ACTA-2 | Aortic aneurism, BAV | (112) | |
| MYH-11 | Aortic aneurism, BAV | (113) | |
| FLN-A | BAV, MVP | (114) |
Abbreviations: BAV bicuspid aortic valve; MVP mitral valve prolapse; SVAS supravalvular aortic stenosis; CAVD calcific aortic valve disease; CVM cardiovascular malformation.
Valve malformation underlies valve disease
Although valve disease has been recognized as a significant cause of morbidity and mortality for a long time, it was not until the 1950s that isolated aortic valve disease in the context of valve malformation was appreciated. Consequently, the idea emerged that latent disease has its origins in subtle developmental abnormalities (75, 76). Subsequently, large scale studies have shown that at all ages, including advanced age, the majority of valve disease cases have a malformed valve (8, 77–80), suggesting valve disease is attributable to aberrant developmental mechanisms (81). In this context, valve disease may develop as a result of predisposing genotypes in combination with maladaptive valve tissue maintenance, that over time leads to valve disease. In addition to the association between valve disease and more severe congenital CVM, valve disease may also be associated with other “acquired” CVM. For example, approximately 20% of patients with aortic valve malformation also have aortopathy, raising fundamental questions about both etiology and therapy. In addition to abnormalities in the aorta, de Sa et al. demonstrated that patients with aortic valve malformation had histologic abnormalities in the pulmonary artery, supporting the idea that developmental abnormalities have multiple effects that may be clinically relevant (82). As more is learned about the pathogenesis of associated diseases, a molecular taxonomy will emerge that will facilitate clinical decision-making.
Valve histopathology identifies two basic disease processes
Valve histopathology tends to conform to one of two patterns, myxomatous change or fibrotic change. Myomatous degeneration is characterized by proteoglycan accumulation, collagen degradation, and elastic fiber fragmentation. These changes result in a “floppy” valve that is prone to prolapse and regurgitation. Conversely, fibrosis is characterized by collagen accumulation, proteoglycan degradation, and elastic fiber fragmentation. These changes result in a “stiff” valve that is prone to restricted movement and stenosis. Aortic valve stenosis is typically characterized by sclerosis (“hardening”) and progressive fibrosis with advanced disease marked by calcification. Calcification is a common late finding. The etiology of calcification is poorly understood; however, this aspect of valve disease has generated substantial interest as a potential avenue to develop new therapeutics. One benefit to studying pediatric valve disease is that the histopathology identified is not confounded by the common comorbidities of adulthood, namely coronary artery disease and hypertension. Since aortic valve disease often occurs in the context of coronary artery disease, there is considerable interest in applying coronary artery disease treatment paradigms to valve disease. For example, statin therapy is hypothetically appealing and showed early in vitro evidence of positive impact; unfortunately, a large clinical trial demonstrated that statin therapy does not appear to impact aortic valve disease incidence or progression (83). Elucidating the genetic and molecular basis of valve malformation will provide opportunities for the development of new therapies.
At the cellular level, heart valve disease is characterized by VIC activation, as well as increased ECM and remodeling enzyme gene expression (Figure 3)(24, 84, 85). VIC activation is apparent in increased cell proliferation and induction of myofibroblast markers, such as vimentin, MMP-13, smooth muscle α-actin (SMA), and embryonic nonmuscle myosin heavy chain (SMemb) (84). These markers also are expressed in valve progenitor cells during development, supporting the idea that activated VICs in diseased valves represent a developmental phenotype. This is supported by the observation that the transcription factor Twist1, critical in endocardial cushion mesenchyme, also is expressed in human diseased heart valves (Chakraborty, Wirrig, Hinton, and Yutzey, unpublished). During human aortic valve calcification, expression of several genes associated with osteogenesis, including Sox9, Runx2, osteocalcin, osteopontin, alkaline phophatase, and bone sialoprotein, is induced (86, 87). There is increasing evidence that calcific valve disease recapitulates gene regulatory interactions characteristic of osteogenesis.
Figure 3.

Valve interstitial cell (VIC) phenotype relates to maladaptive and pathologic signaling pathways. Quiescent VICs show little proliferation or gene expression, while activated VICs demonstrate increased proliferation and increased myofibroblast-associated gene expression. VIC activation may be adaptive or maladaptive, and patterns of signaling pathway gene expression may distinguish these features. Some maladaptive VIC activation and induction of genes associated with bone formation are apparent in valve tissue calcification. SMA smooth muscle α-actin; MMP matrix metalloprotease; OCN osteocalcin; BSP bone sialoprotein; ALP alkaline phosphatase.
The origins and inductive mechanisms of activated VICs in valve disease have not been identified. There is initial evidence from primary cell cultures that the interaction of VICs with the surrounding ECM contributes to VIC activation and osteogenic gene induction (88). Some VICs have been shown to be dynamic and play an active role in ECM maintenance (85, 89). It is possible that activated VICs arise from quiescent VICs resident in the valve leaflets. Alternatively, immature valve progenitors arising during development could remain in the adult valves as potential effectors of regeneration and repair. The presence of an exogenous stem cell population that is recruited to the valves during disease is supported by reports of hematopoietic stem cell derivatives in adult heart valves (90, 91). Further studies are necessary to determine the regenerative potential or pathologic mechanisms associated with VIC activation in valve disease.
Genetic syndromes and animal models of valve disease
Normal heart valve function is dependent on the biomechanical properties of the stratified ECM, and mutations in a variety of ECM genes are associated with human heart valve disease (Table 2). Several genetic syndromes characterized by connective tissue disorders include valve malformations and progressive valve dysfunction. Marfan syndrome, caused by mutations in FIBRILLIN-1, is characterized by thickening of the mitral and aortic valves in addition to the characteristic aortic root and skeletal anomalies (92). Similarly, Williams syndrome, associated with heterozygous ELASTIN mutations, includes arteriopathy manifested as supravalvar aortic stenosis as well as aortic valve disease (93). Many valve phenotypes in human genetic syndromes are recapitulated by targeted mutagenesis animal models (Table 1). Fibrillin-1 insufficient mice develop mitral valve prolapse similar to that seen in humans (94). Likewise, heterozygous elastin (eln +/−) mice develop progressive aortic valve malformation and latent aortic valve disease, similar to humans with degenerative aortic valve disease (56–58). Interestingly, these mice have both valve disease and aortopathy with the annulus region implicated in disease manifestation. These findings raise fundamental questions about both the origin and functional capacity of the aortic valve and aortic root.
Ehlers-Danlos syndrome is caused by a variety of collagen and tenascin gene mutations that affect connective tissue structure and function in multiple organs, including the heart valves (reviewed in (12)). There is currently not a mouse model for valve abnormalities related to Ehlers-Danlos syndrome, but mice lacking collagen 3a1 recapitulate the aortic rupture phenotype (95). In the future, it would be of interest to determine if these mice also have valve abnormalities and dysfunction associated with human Ehlers-Danlos syndrome. Mutations in COL1A1 are associated with the human bone condition ostegenesis imperfecta, and prolapse of aortic and mitral valves can occur in this patient population (96). A mouse model has been generated with a targeted Col1a1 oim mutation, and these animals develop progressive thickening of the semilunar valves with increased proteoglycan deposition as adults (97)(Wirrig, Cheek and Yutzey, unpublished). It is interesting to note that human valve disease related to proteoglycan gene mutations has not been reported. However, mice heterozygous for the versican-degrading protease gene Adamts9 have thickening of the semilunar valves and chondrogenic nodules in the annulus region (98). Mutations in additional isolated ECM genes are associated with human aortic and mitral valve malformations and disease, while dysregulation of the valve leaflet ECM organization and deposition is a general feature of valve disease regardless of etiology.
There is increasing evidence that disruption of the valve ECM induces signaling pathways that lead to maladaptive ECM remodeling and ultimately valve disease. The aortic and mitral valve phenotypes of Marfan syndrome are associated with increased TGF-beta signaling that contributes to the overall collagen dysregulation and loss of matrix integrity of these structures in animal models (94). Strikingly, inhibition of TGF-beta signaling via Losartan treatment reduces pathology in a mouse model of Marfan syndrome, and efficacy in humans also has been demonstrated (99, 100). Likewise in adult mice, heterozygous loss of elastin or homozygous loss of periostin, affects TGF-beta signaling associated with aortic valve degeneration and dysfunction (Table 1) (58, 59). Notch and Wnt signaling also have been reported to be altered in animal models of aortic valve disease, as well as in human patients, but the mechanisms of induction have not yet been defined (70, 101). TGF-beta, Notch and Wnt signaling all are required for normal heart valve development during embryogenesis, and there is increasing evidence that these pathways, in association with ECM dysregulation, contribute to progressive valve pathogenesis resulting in a variety of disease phenotypes later in life.
Valve disease treatment
The treatment of valve disease remains primarily surgical. Any one of the four heart valves can be affected; however, the aortic valve is the most common site of disease (7). Indications for valve replacement include clinical symptoms, ventricular dysfunction or exercise intolerance in asymptomatic patients. Aortic valve replacement is the second most common cardiothoracic procedure, and the need for reintervention is common. Nearly 100,000 valve replacement procedures are performed in the United States annually, and the majority of these are aortic valve replacement procedures (6). Bioprosthetic valve replacement has become increasingly popular, however continues to suffer from longevity issues. There have been exciting advances in interventional cardiac catheterization, including percutaneous insertion of the pulmonary valve (102). This approach was approved in January 2010 by the Food and Drug Administration under the Humanitarian Device Exemption program (www.fda.gov/NewsEvents/ucm198597.htm) and delays the need for open heart surgery. It may also be an appealing alternative in high risk cases. In addition, transcatheter aortic valve implantation using either a trans-femoral retrograde or trans-apical antegrade approach is under investigation in humans, primarily in Europe, and shows early promise (103). Once feasibility is established, clinical trials will be organized.
To improve the care of patients with valve disease, markers of future disease and disease progression need to be identified. Early identification of disease will allow early intervention and potentially preventive approaches to valve disease. Current medical therapy for valve disease treats the symptoms of cardiovascular disease. For example, some medicines are directed at the important symptoms that result from congestive heart failure, but do not impact the underlying cause or the primary problem, valve disease. As the genetic and developmental basis of valve malformation and disease is elucidated, opportunities for novel medical therapies will emerge and potentially preclude or delay the need for surgery. Defining regulation of valve tissue maintenance and homeostasis will provide exciting opportunities for cell-based or molecular therapies for valve disease.
CONCLUSIONS
The mature valve structure, consisting of highly organized ECM and dynamic VICs, maintains valve function. Together, the stratified ECM and VIC homeostatic mechanisms underlie valve structure and function and coordinate maintenance of valve tissue throughout a lifetime. Heart valve disease is characterized by dysregulation of ECM organization and VIC activation with induction of regulatory pathways active in valve development. Valve disease pathogenesis is being elucidated through study of animal models, and a better understanding of these mechanisms will allow the development of novel therapeutics.
Summary points.
Valve structure is dynamic and composed of interacting cells and stratified extracellular matrix
Valve malformation underlies valve disease, suggesting a developmental origin
Valve disease is a common problem that often requires surgery
Valve disease has a genetic basis characterized by aberrant developmental programs and maladaptive ECM remodeling
Developmental pathways modulate valve tissue maintenance
Translational efforts combining basic and clinical research may identify ways to manipulate faulty valve tissue maintenance
Future issues.
Identify links between cell plasticity and disease processes
Determine whether a subset of VICs can regenerate, and if so define repair mechanisms
Identify novel pharmacologic therapeutics using developmental pathways to elucidate valve disease pathogenesis
Develop durable valve bioprostheses using a combination of clinical, molecular and engineering approaches
Identify markers of disease to allow early intervention or preventive strategies
List of definitions.
Valve leaflet/cusp: the mobile region of valve tissue, AV valves have leaflets, SL valves have cusps
Valve annulus: the support structure of valve tissue, AV valves have rings, SL valves have crowns
Sinus of Valsalva: the arterial pouches within the aortic root that function in coordination with the aortic valve annulus
Acknowledgements
We thank Elaine Wirrig for critical reading of the manuscript and advice on figure preparation. We thank Christina Alfieri, Christopher Carruthers, Santanu Chakraborty, Jonathan Cheek, Timothy Mead, Michael Sacks, and Elaine Wirrig for communication of results prior to publication. R.B.H. is supported by the Cincinnati Children’s Research Foundation and NIH HL085122. Research in K.E.Y.’s laboratory is supported by the American Heart Association and NIH HL094319 and HL082716.
List of acronyms
- AV
Atrioventricular
- AVR
Aortic valve replacement
- BAV
Bicuspid aortic valve
- CVM
Cardiovascular malformation
- ECM
Extracellular matrix
- EMT
Epithelial to mesenchymal transition
- MVP
Mitral valve prolapse
- OFT
Outflow tract
- SL
Semilunar
- VIC
Valve interstitial cell
Footnotes
Disclosure Statement
The authors are not aware of any affiliations, memberships, funding or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
- 1.Schoen FJ. Evolving concepts of cardiac valve dynamics. Circulation. 2008;118:1864–1880. doi: 10.1161/CIRCULATIONAHA.108.805911. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong EJ, Bischoff J. Heart valve development: Endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 4.Sacks MS, David Merryman W, Schmidt DE. On the biomechanics of heart valve function. J Biomech. 2009;42:1804–1824. doi: 10.1016/j.jbiomech.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 6.Bonow RO, Carabello BA, Kanu C, de Leon AC, Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O’Gara PT, O’Rourke RA, Otto CM, Shah PM, Shanewise JS, Smith SC, Jr, Jacobs AK, Adams CD, Anderson JL, Antman EM, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Lytle BW, Nishimura R, Page RL, Riegel B. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): developed in collaboration with the Society of Cardiovascular Anesthesiologists: endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. Circulation. 2006;114:e84–e231. doi: 10.1161/CIRCULATIONAHA.106.176857. [DOI] [PubMed] [Google Scholar]
- 7.Supino PG, Borer JS, Preibisz J, Bornstein A. The epidemiology of valvular heart disease: a growing public health problem. Heart Fail Clin. 2006;2:379–393. doi: 10.1016/j.hfc.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;2005:920–925. doi: 10.1161/01.CIR.0000155623.48408.C5. [DOI] [PubMed] [Google Scholar]
- 9.Anderson RH, Ho SY, Becker AE. Anatomy of the human atrioventricular junctions revisited. Anat Rec. 2000;260:81–91. doi: 10.1002/1097-0185(20000901)260:1<81::AID-AR90>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Anderson RH. Clinical anatomy of the aortic root. Heart. 2000;84:670–673. doi: 10.1136/heart.84.6.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misfeld M, Sievers HH. Heart valve macro- and microstructure. Philos Trans R Soc Lond B Biol Sci. 2007;362:1421–1436. doi: 10.1098/rstb.2007.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lincoln J, Lange AW, Yutzey KE. Hearts and bones: Shared regulatory mechanisms in heart valve, cartilage, tendon, and bone development. Dev Biol. 2006;294:292–302. doi: 10.1016/j.ydbio.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 13.Zimmerman J, MBailey C. The surgical significance of the fibrous skeleton of the heart. J Thorac Cardiovasc Surg. 1962;44:701–712. [Google Scholar]
- 14.Yacoub MH, Kilner PJ, Birks EJ, Misfeld M. The aortic outflow and root: a tale of dynamism and crosstalk. Ann Thorac Surg. 1999;68:S37–S43. doi: 10.1016/s0003-4975(99)00745-6. [DOI] [PubMed] [Google Scholar]
- 15.Choo SJ, McRae G, Olomon JP, St George G, Davis W, Burleson-Bowles CL, Pang D, Luo HH, Vavra D, Cheung DT, Oury JH, Duran CM. Aortic root geometry: pattern of differences between leaflets and sinuses of Valsalva. J Heart Valve Dis. 1999;8:407–415. [PubMed] [Google Scholar]
- 16.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 17.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 18.Combs MD, Yutzey KE. Heart valve development: Regulatory networks in development and disease. Circ Res. 2009;105:408–421. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 20.de Lange FJ, Moorman AFM, Anderson RH, Manner J, Soufan AT, deGier-deVries C, Schneider MD, Webb S, Van Den Hoff MJ, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- 21.Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004;230:239–250. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- 22.Schroeder JA, Jackson LF, Lee DC, Camenisch TD. Form and function of developing heart valves: coordination by extracellular matrix growth and signaling. J Mol Med. 2003;81:392–403. doi: 10.1007/s00109-003-0456-5. [DOI] [PubMed] [Google Scholar]
- 23.Butcher JT, McQuinn TC, Sedmera D, Turner D, Markwald RR. Transitions in early embryonic atrioventricular valvular functions correspond with changes in cushion biomechanics that are predictable with tissue composition. Circ Res. 2007;100:1503–1511. doi: 10.1161/CIRCRESAHA.107.148684. [DOI] [PubMed] [Google Scholar]
- 24.Hinton RB, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 25.Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult. Circulation. 2006;113:1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 26.Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–145. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 27.Snarr BS, Kern CB, Wessels A. Origin and fate of cardiac mesenchyme. Dev Dyn. 2008;237:2804–2819. doi: 10.1002/dvdy.21725. [DOI] [PubMed] [Google Scholar]
- 28.Zhou B, von Gise A, Ma Q, Hu YW, Pu WT. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol. 338:251–261. doi: 10.1016/j.ydbio.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gittenberger-de Groot AC, Peeters MPFMV, Mentink MMT, Gourdie RG, Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82:1043–1052. doi: 10.1161/01.res.82.10.1043. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- 31.Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–5611. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- 32.Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Zivkovic D, Plasterk RH, Clevers H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425:633–537. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
- 33.Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. 2004;166:359–367. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, delaPompa JL. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang JH, Weinberg RA. Epithelial-Mesenchymal transition: At the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Shelton EL, Yutzey KE. Twist1 function in endocardial cell proliferation, migration, and differentiation during heart valve development. Dev Biol. 2008;317:282–295. doi: 10.1016/j.ydbio.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chakraborty S, Cheek J, Sakthivel B, Aronow BJ, Yutzey KE. Shared gene expression profiles in developing heart valves and osteoblasts. Physiol Genomics. 2008;35:75–85. doi: 10.1152/physiolgenomics.90212.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 39.Ranger AM, Grusby MJ, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 40.Combs MD, Yutzey KE. VEGF and RANKL regulation of NFATc1 in heart valve development. Circ Res. 2008;105:565–574. doi: 10.1161/CIRCRESAHA.109.196469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lincoln J, Alfieri CM, Yutzey KE. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev Biol. 2006;292:290–302. doi: 10.1016/j.ydbio.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 42.Zhao B, Etter L, Hinton RB, Benson DW. BMP and FGF regulatory pathways in semilunar valve precursor cells. Dev Dyn. 2007;236:971–980. doi: 10.1002/dvdy.21097. [DOI] [PubMed] [Google Scholar]
- 43.Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. 2010;338:127–135. doi: 10.1016/j.ydbio.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kruithof BPT, Krawitz SA, Gaussin V. Atrioventricular valve development during late embryonic and postnatal stages involves condensation and extracellular matrix remodeling. Dev Biol. 2007;302:208–217. doi: 10.1016/j.ydbio.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 45.Gross L, Kugel MA. Topographic anatomy and histology of the valves in the human heart. Am J Path. 1931;7:445–456. [PMC free article] [PubMed] [Google Scholar]
- 46.Broom ND. The observation of collagen and elastin structures in wet whole mounts of pulmonary and aortic leaflets. J Thorac Cardiovasc Surg. 1978;75:121–130. [PubMed] [Google Scholar]
- 47.Missirlis YF, Armeniades CD. Ultrastructure of the human aortic valve. Acta Anat (Basel) 1977;98:199–205. doi: 10.1159/000144794. [DOI] [PubMed] [Google Scholar]
- 48.Kershaw JD, Misfeld M, Sievers HH, Yacoub MH, Chester AH. Specific regional and directional contractile responses of aortic cusp tissue. J Heart Valve Dis. 2004;13:798–803. [PubMed] [Google Scholar]
- 49.Sacks MS, Yoganathan AP. Heart valve function: a biomechanical perspective. Philos Trans R Soc Lond B Biol Sci. 2007;362:1369–1391. doi: 10.1098/rstb.2007.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoen FJ. Aortic valve structure-function correlations: Role of elastic fibers no longer a stretch of the imagination. J Heart Valve Dis. 1997;6:1–6. [PubMed] [Google Scholar]
- 51.Vesely I. The role of elastin in aortic valve mechanics. J Biomech. 1998;31:115–123. doi: 10.1016/s0021-9290(97)00122-x. [DOI] [PubMed] [Google Scholar]
- 52.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro AJ, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mjaatvedt CH, Yamamura H, Capehart AA, Turner D, Markwald RR. The cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev Biol. 1998;202:56–66. doi: 10.1006/dbio.1998.9001. [DOI] [PubMed] [Google Scholar]
- 54.Costell M, Carmona R, Gustafsson E, Gonzalez-Iriarte M, Fassler R, Munoz-Chapuli R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ Res. 2002;91:158–164. doi: 10.1161/01.res.0000026056.81424.da. [DOI] [PubMed] [Google Scholar]
- 55.Wirrig EE, Snarr BS, Chintalapudi MR, O’Neal JL, Phelps AL, Barth JL, Fresco VM, Kern CB, Mjaatvedt CH, Toole BP, Hoffman S, Trusk TC, Argraves WS, Wessels A. Cartilage link protein 1 (Crtl1), an extracellular matrix component playing an important role in heart development. Dev Biol. 2007;310:291–303. doi: 10.1016/j.ydbio.2007.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 57.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RPPS, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783–1787. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hinton RB, Adelman-Brown J, Witt S, Krishnamurthy VK, Osinska H, Sakthivel B, James JF, Narmoneva DA, Mecham RP, Benson DW. Elastin Haploinsufficiency results in progressive aortic valve malformation and latent valve disease in a mouse model. Circ Res. 2010 doi: 10.1161/CIRCRESAHA.110.221358. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Snider P, Hinton RB, Moreno-Rodriguez R, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, Firulli AB, Molkentin JD, Markwald RR, Conway SJ. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102:752–760. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Norris RA, Moreno-Rodriguez R, Sugi Y, Hoffman S, Amos J, Hart MM, Potts JD, Goodwin RL, Markwald RR. Periostin regulates atrioventricular valve maturation. Dev Biol. 2008;316:200–213. doi: 10.1016/j.ydbio.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lincoln J, Florer JB, Deutsch GH, Wenstrup RJ, Yutzey KE. ColVa1 and ColXIa1 are required for ventricular chamber morphogenesis and heart valve development. Dev Dyn. 2006;235:3295–3305. doi: 10.1002/dvdy.20980. [DOI] [PubMed] [Google Scholar]
- 62.Otto CM. Valvular aortic stenosis: disease severity and timing of intervention. J Am Coll Cardiol. 2006;47:2141–2151. doi: 10.1016/j.jacc.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 63.Vesely I, Noseworthy R. Micromechanics of the fibrosa and the ventricularis in aortic valve leaflets. J Biomech. 1992;25:101–113. doi: 10.1016/0021-9290(92)90249-z. [DOI] [PubMed] [Google Scholar]
- 64.Grande KJ, Cochran RP, Reinhall PG, Kunzelman KS. Stress variations in the human aortic root and valve: the role of anatomic asymmetry. Ann Biomed Eng. 1998;26:534–545. doi: 10.1114/1.122. [DOI] [PubMed] [Google Scholar]
- 65.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 66.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 67.Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341:142–147. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- 68.Rajamannan NM, Gersh B, Bonow RO. Calcific aortic stenosis: from bench to the bedside--emerging clinical and cellular concepts. Heart. 2003;89:801–805. doi: 10.1136/heart.89.7.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pierpont ME, Basson CT, Benson DW, Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–3038. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 70.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 71.Martin LJ, Ramachandran V, Cripe LH, Hinton RB, Andelfinger G, Tabangin M, Shooner K, Keddache M, Benson DW. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum Genet. 2007;121:275–284. doi: 10.1007/s00439-006-0316-9. [DOI] [PubMed] [Google Scholar]
- 72.Disse S, Abergel E, Berrebi A, Houot AM, Le Heuzey JY, Diebold B, Guize L, Carpentier A, Corvol P, Jeunemaitre X. Mapping of a first locus for autosomal dominant myxomatous mitral-valve prolapse to chromosome 16p11.2-p12.1. Am J Hum Genet. 1999;65:1242–1251. doi: 10.1086/302624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freed LA, Acierno JSJ, Dai D, Leyne M, Marshall JE, Nesta F, Levine RA, Slaugenhaupt SA. A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. Am J Hum Genet. 2003;72:1551–1559. doi: 10.1086/375452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nesta F, Leyne M, Yosefy C, Simpson C, Dai D, Marshall JE, Hung J, Slaugenhaupt SA, Levine RA. New locus for autosomal dominant mitral valve prolapse on chromosome 13: clinical insights from genetic studies. Circulation. 2005;112:2022–2030. doi: 10.1161/CIRCULATIONAHA.104.516930. [DOI] [PubMed] [Google Scholar]
- 75.Smith DE, Matthews MB. Aortic valvular stenosis with coarctation of the aorta, with special reference to the development of aortic stenosis upon congenital bicuspid valves. Br Heart J. 1955;17:198–206. doi: 10.1136/hrt.17.2.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol. 1970;26:72–83. doi: 10.1016/0002-9149(70)90761-7. [DOI] [PubMed] [Google Scholar]
- 77.Pomerance A. Pathogenesis of aortic stenosis and its relation to age. Br Heart J. 1972;34:569–574. doi: 10.1136/hrt.34.6.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Passik CS, Ackermann DM, Pluth JR, Edwards WD. Temporal changes in the causes of aortic stenosis: a surgical pathologic study of 646 cases. Mayo Clin Proc. 1987;62:119–123. doi: 10.1016/s0025-6196(12)61880-1. [DOI] [PubMed] [Google Scholar]
- 79.Peterson MD, Roach RM, Edwards JE. Types of aortic stenosis in surgically removed valves. Arch Pathol Lab Med. 1985;109:829–832. [PubMed] [Google Scholar]
- 80.Stephan PJ, Henry AC, 3rd, Hebeler RF, Jr, Whiddon L, Roberts WC. Comparison of age, gender, number of aortic valve cusps, concomitant coronary artery bypass grafting, and magnitude of left ventricular-systemic arterial peak systolic gradient in adults having aortic valve replacement for isolated aortic valve stenosis. Am J Cardiol. 1997;79:166–172. doi: 10.1016/s0002-9149(96)00705-9. [DOI] [PubMed] [Google Scholar]
- 81.Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. 2009;2:489–498. doi: 10.1161/CIRCGENETICS.108.820795. [DOI] [PubMed] [Google Scholar]
- 82.de Sa M, Moshkovitz Y, Butany J, David TE. Histologic abnormalities of the ascending aorta and pulmonary trunk in patients with bicuspid aortic valve disease: clinical relevance to the ross procedure. J Thorac Cardiovasc Surg. 1999;118:588–594. doi: 10.1016/S0022-5223(99)70002-4. [DOI] [PubMed] [Google Scholar]
- 83.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 84.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes in interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Disease. 2004;13:841–847. [PubMed] [Google Scholar]
- 85.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathology. Am J Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rajamannan NM, Subramaniam M, Rickard DJ, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg TC. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 89.Filip DA, Radu A, Simionescu M. Interstitial cells of the heart valves posess characteristics similar to smooth muscle cells. Circ Res. 1986;59:310–320. doi: 10.1161/01.res.59.3.310. [DOI] [PubMed] [Google Scholar]
- 90.Deb A, Wnag SH, Skelding K, Miller D, Simper D, Caplice N. Bone marrow-derived myofibroblasts are present in adult human heart valves. J Heart Valve Dis. 2005;14:674–678. [PubMed] [Google Scholar]
- 91.Visconti RP, Ebihara Y, LaRue AC, Fleming PA, McQuinn TC, Masuya M, Minamiguchi H, Markwald RR, Ogawa M, Drake CJ. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006;98:690–696. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- 92.Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet. 2005;139C:4–9. doi: 10.1002/ajmg.c.30068. [DOI] [PubMed] [Google Scholar]
- 93.Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- 94.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JMW, Mecham RP, Judge DP, Dietz HC. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Nat Acad Sci USA. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Starman BJ, Eyre D, Charbonneau H, Harrylock M, Weis MA, Weiss L, Graham JM, Jr, Byers PH. Osteogenesis imperfecta. The position of substitution for glycine by cysteine in the triple helical domain of the pro alpha 1(I) chains of type I collagen determines the clinical phenotype. J Clin Invest. 1989;84:1206–1214. doi: 10.1172/JCI114286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weis SM, Emery JL, Becker KD, McBride DJ, Omens JH, McCulloch AD. Myocardial mechanics and collagen structure in the osteogenesis imperfecta murine (oim) Circ Res. 2000;87:663–669. doi: 10.1161/01.res.87.8.663. [DOI] [PubMed] [Google Scholar]
- 98.Kern CB, Wessels A, McGarity J, Dixon LJ, Alston E, Argraves WS, Geeting D, Nelson CM, Menick DR, Apte SS. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010;29:304–316. doi: 10.1016/j.matbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys B, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. 2005;112:I229–I34. doi: 10.1161/01.CIRCULATIONAHA.104.524306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bonhoeffer P, Boudjemline Y, Qureshi SA, Le Bidois J, Iserin L, Acar P, Merckx J, Kachaner J, Sidi D. Percutaneous insertion of the pulmonary valve. J Am Coll Cardiol. 2002;39:1664–1669. doi: 10.1016/s0735-1097(02)01822-3. [DOI] [PubMed] [Google Scholar]
- 103.Vahanian A, Alfieri O, Al-Attar N, Antunes M, Bax J, Cormier B, Cribier A, De Jaegere P, Fournial G, Kappetein AP, Kovac J, Ludgate S, Maisano F, Moat N, Mohr F, Nataf P, Pierard L, Pomar JL, Schofer J, Tornos P, Tuzcu M, van Hout B, Von Segesser LK, Walther T. Transcatheter valve implantation for patients with aortic stenosis: a position statement from the European Association of Cardio-Thoracic Surgery (EACTS) and the European Society of Cardiology (ESC), in collaboration with the European Association of Percutaneous Cardiovascular Interventions (EAPCI) Eur Heart J. 2008;29:1463–1470. doi: 10.1093/eurheartj/ehn183. [DOI] [PubMed] [Google Scholar]
- 104.Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weyman AE, Scherrer-Crosbie M. Marfan syndrome and mitral valve prolapse. J Clin Invest. 2004;114:1543–1546. doi: 10.1172/JCI23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hanada K, Vermeij M, Garinis GA, de Waard MC, Kunen MGS, Myers L, Maas A, Duncker DJ, Meijers C, Dietz HC, Kanaar R, Essers J. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ Res. 2007;100:738–746. doi: 10.1161/01.RES.0000260181.19449.95. [DOI] [PubMed] [Google Scholar]
- 107.Kuivaniemi H, Tromp G, Prockop DJ. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat. 1997;9:300–315. doi: 10.1002/(SICI)1098-1004(1997)9:4<300::AID-HUMU2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 108.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, Stetten G, Meyers DA, Francomano CA. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 109.Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet. 1997;6:1021–1028. doi: 10.1093/hmg/6.7.1021. [DOI] [PubMed] [Google Scholar]
- 110.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 111.Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a muliti-exon deletion in one of the two COL3A1 allels affecting structure, stability, and processing of type III procollagen. J Biol Chem. 1988;263:6226–6232. [PubMed] [Google Scholar]
- 112.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 113.Pannu H, Tran-Fadulu V, Papke CL, Scherer S, Liu Y, Presley C, Guo D, Estrera AL, Safi HJ, Brasier AR, Vick GW, Marian AJ, Raman CS, Buja LM, Milewicz DM. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16:2453–2462. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, Toquet C, Roy E, McGregor L, Lynch SA, Newbury-Ecob R, Tran V, Young I, Trochu JN, Le Marec H, Schott JJ. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115:40–49. doi: 10.1161/CIRCULATIONAHA.106.622621. [DOI] [PubMed] [Google Scholar]
- 115.Edwards WD. Cardiac anatomy and examination of cardiac specimens. In: Allen HD, Driscoll DJ, Shaddy RE, Felts TF, editors. Moss and Adams’ heart disease in infants, children, and adolescents. Vol. 1. Philadephia: Lippincott, Williams, and Wilkins; 2008. pp. 2–34. [Google Scholar]