

Abstract
Skeletal dysplasias are not uncommon entities and a radiologist is likely to encounter a suspected case of dysplasia in his practice. The correct and early diagnosis of dysplasia is important for management of complications and for future genetic counselling. While there is an exhaustive classification system on dysplasias, it is important to be familiar with the radiological features of common dysplasias. In this article, we enumerate a radiographic approach to skeletal dysplasias, describe the essential as well as differentiating features of common non-lethal skeletal dysplasias and conclude by presenting working algorithms to either definitively diagnose a particular dysplasia or suggest the most likely differential diagnoses to the referring clinician and thus direct further workup of the patient.
Keywords: Skeletal dysplasia, Short limb dwarfism, Rhizomelia, Radiograph, Skeletal survey, Review, Spondylopepiphyseal dysplasia, Multiple epiphyseal dysplasia, Achondroplasia, Algorithm, Approach
Core tip: This article describes the radiographic approach to skeletal dysplasias, reviews the essential radiographic features of various non-lethal epiphyseal, metaphyseal, diaphyseal, osteopenic and sclerosing dysplasias and also describes features to differentiate these entities from other similar dysplasias. In summary, working algorithms for diagnosis of common skeletal dysplasias have also been provided.
INTRODUCTION
Skeletal dysplasias also termed as osteochondrodysplasias are a large heterogeneous group of disorders comprising of abnormalities of bone or cartilage growth or texture. They occur due to genetic mutations and their phenotype continues to evolve throughout life. Skeletal dysplasias thus differ from dysostoses which are malformations of single or multiple bones in combination, are due to abnormal blastogenesis in-utero and phenotypically remain static throughout life[1]. Currently more than 450 different entities have been described based on radiologic, molecular and biochemical criteria[2]. While certain dysplasias individually are quite rare, their overall prevalence as a group has been reported to be 2.3-7.6 per 10000 births in various epidemiologic studies[3-6]. However the actual prevalence may even be higher as concluded by these studies.
Some dysplasias are lethal in perinatal period and are detected on antenatal ultrasound scans while the non-lethal dysplasia present early in infancy or childhood with disproportionate short stature, failure of linear growth or with other physical deformities.
The appropriate diagnosis of a dysplasia is dependent upon the integration of clinical and family history, physical examination, radiologic examination and molecular and biochemical tests. Among these, a radiologic evaluation is an integral part of the diagnostic workup of a dysplasia. A general radiologist will often encounter a set of radiographs of a patient with a suspected skeletal dysplasia. While some dysplasias can be easy to diagnose based on certain characteristic or so-called “text-book” findings, it is also important to have an appropriate approach to diagnosis. Thus in this article, we review the radiologic approach to the diagnosis of a non-lethal dysplasia and thereafter describe the radiologic features of a few important and more common non-lethal dysplasias.
RADIOLOGIC EVALAUTION
The radiologic evaluation begins with a complete skeletal survey ideally comprising of a set of radiographs outlined in Table 1.
Table 1.
Set of radiographs obtained in a skeletal survey[1]
| Skull (AP and lateral) |
| Thoracolumbar spine (AP and lateral) |
| Chest (AP) |
| Pelvis (AP) |
| One upper limb (AP) |
| One lower limb (AP) |
| Left hand (AP) ( for bone age) |
AP: Anteroposterior.
In cases with epiphyseal irregularity or stippling, it is recommended to obtain radiographs of both sides upper and lower limbs. Also, because dysplasias continue to phenotypically evolve throughout life, serial radiographs are recommended and comparison should always be made with previous radiographs to assess evolution of disease and complications[1]. It is also recommended to obtain radiographs early in childhood since the optimal age for recognition of most dysplasias is before the obliteration of growth cartilage. Later when there is epiphyseal fusion and growth ceases, the recognition of many dysplasias becomes difficult and even impossible[7].
Offiah and Hall[1], in their excellent article have enumerated the ABCs of evaluation, comprising of anatomical localisation, analysis of bones and assessment of complications. Anatomically, the abnormalities can be located in the axial skeleton (Table 2) or in the appendicular skeleton.
Table 2.
Dysplasias with involvement of axial skeleton
| Location | Examples |
| Skull | Achondroplasia, Cleidocranial dysplasia |
| Mandible | Pyknodysostosis |
| Clavicle | Cleidocranial dysplasia |
| Ribs | Asphyxiating thoracic dysplasia, Thanatophoric dysplasia |
| Spine | Spondyloepiphyseal dysplasia congenita, Mucopolysaccharidoses |
| Pelvis | Achondroplasia |
In axial skeleton skull and spine are most commonly involved. The skull can either be large (achondroplasia) or can have multiple wormian bones (cleidocranial dysplasia). Involvement of spine is commonly in the form of flattening and decreased vertebral body height termed as platyspondyly or there can be irregularity of end-plates. There may also be abnormal vertebral hooking or beaking which are characteristic for certain dysplasias (central beaking in Morquio’s syndrome, posterior hump-shaped vertebrae in spondyloepiphyseal dysplasia tarda).
The appendicular skeleton has to be assessed for (1) type of bone shortening and (2) location of abnormality, i.e., epiphyseal, metaphyseal or diaphyseal. Shortening of the limbs can be (1) rhizomelic (involving proximal parts of limb, i.e., humerus and femur); (2) mesomelic (involving middle parts of limb, i.e., radius/ulna; tibia/fibula); (3) acromelic (involving hands and feet); or (4) micromelic (generalised shortening of entire limb).
Location of abnormality can be purely epiphyseal involving only the epiphyses, metaphyseal involving the metaphyses or diaphyseal involving only the diaphyses or there can be concomitant involvement of more than one location in appendicular skeleton. The involvement of appendicular skeleton has been summarised in Table 3.
Table 3.
Dysplasias with involvement of appendicular skeleton
| Type of shortening | Examples | Location of abnormality | Examples |
| Rhizomelic | Achondroplasia Spondyloepiphyseal dysplasia congenita | Epiphyseal | Chondrodysplasia punctata Spondyloepiphyseal dysplasia |
| Mesomelic | Mesomelic dysplasia | Metaphyseal | Achondroplasia Chondroectodermal dysplasia |
| Acromelic | Acrodysostosis | Diaphyseal | Progressive diaphyseal dysplasia |
| Micromelic | Achondrogenesis | Combination | Spondylo-epimetaphyseal dysplasia Metatropic dysplasia Mucopolysaccharidoses |
In addition, look at the bone density (decreased in osteopenic and increased in sclerosing dysplasias respectively) and for an abnormal shape of bone (e.g., champagne glass pelvis in achondroplasia).
Thirdly, complications are invariable sequelae of dysplasias because of altered bone shapes. An analysis of complications can also give a clue to the underlying diagnosis. Epiphyseal dysplasias lead to premature osteoarthritis and deformities like coxa vara and genu valga. Spondylo-dysplasias lead to early kyphoscoliosis while fractures are typically noted in dysplasias with altered bone density like osteogenesis imperfecta and osteopetrosis.
COMMON RADIOLOGICAL GROUPINGS
After analysis of the skeletal survey, the radiologic findings can be further grouped into common radiographic groups. These radiographic groups have been created based on common X-ray findings. Within these radiological groups are dysplasias groups conforming to that X-ray appearance and within the dysplasia groups we have enumerated a few common entities. We have basically derived and modified these groups from the 2010 revision of the Nosology and Classification of Genetic Skeletal Disorders framed by the International Skeletal Dysplasia Society[2,8] and from atlases of bone dysplasias[9,10]. By using these groups, we generate radiological differential diagnoses when encountering a common constellation of findings on skeletal surveys.
The groups include: (1) GROUP I: Epiphyseal dysplasias with/without spine involvement (Platyspondyly +/-); (2) GROUP II: Metaphyseal dysplasias with limb shortening/abnormal limb length; (3) GROUP III: Dysplasias with altered bone density; and (4) and GROUP IV: Miscellaneous dysplasias, i.e., those which do not typically have limb shortening or be clearly bracketed anatomically into sponylo-epi/metaphyseal dysplasias.
RADIOGRAPHIC FEATURES OF COMMON DYSPLASIAS
After the radiological grouping of dysplasias, both essential diagnostic and differentiating radiographic features of various common non-lethal skeletal dysplasias in each group have been enumerated below. While we chiefly describe the radiographic appearances of dysplasias, we have also provided the Online Mendelian Inheritance in Man (OMIM) numbers for these dysplasias for reference. OMIM is a comprehensive compilation of all human genes and genetic phenotypes and OMIM numbers are assigned to genetic phenotypes. This is a free resource available on http://www.ncbi.nlm.nih.gov/omim[11]. The OMIM numbers of dysplasias enumerated in this review have been provided for further information about clinical, genetic and phenotypic features of skeletal dysplasias. Since varying underlying genetic mutations can produce a common phenotype, i.e., a common radiographic appearance, more than one OMIM number may also be found within a single dysplasia entity.
Group I-epiphyseal dysplasias
All dysplasias in this group have common radiological findings of abnormal epiphyses and epiphyseal irregularity leading to early osteoarthritis and deformities like coxa vara and genu valga. In addition, there is secondary metaphyseal flaring and irregularity due to epiphyseal abnormality.
Within this broad group, there can be (1) isolated epiphyseal abnormality without platyspondyly as seen in chondrodysplasia punctata group; (2) concomitant involvement of spine (platyspondyly) as seen in Type II collagenopathies such as spondyloepiphyseal dysplasia congenita and tarda, Kniest dysplasia and achondrogenesis type 2; and (3) concomitant metaphyseal involvement as seen in spondyl(epi)metaphyseal dysplasias, multiple epiphyseal dysplasia, pseudoachondroplasia, mucopolysaccharidoses, diastrophic dysplasia and achondrogenesis type 1.
Spondyloepiphyseal dysplasia congenita[9,12-14]: OMIM: 183900[15]. The mode of inheritance is autosomal dominant and is due to a mutation in COL2A1 gene on chromosome locus 12q13.1 affecting Type II collagen protein.
Age of manifestation: At birth with delayed ossification of epiphyses as its hallmark. Essential radiographic features: (1) Bulbous and pear-shaped vertebrae at birth which later flatten leading to severe platyspondyly with thin intervertebral disc spaces (Figure 1A, B). The ensuing complications include kyphoscoliosis, lumbar lordosis and atlanto-axial instability. Atlanto-axial instability is secondary to odontoid hypoplasia and it subsequently endows a greatly increased risk of cervical myelopathy[16,17]; (2) Absent pubic bones at birth with horizontal roofs of acetabula and short and broad iliac wings; (3) Absent epiphyses of calcaneum and knee at birth. Later, there’s delay in the ossification of the heads of femur (Figure 1C). While delayed ossification of carpals and tarsal bones are noted, the hands and feet are typically not involved; and (4) Other features include large and dolicocephalic skull (Figure 1D) and rhizomelic shortening of extremities, more in lower than upper limbs and metaphyseal widening secondary to abnormal epiphyses (Figure 1E and F).
Figure 1.
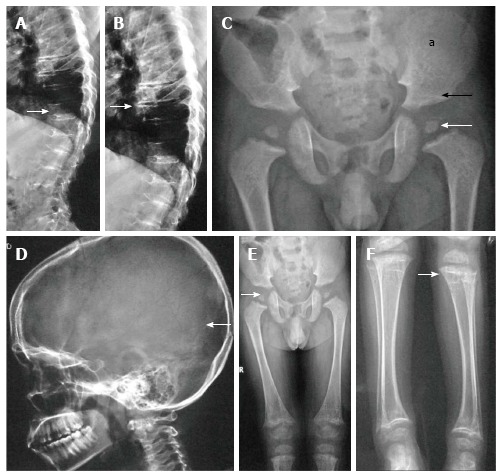
Spondyloepiphyseal dysplasia congenita. Lateral radiographs of dorsolumbar spine show platyspondyly (arrow, A) with severely reduced intervertebral disc spaces (arrow, B). Radiograph of pelvis (C) shows small femoral epiphyses (white arrow), horizontal acetabuli (black arrow) and short iliac wings (a). Radiograph of skull (D) shows relatively enlarged calvarium (arrow). Radiographs of lower limbs (E, F) show relatively short femurs and small epiphyses with secondary metaphyseal irregularity (arrow, F).
Differential diagnoses of SEDC include (1) Spondyloepiphyseal dysplasia tarda, (2) Morquio’s syndrome, (3) Kniest dysplasia and (4) Metatropic dysplasia. The features favouring Morquio’s syndrome include keratosulfaturia clinically and central beaking of spine with increased or maintained intervertebral disc spaces on radiographs. Also hands and feet are always abnormal in Morquio’s syndrome unlike SEDC.
Kniest dysplasia, (OMIM 156550)[18] is also an autosomal dominantly inherited Type II collagenopathy like SEDC and affects the same gene locus. Similar to SEDC, it presents as a short trunk-short limb dwarfism with delayed ossification at birth and in infancy. But in addition to these features, dumbbell shaped femurs and coronal clefting of spine are also noted at birth. Later on, the epiphyses become enlarged giving rise to megaepiphyses with cloud-like calcifications at the growth plate. In hands, there’s characteristic flattening of epiphyses of metacarpals and enlargement at ends of metacarpals and proximal phalanges giving rise to bulbous metacarpophalangeal and proximal interphalalangeal joints[19,20].
Metatropic dysplasia (OMIM: 156530)[21] is also an autosomal dominantly inherited dysplasia due to mutation on gene locus 12q24.1 affecting TRPV4 protein (transient receptor protein channel cation, subfamily V, member 4). It also manifests at birth with severe short limb rhizomelic dysplasia similar to achondroplasia and later evolves into a short trunk dysplasia similar to SEDC. Since metatropic dysplasia also presents with severe platyspondyly and progressive kyphoscoliosis in childhood, it can be mistaken for SEDC radiologically. However unlike SEDC, in metatropic dysplasia the metaphyses are also enlarged and the coccyx is long and resembles a tail. Thus metatropic dysplasia is a differential diagnosis for both achondroplasia because of metaphyseal and rhizomelic involvement and SEDC because of severe platyspondyly[22].
Spondyloepiphyseal dysplasia tarda[9,12-14,23]: OMIM: 313400[24]. The classical spondyloepiphyseal dysplasia tarda (SEDT) has a X-linked recessive inheritance and is noted only in males[25]. This is caused due to a mutation of gene locus X.p22.2 affecting TRAPPC2 gene[24]. However, more recently at least four types of SEDT with autosomal recessive inheritance (OMIM: 271600[26], 271620[27], 609223[28], 600093[29]) and autosomal dominant inheritance (OMIM: 184100)[30] have also been described.
Age of manifestation: The classic age of presentation is between 5-10 years of age, though it can variable, even first manifesting in the second decade of life. However, unlike SEDC, the appearance at birth is normal. Essential radiographic features: (1) Platyspondyly with heaping up and hyperostosis on the posterior two-thirds of end-plates giving rise to a heaped-up or hump-shaped appearance (Figure 2A); (2) Small pelvis with mild-to-moderate epiphyseal irregularity leading to early osteoarthritis at hips (Figure 2B), knees and ankles. However hands, feet and skull are typically not involved; and (3) Other features: In addition to these findings, progressive narrowing of interpedicular distance in lumbar spine have also been described, similar to achondroplasia, in the autosomal recessive forms of SEDT[31,32].
Figure 2.
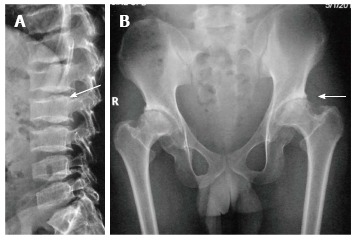
Spondyloepiphyseal dysplasia tarda. Lateral radiograph of lumbar spine (A) shows characteristic posterior hump (arrow). Radiograph of pelvis (B) shows bilateral flattened femoral heads, short necks and premature degenerative changes (arrow).
Differential diagnoses of SEDT include (1) SEDC and (2) Multiple epiphyseal dysplasia/Pseudochondroplasia. In SEDT, the disorder manifests predominantly in young boys and the spine, hips and joint changes are less severe whereas in SEDC, the short-trunk-short-limb dwarfism is apparent at birth itself with more severe and progressive deformities.
In multiple epiphyseal dysplasia/pseudoachondroplasia group, epiphyses of hands and feet are also involved and platyspondyly is typically absent EDM or moderate (pseudochondroplasia).
Multiple epiphyseal dysplasia[9,12,13] [No single OMIM number]: Multiple epiphyseal dysplasia (EDM) is a genetically heterogeneous entity caused by mutations in multiple genes[33,34]. First described in EDM type 1 due to a mutation in cartilage oligomeric matrix protein (COMP) on gene locus 19p13 [OMIM: 132400], currently at least 6 types of MED are described termed as EDM2 [OMIM: 600204], EDM3 [OMIM: 600969], EDM4 [OMIM: 226900], EDM5 [OMIM: 607078] and EDM6[OMIM: 614135][35]. Most cases of EDM are inherited in autosomal dominant manner while EDM4 has autosomal recessive inheritance.
Age of manifestation: Despite the genetic heterogeneity, MED usually presents after the age of 2-4 years when the child begins to walk. Essential radiographic features: (1) Bilateral and symmetric involvement of epiphyses of hips, knees, ankles, shoulders, elbows, wrists and hands and feet (Figure 3A-E); (2) Lateral tibio-talar slant wherein the lateral part of distal tibial epiphyses is thinner than the medial and the trochlea of the talus is shaped to conform to the abnormal ankle joint mortice (Figure 3F); (3) Double-layered patella as seen on a lateral X-ray of knee is considered highly pathognomic of EDM(Figure 3G, H). Double-layered patella is an important diagnostic clue for EDM[36,37]. Initially considered diagnostic only for recessive EDM4, it has now also been found in other dominant forms of EDM[38]. However, more recently, the uncommon occurrence of a double-layered patella in a patient with pseudoachondroplasia was also described[39]. This was attributed to the overlap between both EDM and milder forms of pseudoachondroplasia as both EMD1 and pseudoachondroplasia are caused due to mutations affecting the same COMP gene; and (4) Mild involvement of spine with anterior wedging, mild end plate irregularity and multiple Schmorl’s nodes mimicking Scheuermann’s disease and typical absence of platyspondyly.
Figure 3.
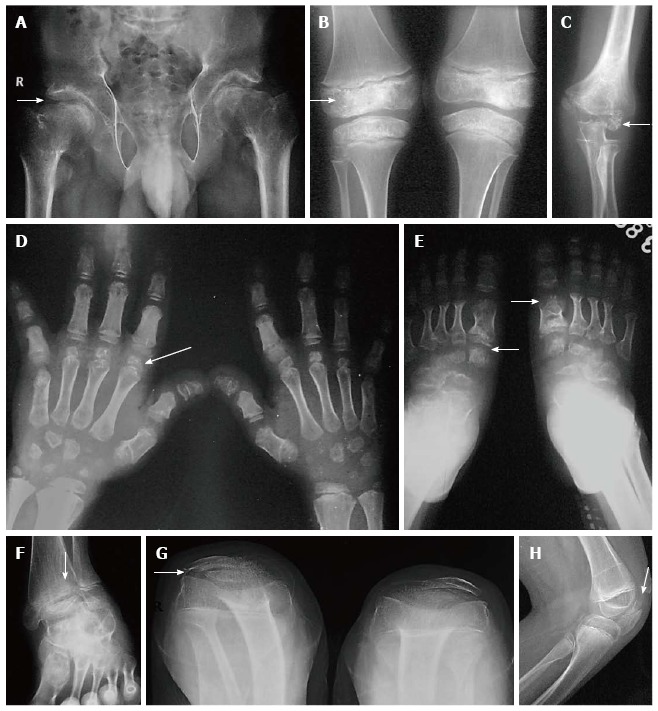
Multiple epiphyseal dysplasia. Radiographs of pelvis, knee and elbow (A-E) show epiphyseal irregularity in proximal femurs (arrow, A), around knee joints (arrow, B), elbow (arrow, C) with involvement of epiphyses of hands and feet (arrows in D, E) suggestive of multiple epiphyseal dysplasia. Radiograph of ankle (F) shows lateral tibio-talar slant. Radiograph of bilateral knees skyline view (G) and lateral view of left knee (H) show double-layered patellae (arrows).
Pseudoachondroplasia[9,11,40]: OMIM: 177170[41]. Pseudoachondroplasia is an autosomal dominantly inherited dysplasia caused due to mutation affecting the COMP gene similar to EDM on gene locus 19p13.11. Since both pseudoachondroplasia and EDM are genotypic alleles, there is considerable overlap in age of presentation and radiographic appearance in both entities. However, pseudoachondroplasia overall has more severe clinical and radiographic involvement as compared to EDM[36].
Essential radiographic features: Vertebrae have a persistent oval shape in childhood with a tongue-like protusion from the anterior aspect of vertebral bodies giving rise to central anterior tongue appearance (Figure 4). The central anterior tongue appearance is pathognomic of this entity. However, this disappears at older age and is replaced by platyspondyly giving rise to the short limb-short trunk dwarfism. Hence it is important to obtain early radiographs of spine to substantiate the diagnosis of pseudochondroplasia.
Figure 4.

Pseudochondroplasia. Lateral radiograph of spine shows typical central anterior tongue (arrow, A) in lumbar vertebrae. Radiograph of both hands (B) also show multiple abnormalities of epiphyses of metacarpals and phalanges with secondary metaphyseal widening (arrow).
Differential diagnoses of pseudoachondroplasia include (1) EDM and (2) achondroplasia. EDM and pseudoachondroplasia may be differentiated on basis of presence of central anterior tongue and platyspondyly in former and double layered patella in EDM. Clinically too, in pseudochondroplasia, there is joint and ligamentous laxity while in EDM, joints show restricted and painful movements[9]. Secondly, in pseudoachondroplasia, the dwarfism and shortening of the extremities is quite dramatic as compared to EDM as pseudoachondroplasia is considered a more severe manifestation of mutation on same gene.
The radiographic differences between pseudoachondroplasia and achondroplasia[13] have been enumerated in Table 4.
Table 4.
Differences between pseudoachondroplasia and achondroplasia
| Pseudoachondroplasia | Achondroplasia |
| Skull: Normal: “Achondroplasia with normal face” | Skull : Abnormal |
| Spine: Platyspondyly + | Spine: Platyspondyly – |
| Interpedicular distance normal | Interpedicular distance decreased in lumbar spine |
| Epiphyses and metaphyses abnormal | Only metaphyses abnormal |
| Trident hand and champagne-glass pelvis absent | Trident hand and champagne-glass pelvis present |
Chondrodysplasia punctata[9,12,13]: Chondrodysplasia punctata (CDP) is another genetically heterogeneous dysplasia. The most common type is the X-linked dominant type also termed as Conradi-Hunermann type [OMIM: 302960] due to mutation on Xp11[42]. Another type is the rhizomelic type chondrodysplasia punctata (RCDP) associated with peroxisomal enzyme disorder and has an autosomal recessive inheritance. RCDP is further divided into 3 sub-types, namely RCDP1 [OMIM: 215100], RCDP2 [OMIM: 222765]and RCDP3 [OMIM:600121] caused mutations affecting various genes encoding for peroxisomal enzymes[43-45]. A third very uncommon type of CDP is the brachytelephalangic type which has X-linked recessive inheritance [OMIM:302950][46]. In addition to genetically inherited forms of CDP, CDP can also be seen in warfarin embryotoxicity with features similar to Conaradi-Hunnermann type of CDP and in babies born to mothers with auto-immune diseases like systemic lupus erythrematosus[47] who present with features similar to RCDP. Unlike the genetically inherited rhizomelic type which is usually lethal in the first year of life, babies born to mothers with auto-immune disorders survive longer and do not have any underlying peroxisomal disorder. Stippling can also be seen in Zellweger’s syndrome which is a separate peroxisomal enzyme biogenesis disorder[43].
Essentially, the hallmark of CDP is stippling of epiphyses at birth. Later on, the stippling disappears and epiphyses become irregular with limb asymmetry (Figure 5 A-F). It is important to identify the radiologic type of chondrodysplasia punctata, namely rhizomelic/lethal or X-linked dominant type to prognosticate the patient. The differences between these two types have been summarised in Table 5.
Figure 5.

Chondrodysplasia punctata. Oblique view radiograph of dorsal spine (A) shows coronal clefting (arrow). Radiographs of another patient with chondrodysplasia punctata show stippling of vertebral bodies (arrows B, C), in toes (D), tarsal bones (E) and in carpals (F).
Table 5.
Differences between Rhizomelic and Conradi-Hunermann type Chondrodysplasia punctata
| Rhizomelic/lethal type | Conradi-Hunermann type |
| Inheritance: Autosomal recessive | Inheritance: X-linked dominant |
| Symmetric rhizomelic limb shortening | Asymmetric and occasional limb shortening |
| Stippling in spine absent/coronal clefts present | Spine: stippling present at endplates and bodies, later leads to kyphoscoliosis |
| Stippling noted in large joints, sparing hands and feet Laryngeal and tracheal cartilage stippling also present | Hands and feet also involved in addition to large joints. No extracartilaginous stippling |
| Mental retardation present and death in infancy | Compatible with normal intelligence and normal life span |
Mucopolysaccharidoses (Lysosomal disorders with skeletal involvement): Mucopolysaccharidoses (MPS) or lysosomal storage disorders are associated with absence of lysososomal enzymes required for degradation of glycosaminoglycans (GAGs) or mucopolysaccharides. There is secondary deposition of GAGs in various tissues causing coarse facies, mental retardation and hepatosplenomegaly. These disorders are also called dysostoses multiplex as they have multiple common skeletal abnormalities. The common skeletal features in this group include epiphyseal abnormalities, proximal pointed metacarpals and beaking in spine. The skeletal features of the two representative entities in this group namely Hurler’s and Morquio’s syndrome are enumerated as follows.
Hurler’s syndrome (MPS I)[9,12,13]: OMIM: 607014. Hurler’s syndrome is an autosomal recessive disorder due to mutation on 4p16.3 causing deficiency of alpha--L-iduronidase enzyme[48].
Age of manifestation: Babies with MPS 1 appear normal t birth with both clinical and radiographic features manifesting over first two years of life. Specific non-skeletal features of MPS 1 include corneal clouding, coronary artery narrowing, endocardial fibroelastosis and cardiac valvular disease[49,50].
Essential radiographic features: (1) Macrocephalic skull with frontal bossing and J-shaped sella (Figure 6A). The sinuses and facial bones are small and angle of mandible is increased. The J-shaped sella is secondary to pituitary gland enlargement due to deposition of GAGs in the gland; (2) Paddle or oar-shaped ribs in which the ribs are thin posteriorly and broad anteriorly (Figure 6B); (3) The lateral ends of clavicles are hypoplastic with small scapulae are small and there’s associated cardiomegaly (Figure 6B and E); (4) While overall length of limbs is maintained, there is diaphyseal widening, more in upper than lower limbs. In older children distal radius and ulna may slope towards each other (Figure 6C). In hands, the tubular bones are typically short and wide and metacarpals appear broad distally and tapered proximally. In addition, osteoporosis and flexion deformities are noted (Figure 6D); (5) In spine, typically, the L2 or L1 vertebra is hypoplastic and set slightly posteriorly giving rise to dorsolumbar kyphosis at that level with antero-inferior beaking of vertebrae (Figure 6E). Atlanto-axial instability is present while platyspondyly is absent; and (6) Other features include flared out iliac wings with sloping, shallow acetabular roofs and delayed ossification of femoral heads.
Figure 6.
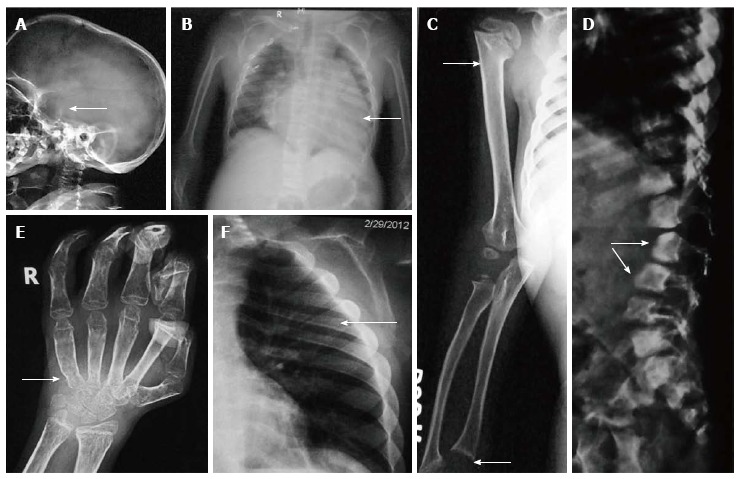
Hurler’s syndrome. Radiographs of patient with Hurler’s syndrome show macrocephaly with enlarged J-shaped sella (arrow, A), cardiomegaly (arrow, B) and paddle-shaped ribs (arrow, E). Also note relative diaphyseal widening in humerus (upper arrow, C) and sloping lower ends or radius and ulna (lower arrow, C). Radiograph of hands (D) shows proximal pointing (arrow), osteopenia and flexion deformities in distal interphalangeal joints, Radiograph of spine (F) shows hypoplastic L1 and antero-inferior beaking (arrows).
Morquio’s syndrome (MPS IV)[9,13]: Morquio’s syndrome of MPS IV is caused by mutations in two genes, Type IVA [OMIM: 253000]; mutation on 16q24.3; enzyme galactosamine-6-sulfate sulfatase[51] and type IVB [OMIM: 253010]; mutation on 3p21.33; enzyme beta-galactosidase[52].
MPS IV shows similarities to MPS I such as enlarged skull, dorsolumbar kyphosis in spine and atlanto-axial instability, Features specific to MPS IV include normal sized sella (unlike MPS 1) and platyspondyly with maintained/increased intervertebral disc spaces with central beaking (Figure 7A, B).
Figure 7.
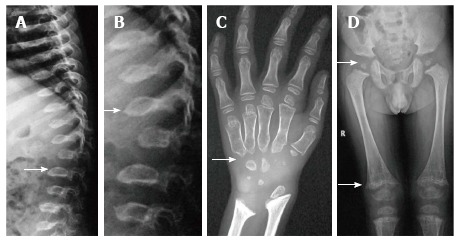
Morquio’s syndrome. Radiographs of spine (A, B) show platyspondyly with maintained intervertebral disc height (arrow, A) and central beaking (arrow, B). Radiograph of hand (C) shows proximal pointing of metacarpals. Radiograph of pelvis and lower limbs (D) show delayed ossification of femoral heads, irregular epiphyses and secondary metaphyseal widening in proximal femur and around knee joint (arrows).
The hands typically show pointing of the base of second to fifth metacarpals and distal ends of phalanges. There is additional delayed and irregular ossification of carpals and tarsals (Figure 7C).
In limbs, along with epiphyseal irregularity, metaphyses are widened to accommodate the enlarged epiphyses. There is also delayed ossification of femoral heads with poorly developed acetabula leading to premature arthropathy mimicking SEDC (Figure 7D).
Recently, attenuated or non-classical skeletal phenotypes of MPS IV and VI have been described and contrasted with the classical phenotype mentioned[53,54]. In attenuated form, involvement is usually limited to femoral epiphyses without the complete spectrum of skeletal involvement.
Initial diagnosis of MPS is made by qualitiative and quantitative urine analysis for elevated GAGs and confirmed by decreased enzyme activity in leucocytes or cultured skin fibroblasts. Definitive diagnosis can be made by identifying the underlying genetic mutation[55].
Group II-metaphyseal dysplasias
In this group, there is (1) predominant metaphyseal irregularity/widening and (2) abnormal limb length. Thus limb shortening can either be (1) rhizomelic as seen in achondroplasia group comprising of achondroplasia, hypochondroplasia and thanatophoric dysplasia (lethal) and metaphyseal chondrodysplasias or (2) mesomelic or acromelic as seen in chondroectodermal dysplasia (Ellis-Van-Creveld syndrome), Jeune’s/Asphyxiating Thoracic Dysplasia (ATD) (lethal) and short rib polydactyly dysplasias.
Achondroplasia[9,12,13,56]: OMIM: 100800; achondroplasia is the most common non-lethal dysplasia and is the prototype of rhizomelic dwarfism. It is inherited in an autosomal dominant fashion, with 80% occurring sporadically, attributable to spontaneous mutation on locus 4p16.3 affecting Fibroblast Growth Factor Receptor 3 (FGFR3) gene[57]. Increased incidence of sporadic mutations have also been associated with increasing paternal age[58].
Age of manifestation: The typical features of achondroplasia are obvious at birth. The most characteristic changes are found in the spine, especially in the lumbar region, pelvis, limbs and skull.
Essential radiological features: (1) Symmetric shortening of all long bones, with proximal portions being more affected and lower limb involvement being more than the upper limb (rhizomelia). There’s relative flaring and splaying of metaphyses with normal epiphyses (Figure 8A); (2) In children, the epiphysis is located closer to metaphyses leading to an apparent increase in the depth of the articular cartilage space. The two limbs of the V of metaphysis appear to embrace the epiphysis giving rise to a ball and socket relationship/chevron deformity (Figure 8B). This appearance is more common at lower end of femur and tends to normalise with increasing age; (3) The hand bones appear thick and tubular with widely separated 2nd and 3rd digits of the hands and inability to approximate them in extension, leading to appearance of trident hand (Figure 8C); (4) The pelvic cavity is short and broad, also called as champagne-glass appearance. There’s squaring of iliac wings with some roundening of corners on a frontal projection (elephant ear shaped iliac wings). The inferior margins of iliac wings and the roofs of acetabulum are flat and horizontal (Figure 8D). The sacrosciatic notches are small with an exaggerated sacral tilt and large, anteriorly protruding sacral promontory; (5) In spine, there is progressive decrease in the interpedicular distance cranio-caudally in the lumbar spine, the decrease in distance becoming more conspicuous with age (Figure 8E and F). Posterior scalloping of vertebral bodies is also common while anteriorly they may appear rounded giving rise to a bullet-shaped configuration. But the overall length of vertebral column and the vertebral heights are normal. There’s associated dorso-lumbar kyphoscoliosis in sitting position with exaggerated lumbar lordosis on standing up. Achondroplasts are prone for premature and severe spinal canal stenosis; and (6) The skull shows narrowed skull base with narrowing of foramen magnum. There is compensatory over-expansion of the skull vault and frontal regions to accommodate the expanding brain (Figure 8G). There’s relative mid-face hypoplasia and depressed nasal bones.
Figure 8.
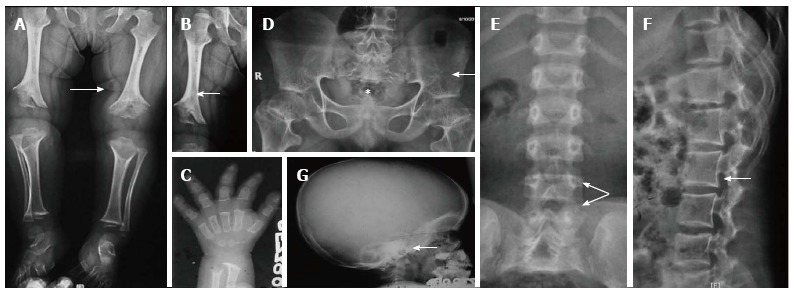
Achondroplasia. Radiograph of lower limbs (A, B) shows bilateral rhizomelic shortening with metaphyseal flaring (arrow, A) and chevron deformity in femur (arrow, B). Note trident hand appearance in (C). Radiograph of pelvis (D) shows short and broad pelvis (*), horizontal acetabuli (arrow) and round iliac wings. Radiographs of spine (E, F) show narrow interpedicular distance in lumbar spine (arrow, E) and posterior scalloping and thick, short pedicles (arrow, F). Radiograph of skull (G) shows enlarged cranial vault with narrowed foramen magnum (arrow).
In general there’s little difficulty in diagnosing achondroplasia. At birth, it must be distinguished from thanatophoric dysplasia and in adulthood from hypochondroplasia.
Hypochondroplasia[9,12]: OMIM: 146000; Hypochondroplasia, a milder form of achondroplasia, is caused due to a mutation of the same FGF receptor gene on locus on 4p16.3[59]. Recently, non-FGFR3 mutations such as those affecting short stature homeobox gene (SHOX), also located on chromosome 4 have been identified and the molecular criteria for diagnosis of hypochondroplasia have been expanded[60].
Age of presentation: It usually manifests after 2-4 years of age as short stature and limb shortening. Hence the clinician should be wary of diagnosing this condition in newborns.
Radiographic features: Spine and limb changes are similar to achondroplasia with decreased interpedicular distance in lumbar spine. But other vertebral changes are mild and spinal stenosis is less common. Limbs also show shortening but in addition to rhizomelia, mesomelia can also be seen[61].
In contrast to achondroplasia, the skull, pelvis and hands are essentially normal. There may be slight enlargement of skull in frontal region (macrocephaly). There’s mild symmetric brachydactyly involving all metacarpals and phalanges whereas in achondroplasia, the 2nd to 5th metacarpals and proximal phalanges are more affected. Thus the trident hand of achondroplasia is not seen in hypochondroplasia[62].
Chondroectodermal dysplasia[9,12]: OMIM: 225500, Chondrodysplasia punctata or Ellis-Van Creveld syndrome (EVC) is an autosomal recessively inherited dysplasia caused due to mutation affecting EVC gene on locus 4p16[63].
Age of presentation: The condition can be noted at birth with dysplastic nails, teeth, polydactyly and congenital cardiac defects, most common being common atrium and atrioventricular cushion defects[64].
Essential radiographic features: There is progressive distal shortening of limbs leading to mesomelia and acromelia (Figure 9A) with postaxial hexadactyly in hands and feet, carpal fusion (syncarpalism) involving capitate and hamate (Figure 9B), premature ossification of femoral heads and narrow thorax with short ribs (Figure 9C). The pelvis is short with flared iliac wings, narrow base and hook like projection from acetabulum forming trident acetabula (Figure 9D). The pelvic changes normalise later while spine remains normal throughout.
Figure 9.

Chondroectodermal Dysplasia (Ellis Van-Creveld Syndrome). Multiple radiographs of patient with chondroectodermal dysplasia show mesomelia (arrow, A), polydactyly on ulnar aspect with fused metacarpals (arrow, B), cardiomegaly with right side enlargement due to atrial septal defect (arrow, C) and acetabular hook (arrow, D). Also note flared iliac wings in pelvis.
Other features described recently in two patients with chondroectodermal dysplasia (CED) include genu valgum deformity, acroosteolysis (resorption of tips of phalanges), synmetacarpalism and synphalangism presenting at later age due to progressive skeletal involvement[65].
Differential diagnoses: Other dysplasias with similar radiological features include Jeune’s dysplasia and short rib dysplasia with/without polydactyly. But the combination of non-skeletal involvement of hair, nail, teeth and cardiac abnormalities with these radiologic findings are diagnostic of CED[66].
Group III-dysplasias with altered bone density: osteopenic or osteosclerotic: Osteopenic dysplasias are dysplasias with decreased bone density; of which osteogenesis imperfecta is the prototype. Likewise sclerosing or osteosclerotic dysplasias are dysplasias with increased bone density; of which osteopetrosis is the prototype. Both OI and osteopetrosis are genetically heterogeneous diseases, caused by multiple genetic mutations that phenotypically have a common appearance of decreased or increased bone density respectively. Simultaneously both these entities are also phenotypic alleles, namely single genetic mutation causes variable phenotypic manifestations. Due to the underlying genetic complexity for both these conditions, the clinical classification system is more commonly used to describe and prognosticate these patients. Therefore, all the OMIM numbers for these entities have been not been enumerated and we have mentioned only the OMIM number of common subtypes.
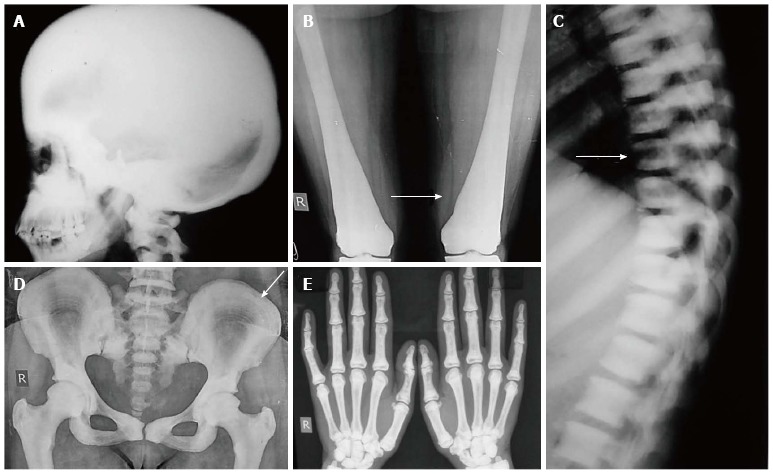
Osteogenesis imperfecta: Osteogenesis imperfecta (OI) is an autosomal dominantly or recessively inherited genetic disorder due to mutations in type 1 procollagen genes, characterised by decreased bone mass and increased bone fragility. Severity varies widely from perinatal lethality (type II) to milder forms with minimal fractures. Extraskeletal manifestations like blue sclerae, dentinogenesis imperfecta and deafness are also seen. Initially, Sillence et al[67] divided OI into four subtypes based on clinical features and disease severity: OI type I, with blue sclerae (OMIM: 166200); OI type II, perinatal lethal or congenital type (OMIM: 166210); OI type III, a progressively deforming form with normal sclerae (OMIM: 259420); and OI type IV, with normal sclerae (OMIM: 166220) which has been further expanded to eight types(46-48)[68-70]. The bone fragility increases in severity from type I < type IV< V < VI < VII < Type III < Type VIII < Type II[68].
Essential radiological features: (1) Radiologically, OI is characterised by a triad of diffuse osteopenia, pencil-thin cortices, and multiple bony fractures. The fractures are usually multiple and heal with exuberant callus formation giving rise to “pseudotumour” formation. Associated findings include deformities and pseudoarthrosis; (2) The vertebrae are also osteopenic, have a biconcave “cod-fish vertebrae” appearance with areas of collapse (Figure 10 A-C); (3) The skull shows multiple wormian bones, lucent calvarium, enlarged sinuses and platybasia; and (4) The pelvis is also abnormal in shape with deformities like protusio acetabuli and “shepherd crook” femurs.
Figure 10.

Osteogenesis Imperfecta. Infantogram of 1-mo baby shows diffuse osteopenia with multiple fractures in extremities (arrow). Radiograph of another patient shows fractures in bilateral femurs with callus formation (arrow). Radiograph of spine (C) shows osteopenia with codfish vertebrae.
Differential diagnoses include battered baby syndrome, hypophosphatasia, juvenile idiopathic osteoporosis, all of which can be excluded by careful analysis of X-rays, clinical and biochemical evaluation[68]. In addition, multiple new syndromes with congenital brittle bones have been elucidated which are similar to OI but have additional clinical features and are due to mutations in other than type 1 procollagen genes. These are referred to Syndromes Resembling Osteogenesis Imperfecta and should not be mistakenly labelled as OI without a complete evaluation[71].
Osteosclerotic or sclerosing dysplasias: Based on a target site approach, these anomalies are classified into three groups, namely (1) dysplasias of endochondral bone formation: osteopetrosis, pyknodysostosis, bone islands, osteopoikilosis and osteopathia striata; (2) dysplasias of intramembranous bone formation: progressive diaphyseal dysplasia; and (3) mixed sclerosing dysplasias: melorheostosis and overlap syndromes[72,73].
Osteopetrosis[9,13]: Considered to be the prototype of sclerosing dysplasia, it is characterised by wide clinical and genetic heterogeneity with a common end-pathway of failure of normal osteoclastic resorption of bone and increased density in medullary portions of bones with sparing of cortices[74]. The most severe form, termed as autosomal recessive type [OMIM:259700] is characterised by early onset of symptoms, obliteration of medullary canals with bone marrow failure leading to anemia, thrombocytopenia, hepatosplenomegaly and early death. On other hand, in dominant form, the onset occurs in adulthood with variable penetrance. These patients have mild anemia and present more with fractures and deformities [OMIM: 607364][2,75].
Essential radiological features: (1) There is diffuse sclerosis involving both the skull vault and base (Figure 11A) with progressive narrowing of foramina causing cranial nerve impingement, more so in the recessive type. In addition, there is prognathism with predisposition to mandibular osteomyelitis; (2) In limbs, despite increased density, there are multiple fractures. Fracture healing rate is normal but callus formation is defective comprising of osteoporotic bone. In addition, there is metaphyseal flaring leading to Erlenmeyer flask deformity[76] (Figure 11B); (3) “Bone-within-bone” appearance typically noted in spine, pelvis and short tubular bones. In spine, this is termed as a sandwich vertebrae appearance due to end-plate sclerosis and relative lucency of centre of body. In pelvis, they appear as multiple dense white lines parallel to the iliac crest (Figure 11C-E).
Figure 11.
Osteopetrosis. Radiograph of skull shows diffusely increased density (A). Radiograph of bilateral femurs show obliteration of medullary cavity and Erlenmeyer flask deformity (arrow, B). Also note sandwich vertebrae (arrow, C) bone-within-bone appearance in pelvis (arrow, D) and increased density in hand bones (E).
Differential diagnoses include pycnodysostosis and craniotubular dysplasias. Craniotubular dysplasias comprising of disorders with concomitant involvement of long bones and skull further comprise of craniodiaphyseal dysplasias, craniometaphyseal dysplasias and craniometadiaphyseal dysplasias. These can mimic osteoporosis radiologically with sclerosis of skull, foraminal narrowing, cranial nerve impingement and tubulation defects in long bones leading to Erlenmeyer flask appearance[76,77]. However in craniotubular dysplasia, there is an apparent increase in bone density that normalizes later, the vertebrae are normal, hematopoeisis is maintained and pattern of tubular bone sclerosis and long bone involvement is different[13].
Pkynodysostosis[9,13]: OMIM: 265800. First described in 1965 by Maroteaux and Lamy[36], Pycnodysostosis (PKND) is an autosomal recessive disorder due to mutation involving cathepsin K gene on locus 1q21[78].
Age of presentation: They present early in childhood with a triad of increased bone density, short limb dwarfism and increased propensity for fractures.
Essential radiological features: (1) Skull shows widely open sutures and fontanelles with multiple wormian bones, mandibular hypoplasia with obtuse angle and increased sclerosis of vault, base and orbital rims (Figure 12A, C); (2) There is increased bone density involving both limb bones and pelvis (Figure 12B). The limb length is decreased and pelvis is also small with shallow acetabulae (Figure 12B); (3) In hands, there is typically acro-osteolysis, i.e., resorption and tufting of terminal phalanges[79] (Figure 12D); and (4) In limbs, medullary cavity is maintained while bowing of radius and Madelung’s deformity can be occasionally seen.
Figure 12.
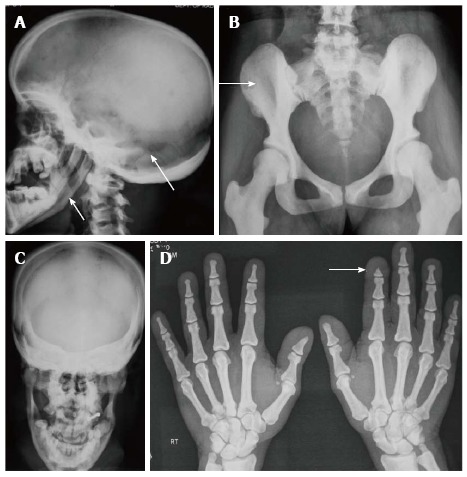
Pyknodysostosis. Radiographs of skull (A, C) show hypoplastic mandible, open sutures and increased bone density (arrows, A). Increased bone desnity also noted in pelvis (B) and hands (D). Also note acroosteolysis (arrow, D).
Other radiological features include hypoplasia of lateral ends of clavicles similar to cleidocranial dysplasia, and occasional spool-shaped vertebrae[13] (Figure 12A-D).
Pyknodyostosis can be differentiated from osteopetrosis by its typical appearance of skull, mandible and hands[72,80].
Osteopoikilosis [OMIM: 166700][81]: It is a benign condition with autosomal dominant inheritance, more common in males characterised by multiple small (1-10 mm), symmetric, uniform radiopaque densities located at ends of long bones, carpals, tarsals and periacetabular and subglenoid areas[72,74] (Figure 13A, B). An important differential can be osteoblastic metastases which can be differentiated by the variable size of lesions and by radionuclide scintigraphy[82].
Figure 13.
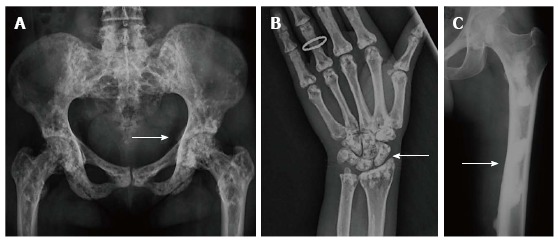
Osteopoikilosis (A,B) and Melorheostosis (C). Radiographs of pelvis (A) and hand (B) of a patient with osteopoikilosis show multiple bilateral symmetrical sclerotic lesions in periarticular location (arrows, A and B). Similar changes were also noted in knees, elbows and vertebral bodies (not shown). Radiograph of lower limb (C) of a young patient with melorheostosis shows “flowing wax appearance“ (arrow, C).
Osteopathia striata [OMIM: 300373][83]: Another benign condition with a X-linked dominant inheritance, it is more commonly seen in females and is characterised by bilateral symmetric involvement of long bones, pelvis and scapulae in the form of multiple vertical radio opaque lines in the metaphysis extending into the diaphysis[72]. In the pelvis, this gives a sunburst effect[13]. Other findings include osteosclerosis of long bones and skull leading to foraminal narrowing and cranial nerve compression[83].
Melorheostosis: Melorheostosis can be both a sporadic, non-inherited disorder or an inherited disorder presenting with melorheostosis and osteopoikilosis and assigned an OMIM: 155950[84]. Melorheostosis is a benign condition characterised clinically by pain and soft-tissue contractures. The distribution is asymmetric, can be monostotic (involving single bone) or polyostotic (involving multiple bones) or monomelic (involving one limb), most typically the lower limb. Other bones like skull,ribs, spine and short tubular bones can be affected at times. There is typically cortical thickening in a streaky or wavy pattern extending from the proximal to distal part of bone giving a “flowing wax candle appearance”(Figure 13C). The distribution in children is usually endosteal (which can mimic bone islands and osteopoikilosis) but this evolves to a periosteal pattern in adults[13,72,74].
Other osteosclerotic conditions include progressive diaphyseal dysplasia (Camurati-Engelmann’s disease) and infantile cortical hyperostosis (Caffey’s disease).
Progressive diaphyseal dysplasia: Progressive diaphyseal dysplasia, also called Camurati-Engelmann’s disease [OMIM: 131300] is an autosomal dominant disorder (locus 19q13). There is bilateral symmetric, fusiform enlargement with increased density of diaphysis of long bones beginning mid-shaft and progressing towards both ends[85]. In severe cases metaphyses may also be involved but typically, epiphyses are spared[74] (Figure 14).
Figure 14.
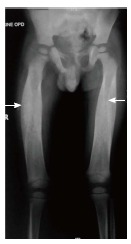
Progressive diaphyseal dysplasia. Radiograph of patient with progressive diaphyseal dysplasia shows symmetrical thickening along bilateral femoral diaphysis (arrows) with sparing of epi- and metaphyses. The pelvis also shows increased bone density.
Caffey’s disease: Caffey’s disease [OMIM: 114000] is an inherited disorder with both autosomal dominant and recessive inheritance characterized by a clinical triad of (1) narrow age group of presentation (before 5th month of age); (2) hyperirritability, soft tissue swelling, bone lesions; and (3) mandible involvement[86]. There is diffuse cortical thickening of mandible due to subperiosteal new bone formation. Other bones such as ulna, tibia, clavicle, scapulae and ribs can also be involved and radiographs show periosteal new bone formation in diaphysis sparing epiphyses and metaphyses[13].
Group IV-miscellaneous entities
Cleidocranial dysplasia: OMIM:119600 is an autosomal dominant dysplasia with predominant membranous bone involvement[87]. Due to its dominant mode of inheritance it can be seen in multiple members of the same family and can present in childhood to as late as 30 years of life[87].
Essential radiological features: (1) The skull shows delayed ossification of calvarium, multiple wormian bones, persistently open sutures and fontanelles giving a hot cross bun appearance. However the mandible is normal with maintained angle (Figure 15A, B); (2) The clavicles are either absent (10%) or hypoplastic (90%), hypoplasia affecting the lateral ends more than middle or medial ends (Figure 15C). Also the scapulae may be small and thoracic cage cone-shaped; (3) In hands and feet, the 2nd digit is elongated due to presence of accessory epiphyses for the second metacarpal while the distal phalanges are small and pointed (Figure 15D); and (4) The pelvis is small with widened symphysis pubis and abnormal shape of femoral heads called “chef-hat” appearance (Figure 15E).
Figure 15.

Cleidocranial dysplasia. Radiographs of skull (A, B) show open fontanelles and wormian bones (arrows, A) and hot cross bone appearance (arrow, B). Radiograph of chest(C) shows hypoplastic right clavicle (arrow). Radiograph of hand (D) shows elongated second digit with an accessory epiphyseal centre (arrow) Radiograph of pelvis (E) shows “chef-hat” shaped femoral heads (arrow) and widened pubis symphysis.
Differential diagnosis: The appearance of skull and hypoplasia of clavicle may be confused with pyknodysostosis; however the bone density and mandibular angle is maintained in cleidocranial dysplasia and short stature is absent. Another differential diagnosis can be manibuloacral dysplasia[88].
WORKING ALGORITHMIC APPROACH TO COMMON DYSPLASIAS
We present a working algorithm for radiological diagnosis of the commonly encountered dysplasias. This algorithm can help in initial diagnosis and lead the clinician to further work-up. Firstly, analyse the skeletal survey for spine and skull involvement. In spine, look for platyspondyly. And then look at the limbs for involvement of epiphyses and metaphyses. The involvement of these two regions can lead to diagnoses of many dysplasias as enumerated in Figure 16. After looking at the spine, look at the skull for either wormian bones or increased density of skull bones. The dysplasias with skull involvement have been in Figure 17.
Figure 16.
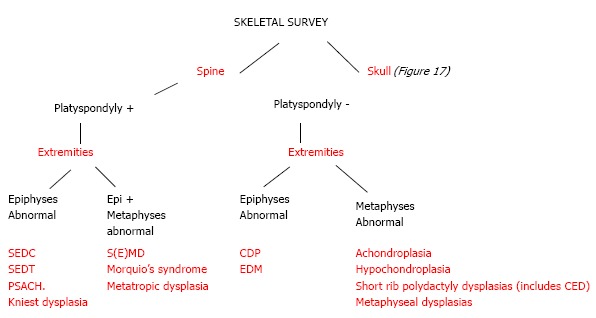
An algorithmic approach to skeletal dysplasias with spine and limb involvement. SEDT: Spondyloepiphyseal dysplasia tarda; CDP: Chondrodysplasia punctata; PSACH: Pseudoachondroplasia; EDM: Multiple epiphyseal dysplasia; CED: Chondroectodermal dysplasia.
Figure 17.
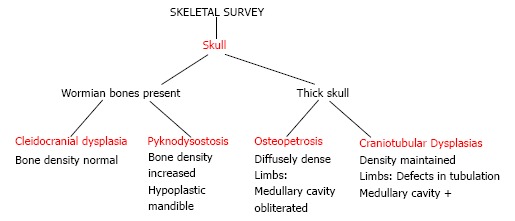
An algorithmic approach to skeletal dysplasias with skull involvement.
CONCLUSION
To conclude, dysplasias are not as uncommon as once thought. A general radiologist is very likely to encounter a set of radiographs of a patient with suspected dysplasia. In such a case, the radiologist should have an algorithmic, step-wise approach to either definitely diagnose a certain dysplasia or to lead the clinician to an appropriate diagnosis and direct further workup. The correct label is essential for prognostication, clinical and orthopaedic management of the present child and also to counsel parents about future pregnancies and their outcome. Undoubtedly, the diagnosis and management of a dysplasia needs teamwork between paediatrician, geneticist, radiologist and orthopaedist. But at the same time, a complete skeletal survey is an essential component of workup and hence it is important for radiologists to carefully analyse the bones, appearance and distribution of abnormalities on the survey and be familiar with the descriptions of common skeletal dysplasias.
Footnotes
P- Reviewer: Imashuku S, Kan L, Sawai H S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
References
- 1.Offiah AC, Hall CM. Radiological diagnosis of the constitutional disorders of bone. As easy as A, B, C? Pediatr Radiol. 2003;33:153–161. doi: 10.1007/s00247-002-0855-8. [DOI] [PubMed] [Google Scholar]
- 2.Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbosa-Buck CO, Orioli IM, da Graça Dutra M, Lopez-Camelo J, Castilla EE, Cavalcanti DP. Clinical epidemiology of skeletal dysplasias in South America. Am J Med Genet A. 2012;158A:1038–1045. doi: 10.1002/ajmg.a.35246. [DOI] [PubMed] [Google Scholar]
- 4.Orioli IM, Castilla EE, Barbosa-Neto JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet. 1986;23:328–332. doi: 10.1136/jmg.23.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen PE, Hauge M. Congenital generalised bone dysplasias: a clinical, radiological, and epidemiological survey. J Med Genet. 1989;26:37–44. doi: 10.1136/jmg.26.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen SA, Bieber FR, Benacerraf BR, Lachman RS, Rimoin DL, Holmes LB. Epidemiology of osteochondrodysplasias: changing trends due to advances in prenatal diagnosis. Am J Med Genet. 1996;61:49–58. doi: 10.1002/(SICI)1096-8628(19960102)61:1<49::AID-AJMG10>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 7.Kozlowski K. The radiographic clues in the diagnosis of bone dysplasias. Pediatr Radiol. 1985;15:1–3. doi: 10.1007/BF02387842. [DOI] [PubMed] [Google Scholar]
- 8.Alanay Y, Lachman RS. A review of the principles of radiological assessment of skeletal dysplasias. J Clin Res Pediatr Endocrinol. 2011;3:163–178. doi: 10.4274/jcrpe.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spranger JW, Langer LO, Wiedemann HR. Bone Dysplasias: An Atlas of Constitutional disorders of Skeletal Development. Philadelphia (PA): WB Saunders Co; 1974. [Google Scholar]
- 10.Taybi H, Lachman RS. Radiology of Syndromes, Metabolic disorders and Skeletal Dysplasias. 4th ed. Philadelphia: Mosby Elsevier;; 1996. [Google Scholar]
- 11.OMIM [Internet] Cited: 2014-06-24. Available from: http: //www.ncbi.nlm.nih.gov/omim.
- 12.Lachman RS. Skeletal Dysplasias. Caffey’s Pediatric Diagnostic Imaging. 11th ed. In: Slovis TL, editor. Philadelphia: Mosby Elsevier; 2008. pp. 2613–2671. [Google Scholar]
- 13.Tachdjian MO. Pediatric Orthopedics. 2nd ed. Philadelphia: W B Saunders Co; 1990. pp. 690–843. [Google Scholar]
- 14.Amirfeyz R, Clark C, Gargan M. Spondyloepiphyseal dysplasia. Current Orthopaedics. 2005;19:309–313. [Google Scholar]
- 15.OMIM Entry # 183900 Spondyloepiphyseal dysplasia congenital (SEDC) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/1839004.
- 16.McKay SD, Al-Omari A, Tomlinson LA, Dormans JP. Review of cervical spine anomalies in genetic syndromes. Spine (Phila Pa 1976) 2012;37:E269–E277. doi: 10.1097/BRS.0b013e31823b3ded. [DOI] [PubMed] [Google Scholar]
- 17.Veeravagu A, Lad SP, Camara-Quintana JQ, Jiang B, Shuer L. Neurosurgical interventions for spondyloepiphyseal dysplasia congenita: clinical presentation and assessment of the literature. World Neurosurg. 2013;80:437.e1–8. doi: 10.1016/j.wneu.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 18.OMIM Entry # 156550 Kniest dysplasia [Internet] Cited: 2014-06-24. Available from: http://omim.org/entry/156550.
- 19.Lachman RS, Rimoin DL, Hollister DW, Dorst JP, Siggers DC, McAlister W, Kaufman RL, Langer LO. The Kniest syndrome. Am J Roentgenol Radium Ther Nucl Med. 1975;123:805–814. doi: 10.2214/ajr.123.4.805. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian S, Gamanagatti S, Sinha A, Sampangi R. Kniest syndrome. Indian Pediatr. 2007;44:931–933. [PubMed] [Google Scholar]
- 21.OMIM Entry # 156530 Metatropic Dysplasia [Internet] Cited: 2014-06-24. Available from: http://omim.org/entry/156530.
- 22.Kannu P, Aftimos S, Mayne V, Donnan L, Savarirayan R. Metatropic dysplasia: clinical and radiographic findings in 11 patients demonstrating long-term natural history. Am J Med Genet A. 2007;143A:2512–2522. doi: 10.1002/ajmg.a.31941. [DOI] [PubMed] [Google Scholar]
- 23.MacKenzie JJ, Fitzpatrick J, Babyn P, Ferrero GB, Ballabio A, Billingsley G, Bulman DE, Strasberg P, Ray PN, Costa T. X linked spondyloepiphyseal dysplasia: a clinical, radiological, and molecular study of a large kindred. J Med Genet. 1996;33:823–828. doi: 10.1136/jmg.33.10.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.OMIM Entry # 313400 Spondyloepiphyseal dysplasia tarda, X-Linked (SEDT) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/313400.
- 25.Langer LO. Spondyloepiphysial dysplasia tarda. Hereditary chondrodysplasia with characteristic vertebral configuration in the adult. Radiology. 1964;82:833–839. doi: 10.1148/82.5.833. [DOI] [PubMed] [Google Scholar]
- 26.OMIM Entry # 271600 Spondyloepiphyseal dysplasia tarda, autosomal recessive. [Internet]. Cited: 2014-06-24. Available from: http: //omim.org/entry/271600.
- 27.OMIM Entry # 271620 Spondyloepiphyseal dysplasia tarda with mental retardation [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/271620.
- 28.OMIM Entry # 609223 Spondyloepiphyseal dysplasia tarda, autosomal recessive, Leroy-Spranger type [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/609223.
- 29.OMIM Entry # 600093 Spondyloepiphyseal dysplasia tarda with characteristic facies [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/600093.
- 30.OMIM Entry # 184100 Spondyloepiphyseal dysplasia tarda, autosomal dominant [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/184100.
- 31.Huson SM, Crowley S, Hall CM, Supramaniam G, Winter RM. Previously unrecognized form of familial spondyloepiphyseal dysplasia tarda with characteristic facies. Clin Dysmorphol. 1993;2:20–27. [PubMed] [Google Scholar]
- 32.Leroy JG, Leroy BP, Emmery LV, Messiaen L, Spranger JW. A new type of autosomal recessive spondyloepiphyseal dysplasia tarda. Am J Med Genet A. 2004;125A:49–56. doi: 10.1002/ajmg.a.20419. [DOI] [PubMed] [Google Scholar]
- 33.Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: New etiologic developments. Am J Med Genet. 2001;106:244–250. [PubMed] [Google Scholar]
- 34.Lachman RS, Krakow D, Cohn DH, Rimoin DL. MED, COMP, multilayered and NEIN: an overview of multiple epiphyseal dysplasia. Pediatr Radiol. 2005;35:116–123. doi: 10.1007/s00247-004-1323-4. [DOI] [PubMed] [Google Scholar]
- 35.OMIM Entry # 132400 Epiphyseal dysplasia, multiple, 1 (EDM1) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/132400.
- 36.Ramachandran G, Mason D. Double-layered patella: marker for multiple epiphyseal dysplasia. Am J Orthop (Belle Mead NJ) 2004;33:35–36. [PubMed] [Google Scholar]
- 37.Sheffield EG. Double-layered patella in multiple epiphyseal dysplasia: a valuable clue in the diagnosis. J Pediatr Orthop. 1998;18:123–128. [PubMed] [Google Scholar]
- 38.Nakashima E, Ikegawa S, Ohashi H, Kimizuka M, Nishimura G. Double-layered patella in multiple epiphyseal dysplasia is not exclusive to DTDST mutation. Am J Med Genet A. 2005;133A:106–107. doi: 10.1002/ajmg.a.30481. [DOI] [PubMed] [Google Scholar]
- 39.Vatanavicharn N, Lachman RS, Rimoin DL. Multilayered patella: similar radiographic findings in pseudoachondroplasia and recessive multiple epiphyseal dysplasia. Am J Med Genet A. 2008;146A:1682–1686. doi: 10.1002/ajmg.a.32313. [DOI] [PubMed] [Google Scholar]
- 40.Heselson NG, Cremin BJ, Beighton P. Pseudoachondroplasia, a report of 13 cases. Br J Radiol. 1977;50:473–482. doi: 10.1259/0007-1285-50-595-473. [DOI] [PubMed] [Google Scholar]
- 41.OMIM Entry # 177170 Pseudoachondroplasia (PSACH) [Internet]. Cited: 2014-06-24. Available from: http: //omim.org/entry/177170.
- 42.OMIM Entry # 302960 Chondrodysplasia punctata 2, X-linked dominant [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/302960.
- 43.OMIM Entry # 215100 Rhizomelic chondrodysplasia punctata, Type 1 (RCDP1) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/215100.
- 44.OMIM Entry # 222765 Rhizomelic chondrodysplasia punctata, type 2 (RCDP2) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/222765.
- 45.OMIM Entry # 600121 Rhizomelic chondrodysplasia punctata, Type 3 (RCDP3) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/600121.
- 46.OMIM Entry # 302950 Chondrodysplasia punctata 1, X-linked recessive (CDPX1) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/302950.
- 47.Shanske AL, Bernstein L, Herzog R. Chondrodysplasia punctata and maternal autoimmune disease: a new case and review of the literature. Pediatrics. 2007;120:e436–e441. doi: 10.1542/peds.2006-2997. [DOI] [PubMed] [Google Scholar]
- 48.OMIM Entry # 607014 Hurler syndrome [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/607014.
- 49.Braunlin E, Orchard PJ, Whitley CB, Schroeder L, Reed RC, Manivel JC. Unexpected coronary artery findings in mucopolysaccharidosis. Report of four cases and literature review. Cardiovasc Pathol. 2014;23:145–151. doi: 10.1016/j.carpath.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Schroeder L, Orchard P, Whitley CB, Berry JM, Tolar J, Miller W, Braunlin EA. Cardiac Ultrasound Findings in Infants with Severe (Hurler Phenotype) Untreated Mucopolysaccharidosis (MPS) Type I. JIMD Rep. 2013;10:87–94. doi: 10.1007/8904_2012_208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.OMIM Entry # 253000 Mucopolysaccharidosis, Type IVA (MPS4A) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/253000.
- 52.OMIM Entry # 253010 Mucopolysaccharidosis Type IV [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/253010.
- 53.Lachman RS, Burton BK, Clarke LA, Hoffinger S, Ikegawa S, Jin DK, Kano H, Kim OH, Lampe C, Mendelsohn NJ, et al. Mucopolysaccharidosis IVA (Morquio A syndrome) and VI (Maroteaux-Lamy syndrome): under-recognized and challenging to diagnose. Skeletal Radiol. 2014;43:359–369. doi: 10.1007/s00256-013-1797-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hendriksz CJ, Harmatz P, Beck M, Jones S, Wood T, Lachman R, Gravance CG, Orii T, Tomatsu S. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54–64. doi: 10.1016/j.ymgme.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mendelsohn NJ, Wood T, Olson RA, Temme R, Hale S, Zhang H, Read L, White KK. Spondyloepiphyseal dysplasias and bilateral legg-calvé-perthes disease: diagnostic considerations for mucopolysaccharidoses. JIMD Rep. 2013;11:125–132. doi: 10.1007/8904_2013_231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langer LO, Baumann PA, Gorlin RJ. Achondroplasia. Am J Roentgenol Radium Ther Nucl Med. 1967;100:12–26. doi: 10.2214/ajr.100.1.12. [DOI] [PubMed] [Google Scholar]
- 57.OMIM Entry # 100800 Achondroplasia (ACH) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/100800.
- 58.Orioli IM, Castilla EE, Scarano G, Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. Am J Med Genet. 1995;59:209–217. doi: 10.1002/ajmg.1320590218. [DOI] [PubMed] [Google Scholar]
- 59.OMIM Entry # 146000 Hypochondroplasia (HCH) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/146000.
- 60.Song SH, Balce GC, Agashe MV, Lee H, Hong SJ, Park YE, Kim SG, Song HR. New proposed clinico-radiologic and molecular criteria in hypochondroplasia: FGFR 3 gene mutations are not the only cause of hypochondroplasia. Am J Med Genet A. 2012;158A:2456–2462. doi: 10.1002/ajmg.a.35564. [DOI] [PubMed] [Google Scholar]
- 61.Bober MB, Bellus GA, Nikkel SM, Tiller GE. Hypochondroplasia. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, et al., editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle, 1993-; 2014. [Google Scholar]
- 62.Hall BD, Spranger J. Hypochondroplasia: clinical and radiological aspects in 39 cases. Radiology. 1979;133:95–100. doi: 10.1148/133.1.95. [DOI] [PubMed] [Google Scholar]
- 63.OMIM Entry # 225500 Ellis-Van Creveld syndrome (EVC) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/225500.
- 64.Hills CB, Kochilas L, Schimmenti LA, Moller JH. Ellis-van Creveld syndrome and congenital heart defects: presentation of an additional 32 cases. Pediatr Cardiol. 2011;32:977–982. doi: 10.1007/s00246-011-0006-9. [DOI] [PubMed] [Google Scholar]
- 65.Rudnik-Schöneborn S, Zerres K, Graul-Neumann L, Wiegand S, Mellerowicz H, Hehr U. Two Adult Patients with Ellis-van Creveld Syndrome Extending the Clinical Spectrum. Mol Syndromol. 2011;1:301–306. doi: 10.1159/000331338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baujat G, Le Merrer M. Ellis-van Creveld syndrome. Orphanet J Rare Dis. 2007;2:27. doi: 10.1186/1750-1172-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 69.Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.OMIM Entry # 610915 Osteogenesis imperfecta, Type VIII (OI8) [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/610915.
- 71.Plotkin H. Syndromes with congenital brittle bones. BMC Pediatr. 2004;4:16. doi: 10.1186/1471-2431-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vanhoenacker FM, De Beuckeleer LH, Van Hul W, Balemans W, Tan GJ, Hill SC, De Schepper AM. Sclerosing bone dysplasias: genetic and radioclinical features. Eur Radiol. 2000;10:1423–1433. doi: 10.1007/s003300000495. [DOI] [PubMed] [Google Scholar]
- 73.Greenspan A. Sclerosing bone dysplasias--a target-site approach. Skeletal Radiol. 1991;20:561–583. doi: 10.1007/BF01106087. [DOI] [PubMed] [Google Scholar]
- 74.Ihde LL, Forrester DM, Gottsegen CJ, Masih S, Patel DB, Vachon LA, White EA, Matcuk GR. Sclerosing bone dysplasias: review and differentiation from other causes of osteosclerosis. Radiographics. 2011;31:1865–1882. doi: 10.1148/rg.317115093. [DOI] [PubMed] [Google Scholar]
- 75.Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis. 2009;4:5. doi: 10.1186/1750-1172-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Faden MA, Krakow D, Ezgu F, Rimoin DL, Lachman RS. The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am J Med Genet A. 2009;149A:1334–1345. doi: 10.1002/ajmg.a.32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beighton P. Craniometaphyseal dysplasia (CMD), autosomal dominant form. J Med Genet. 1995;32:370–374. doi: 10.1136/jmg.32.5.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.OMIM Entry # 265800 Pycnodysostosis [Internet] Cited: 2014-06-24. Available from: http: //omim.org/entry/265800.
- 79.Lamy M, Maroteaux P. [Pycnodysostosis] Rev Esp Pediatr. 1965;21:433–437. [PubMed] [Google Scholar]
- 80.Balthazar E, Smith EH, Moskowitz H. Pycnodysostosis: an unusual case. Br J Radiol. 1972;45:304–307. doi: 10.1259/0007-1285-45-532-304. [DOI] [PubMed] [Google Scholar]
- 81.OMIM Entry # 166700 Buschke-ollendorff syndrome (BOS) [Internet] Cited: 2014-06-24. Available from: http://omim.org/entry/166700.
- 82.McArdle A, O’Riordan C, Connolly EM. Osteopoikilosis masquerading as osseous metastases in breast cancer [Internet].Cited: 2014-06-13. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21990037. [DOI] [PubMed]
- 83.OMIM Entry # 300373 Osteopathia striata with cranial sclerosis (OSCS) [Internet] Cited: 2014-06-28. Available from: http://omim.org/entry/300373.
- 84.OMIM Entry # 155950 Melorheostosis, Isolated[Internet] Cited: 2014-06-28. Available from: http://omim.org/entry/155950.
- 85.OMIM Entry # 131300 Camurati-Engelmann disease (CAEND) [Internet] Cited: 2014-06-28. Available from: http://omim.org/entry/131300.
- 86.OMIM Entry # 114000 Caffey disease [Internet] Cited: 2014-06-28. Available from: http://omim.org/entry/114000.
- 87.OMIM Entry # 119600 Cleidocranial dysplasia (CCD) [Internet] Cited: 2014-06-28. Available from: http://omim.org/entry/119600.
- 88.OMIM Entry # 248370 Mandibuloacral dysplasia with Type A lipodystrophy (MADA) [Internet] Cited: 2014-06-28. Available from: http: //omim.org/entry/248370.