

Abstract
Mitochondria are one of the major sites for the generation of reactive oxygen species (ROS) as an undesirable side product of oxidative energy metabolism. Damaged mitochondria can augment the generation of ROS. Dysfunction of mitochondria increase the risk for a large number of human diseases, including cardiovascular diseases (CVDs). Heart failure (HF) following ischemic heart disease, infantile cardiomyopathy and cardiac hypertrophy associated with left ventricular dilations are some of the CVDs in which the role of mitochondrial oxidative stress has been reported. Advances in mitochondrial research during the last decade focused on the preservation of its function in the myocardium, which is vital for the cellular energy production. Experimental and clinical trials have been conducted using mitochondria-targeted molecules like: MnSOD mimetics, such as EUK-8, EUK-134 and MitoSOD; choline esters of glutathione and N-acetyl-L-cysteine; triphenylphosphonium ligated vitamin E, lipoic acid, plastoquinone and mitoCoQ10; and Szeto-Schiller (SS)- peptides (SS-02 and SS-31). Although many results are inconclusive, some of the findings, especially on CoQ10, are worthwhile. This review summarizes the role of mitochondria-targeted delivery of agents and their consequences in the control of HF.
Keywords: Cardiovascular diseases, Oxidative stress, Antioxidant, Electron transport chain, Mitochondrial medicine, Heart failure
Core tip: Dysfunction of mitochondria increases the risk for a large number of human diseases, including cardiovascular diseases. Heart failure (HF) following ischemic heart disease, infantile cardiomyopathy and cardiac hypertrophy associated with left ventricular dilations are some of the cardiovascular diseases in which the role of mitochondrial oxidative stress has been reported. Recent reports on chronic HF followed by ischemic heart disease suggested a reduced supply of energy necessary for the contractile function of cardiomyocytes. Since mitochondrial damages are central to the pathophysiology of HF, various approaches are used to target compounds at mitochondria alone or adjunct to standard therapies.
INTRODUCTION
Although substantial improvements were made in the treatment of cardiovascular events during the last decade, cardiovascular disease (CVD), such as atherosclerosis, ischemic heart disease (IHD), heart failure (HF), stroke and hypertension, still remain one of the major challenges to humans. HF is a leading cause of morbidity and mortality in industrialized countries. It is also a growing public health problem, mainly because of the aging of population and an increase in prevalence in the elderly. In developing countries, around 2% of adults suffer from HF; the prevalence is found to be increased to approximately 6%-10% over the age of 65[1]. The mechanisms of HF are complex and multifactorial. Common causes of HF include myocardial infarction (MI) and other forms of IHD, valvular heart disease and different types of cardiomyopathies. A study of healthy adults in the United States reported that IHD increases the risk factors of HF by approximately 62%[2]. No curative treatment is currently available for HF. The existing therapies for HF are able to relieve symptoms but are unable to reverse molecular changes that occur in the cardiomyocytes. A reduced supply of energy necessary for the contractile function of cardiomyocytes can explain the chronic HF followed by IHD[3]. This may probably be due to the increased production of oxygen radicals with or without preserving the antioxidant status in the cardiomyocytes[4].
The primary factor that initiates the dysfunction of mitochondria has been proposed to be the defects in oxidative phosphorylation (OXPHOS) which can further enhance the production of reactive oxygen species (ROS) and eventually destroy the mtDNA[5]. Since slowly dividing/postmitotic cardiac myocytes are highly dependent on energy from OXPHOS, the cardiac myocardium will be affected, especially when the proportion of the damaged mitochondria is considerably high, as evidenced in HF[3]. Hence, challenging mitochondrial dysfunction remains one of the main streams of mitochondrial research that is primarily focussed on alleviating the organ damage associated with CVD. In spite of experimental evidence to support the role of mitochondria-mediated antioxidant therapy to alleviate the ROS-mediated injury in CVD, clinical studies are fragmentary. Many antioxidant molecules are designed and evaluated in clinical and experimental trials to stop the deterioration of mitochondrial function but only a few achieve success. Hence, mitochondrial-targeted antioxidant therapy for CVDs is a controversial field and warrants further research. It is worth knowing the scope for mitochondrially-mediated interventions in the conventional therapeutic regimen in order to render complete protection for early stages of CVD to result in protection for HF. This review discusses the mitochondria-targeted delivery of agents to alleviate the decline of myocardial function in CVD.
FORMATION AND DAMAGE INDUCED BY REACTIVE OXYGEN SPECIES IN THE MITOCHONDRIA OF CARDIOMYOCYTES
Mitochondria play a key role in cardiac energy balance. Energy for cardiomyocytes is solely met from mitochondrial OXPHOS. Moreover, mitochondria are involved in maintaining the fine regulatory balance between Ca2+ concentration and production of ROS and nitric oxide (NO). The majority of cellular oxygen (O2) that enters into mitochondria is reduced to water in the mitochondrial respiratory chain, whereas a fraction of all O2 consumed can be converted to potentially cytotoxic ROS, such as superoxide anion radical (O2-.), indicating that the mitochondrion itself is the source of ROS[6]. Any factor that affects the flow of electrons (e-) in the electron transport chain (ETC) can result in the leakage of e- to O2, leading to the formation of O2-. The O2- is a primary radical that could produce other ROS, such as hydrogen peroxide (H2O2) and hydroxyl radicals (·OH), in the failing myocardium. The .OH is generated by the reduction of H2O2 in the presence of endogenous iron and copper by means of the Fenton reaction. Copper and iron are found to be mobilized following myocardial ischemia. Chevion et al[7] reported a 8 to 9-fold higher level of copper and iron in the first coronary flow fraction of reperfusion after 35 min of ischemia compared to the pre-ischemic value in isolated rat heart. This was further supported by the obervation of Reddy et al[8] that early treatment with deferoxamine, a potent iron chelator, limits the injury related to myocardial ischemia/reperfusion in dogs, probably due to the lesser availability of iron for the Fenton reaction. The production of various ROS in the mitochondrion is given in Figure 1.
Figure 1.
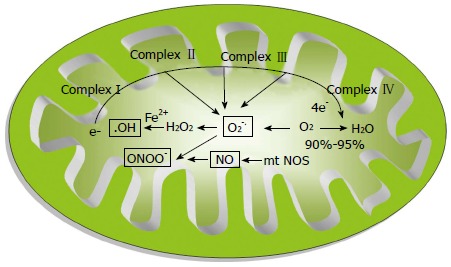
Formation of various reactive oxygen species in mitochondria. HO.: Hydroxyl radical; O2.-: Super oxide anion radical; ONOO-: Peroxynitrite; mtNOS: Mitochondrial-specific nitric oxide synthase; NO: Nitric oxide.
The drugs being used in clinical practice, such as statins (decreases ubiquinone), aspirin and valproic acid (sequesters of CoA), doxorubicin and daunorubicin (releases ROS), and acetaminophen (decreases reduced glutathione), will affect mitochondrial energy production and may play a critical role in the development of cardiomyopathy[6]. Physiologically, increased demand of the organ can favor the generation of free radicals. Sudheesh et al[9] recently reported that isoproterenol-induced acute MI in rat affected the respiratory chain complexes I-IV, mediated through an increase in the ROS level in the cardiomyocytes. Furthermore, the declined antioxidant status in the mitochondria during aging can also provoke mitochondrial dysfunction in cardiomyocytes[10]. Hypercholesterolemia can also affect mitochondrial functions by declining the mitochondrial membrane potential mediated through the generation of ROS and activation of mitochondrial apoptotic pathway[11].
Evidence shows that cytokines, such as tumor necrosis factor-alpha (TNF-alpha) and interleukin-6, are important pathological factors in inflammatory responses during the pathological progression of myocardial ischemia/reperfusion and hypertrophy. They are released during chronic inflammation, either in endothelial cells or cardiomyocytes, and inhibit the electron transport through the complex I and complex III-ubiquinone cycle, facilitating the generation of ROS[12]. Elevated activities of certain mitochondrial enzymes are also directly correlated with the excess production of ROS (Figure 2). The generated ROS is known to induce oxidation of low-density lipoproteins (LDL) in the coronary sinus of patients with dilated cardiomyopathy[13]. The oxidized LDL is abrogated by binding to the lectin-like oxidized LDL scavenger receptor-1 (LOX-1) on the arterial wall[14]. Activation of LOX-1 has been related to many pathophysiological events that lead to IHD.
Figure 2.
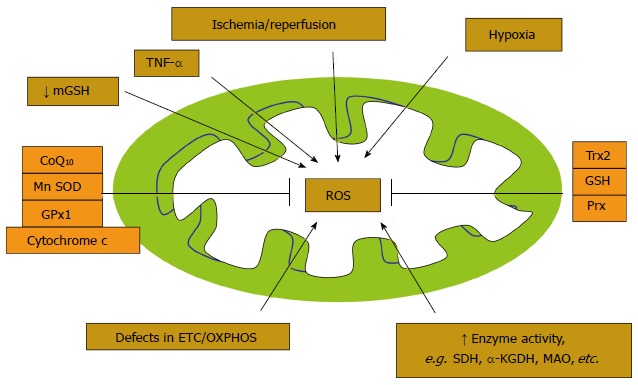
Factors that form and attenuate (antioxidants) reactive oxygen species in mitochondria. SDH: Succinate dehydrogenase; αKGDH: Alpha ketoglutarate dehydrogenase; MAO: Monoamine oxidase; Trx: Thioredoxins; Prx: Peroxiredoxin; OXPHOS: Oxidative phosphorylations; TNF-α: Tumor necrosis factor-alpha; GSH: Reduced glutathione; MnSOD: Manganese containing superoxide dismutase; GPx1: Glutathione peroxidase; CoQ10: Co-enzyme Q10.
The generated ROS under oxidative stress may contribute to potential mitochondrial damage that induces endothelial dysfunction and promotes leukocyte adhesion, inflammation, thrombosis and smooth muscle cell proliferation[15]. Among the damage induced by generated ROS at the cellular level, mtDNA remains the major target (Figure 3). mtDNA contains about 16.5 kb of circular double-stranded DNA to encode 13 protein components of the ETC. Mitochondrial function is controlled by the mtDNA, as well as factors that regulate mtDNA transcription and/or replication. A large part of the O2- that is formed inside the mitochondria cannot pass through the membrane and hence affect the DNA. Since 1988 when the first mutation in mtDNA was established, more than 400 mutations have been identified. The mutations described are either typically 50% to 60% for single, large-scale deletions or 80% to 90% for point mutations in patients with mitochondrial myopathy and encephalomyopathy[16]. In general, the majority of pathogenic point mutations are maternally transmitted, whereas large-scale deletions of mtDNA are mostly sporadic. More than 10 different types of deletions have been identified in the mtDNA among these; the 4977-bp deletion is the most prevalent in skeletal muscle, whereas the 7436-bp deletion was detected in the heart of human subjects in their late thirties, with no apparent sex difference[17]. However, the clinical severity of the disease is usually correlated with the presence of > 80% of the mutated mtDNA in the target tissues[18]. Furthermore, at the same level, large-scale deletions cause much more severe pathologies than point mutations. The patterns of distribution of the mutated mtDNA and the energy demand of the target tissues are two important factors that determine the pathological outcome of the mutation. HF is frequently associated with qualitative and quantitative defects in mtDNA and is found to increase with the age of human subjects. Recent evidence has suggested that mitochondria have enzymes to proofread mtDNA and fix mutations that may occur due to free radicals[19].
Figure 3.
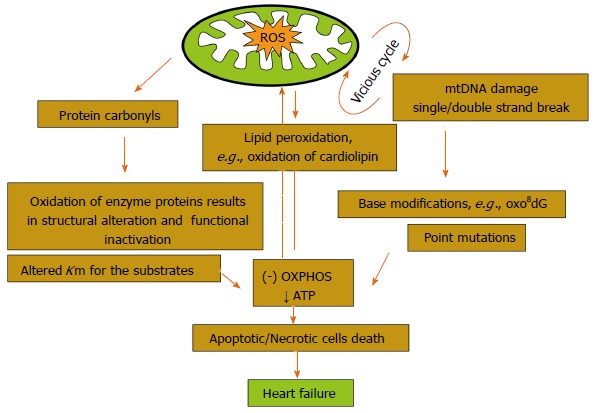
Damage induced by reactive oxygen species in mitochondria. OXPHOS: Oxidative phosphorylations; ROS: Reactive oxygen species.
Often, the damaged mtDNA is degraded by autophagy, whereas mtDNA that escapes the process of autophagy, as observed in atherosclerosis, can induce a potent inflammatory response. Ding et al[14] demonstrated that the damaged mitochondria induced by ox-LDL can result in the expression of toll-like receptor-9 (TLR-9) on the cell membrane. TLR-9 senses the unmethylated CpG motifs in damaged mt DNA and induces inflammation which is mediated through the pro-inflammatory cytokines. Ding et al[14] also demonstrated an intense autophagy, TLR-9 expression and inflammatory signals in the aorta of LDL receptor knockout mice when fed with a high cholesterol diet. Use of LOX-1 antibody or the ROS inhibitor apocynin attenuated ox-LDL-mediated autophagy, mtDNA damage and TLR-9 expression. Experiments using siRNA to DNase II suggested that the DNase II digested mtDNA and protected the tissue from inflammation.
In addition to the mtDNA mutations, damage to protein and lipid molecules in the mitochondrial membrane can contribute to the declined OXPHOS. Cardiolipin, an essential phospholipid present in the inner membrane of mitochondria that serves as a cofactor for a number of critical mitochondrial transport proteins and retains cytochrome c at the inner mitochondrial membrane through the electrostatic interaction, declines during the oxidative damage. Peroxidation of cardiolipin and its release into the cytosol can execute apoptotic cell death[20]. Amino acids, such as lysine, arginine, glutamic acid, histidine, proline and threonine present in the protein, favor the formation of protein carbonyl or nitration of the tyrosine residues, either by direct oxidation or by the binding of aldehydes that formed from the peroxidation of lipids. Mitochondrial aconitase and adenine nucleotide translocase are highly sensitive to O2-[21]. ROS-derived lipid hydroperoxide can also initiate the strand breaks and base modifications in mt DNA. Many cardiotoxic stimuli can lead to ROS generation, Ca2+ overload of the mitochondrial matrix, and opening of a large, nonspecific channel in the inner mitochondrial membrane, such as permeability transition pore (PTP), finally alter the mitochondrial permeability transition (MPT). Ca2+ overload to the mitochondrial matrix can further enhance the generation of ROS. Although the exact mechanism of ROS production is debatable, the effect is probably mediated through Ca2+ mediated inhibition on the complex I[22], III[23] and IV[23] of ETC (Figure 4). Ca2+ can stimulate the TCA cycle dehydrogenases to increase the production of reduced substrate for OXPHOS[24] and further increase the rate of respiration as well. Ca2+ can also activate mitochondrial nitric oxide synthase to produce NO which in turn inhibits the complex IV[25]. The simultaneous generation of NO with O2- favors the formation of peroxynitrite, one of the major agents to induce conformational change in many proteins[26]. MPT dissipates the proton electrochemical potential gradients, depletion of ATP and swelling, as well as rupture of the mitochondria that leads to the release of pro-apoptotic proteins into the cytosol and eventually results in death of cardiomyocytes. Evidence indicates that the activity of complex II is not affected as it is entirely encoded by nuclear DNA, whereas complex IV activity (cytochrome C oxidase), along with complex I, partially encoded by mtDNA genes, are frequently reduced in patients with mtDNA or tRNA mutations[19]. Mt tRNA gene mutations can also variably affect the activity of respiratory chain complexes.
Figure 4.
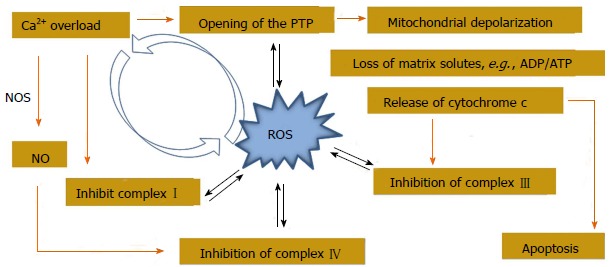
Crosstalk between mitochondrial Ca2+ handling and reactive oxygen species generation. PTP: Permeability transition pore; NOS: Nitric oxide synthase; ADP: Adenosine diphosphate; ATP: Adenosine triphosphate.
ANTIOXIDANTS AND PROTECTION OF MITOCHONDRIA IN THE CARDIOMYOCYTES
Mitochondrial oxidative stress resulting from an imbalance between the generation of ROS and the existing mitochondrial antioxidant mechanisms has been described in the pathogenesis of CVDs, including HF[27]. HF followed by MI can be initiated with the mitochondrial damage and dysfunction that can be ascribed to: (1) increased lipid peroxidation; (2) reduced mitochondrial gene replication, mtDNA copy number and mitochondrial gene transcription; and (3) reduced OXPHOS due to low respiratory chain complex enzyme activities (Figure 5). Therefore, preservation of mitochondrial function is essential. Therapies that are designed to interfere with mitochondrial oxidative stress could be beneficial.
Figure 5.
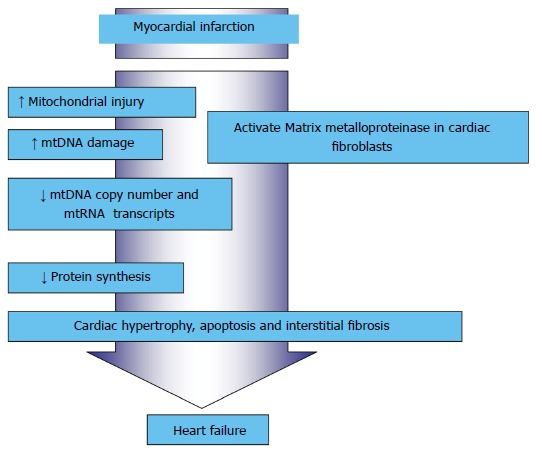
Myocardial infarction-induced mitochondrial damage and dysfunction that resulted in heart failure.
Various molecules are involved in the mitochondrial protection for the myocardium. Among them, tumor necrosis factor receptor-associated protein 1 (TRAP1), a member of the mitochondrial heat-shock family of proteins (70 kDa), has a central role. Overexpressed TRAP1 in the ischemia-like condition preserves ATP levels and cell viability during oxygen-glucose deprivation. The protective effects of TRAP1 against oxidative stress-induced cell death can be ascribed to translocation of cytosolic serine/threonine protein kinase, PTEN-induced putative kinase 1, to mitochondria and phosphorylation of TRAP1 that will prevent the release of cytochrome c and thus preserves MPT. TRAP1 expression is found to be elevated in the cardiomyocytes during hypoxia. However, the excess production of ROS in reperfusion/ischemic injury can inhibit the TRAP1 mediated protection that eventually results in the death of cardiomyocytes[28]. Hence, the role of enzymatic and non-enzymatic antioxidants in the mitochondria has been inevitable to protect the mitochondrial damage. Various mitochondrial antioxidants are useful in alleviating the oxidative stress and are depicted in Figure 2.
The first line of defense against ROS-mediated cardiac injury comprises several antioxidant enzymes, including Mn-superoxide dismutase (MnSOD) and glutathione peroxidase (mtGPx). Among these, mtGPx is an essential enzyme that performs several vital functions. Experimental studies reported the declined cardiac mitochondrial antioxidants, such as activity of Mn-SOD, mtGPx and level of reduced glutathione (GSH) in the myocardium of the aged as well as MI-induced rats[10]. Besides, the activities of the respiratory chain complexes I-IV and Krebs cycle dehydrogenases also declined[9]. Several dietary supplements, including the mitochondrial cofactor and antioxidant lipoic acid (LA), can increase the endogenous antioxidants as well as mitochondrial bioenergetics[6]. Overexpression of the genes for peroxiredoxin-3, a mitochondrial antioxidant, or mitochondrial transcription factor A (TFAM) could ameliorate the decline in mtDNA copy number in failing hearts[28]. Overexpression of TFAM may protect mtDNA from damage by direct binding and stabilizing of mtDNA. Similarly, overexpression of mtGPx inhibit the development of left ventricular remodeling and failure after MI[29].
Co-enzyme Q10 (CoQ10) and L-carnitine can be considered to be a safe adjunct to standard therapies in CVD[30]. CoQ10 is an endogenous compound found in the inner mitochondrial membrane that is essential for electron transport in the ETC and thus for the production of ATP. In addition to its role in bioenergetics, CoQ10 is demonstrated to be an inhibitor of thrombus formation and able to reduce ROS in mitochondria. Both pre-clinical and clinical studies have shown moderately beneficial effects of CoQ10 in reducing blood pressure, blood glucose and myocardial damage[31]. Besides the application of CoQ10 in CVD, its use against the adverse effect of drugs, mainly statins, in the intervention of CVD has recently attracted attention. Nevertheless, the antioxidant property of statins[32,33] can block the endogenous biosynthesis of CoQ10 required for the ETC, resulting in cardiomyopathy and muscle pain[34]. CoQ10 supplementation (100 mg/d) for 30 d has been found to decrease the muscle pain associated with statin treatment[35]. In another study, fifty consecutive new patients discontinued 28 mo of statin therapy due to side effects and began CoQ10 supplementation at an average of 240 mg/d[36] and were followed for an average of 22 mo (84% for more than 12 mo). The prevalence of fatigue from 84% on the initial visit decreased to 16%, the rate of myalgia from 64% to 6%, dyspnea from 58% to 12%, memory loss from 8% to 4% and peripheral neuropathy from 10% to 2%. Moreover, statin-induced cardiomyopathy was found to be reversed with the combination of statin discontinuation and supplementation with CoQ10.
L-carnitine therapy in HF patients (2 g/d, orally) showed improved survival[37]. A recent study in patients with mild diastolic HF treated with L-carnitine (1.5 g/d, p.o for 3 mo) showed improvement in diastolic function[38]. Therapy with L-carnitine 9 g/d, intravenously for 5 d followed by 6 g/d orally for 12 mo along with the standard medical therapy may limit the adverse effects of acute MI on the heart muscle[39,40]. Tolerance to exercise was significantly improved in patients with higher left ventricular ejection fraction volume (greater than 30%) when treated with the propionyl-L-carnitine adjunct to appropriate medical therapy[41].
Carvedilol, both the beta blocker (β1, β2) and alpha blocker (α1), is indicated in the management of congestive HF. It is used as an adjunct to conventional treatments with its effect probably mediated through the potent antioxidant and anti-apoptotic activities[42]. The Japanese Diastolic Heart Failure Study has recently suggested the beneficial effects of standard dose prescriptions of carvedilol (> 7.5 mg/d) in HF without affecting the ejection fraction[43]. An ACE inhibitor, captopril, was also shown to increase the mitochondrial content in the hearts of dogs following coronary ligation[44], suggesting that some of its beneficial effects may be due to the stimulation of mitochondrial biogenesis[45]. However, many extensive clinical trials using conventional antioxidants such as Vitamin E or Vitamin C yielded disappointing results[46,47]. According to Murphy and Smith[48], a possible explanation for this may be that the antioxidants are distributed widely in the body with only a small fraction being taken up by mitochondria. Therefore, targeting biologically active molecules to mitochondria will open up avenues for manipulating mitochondrial functions.
FUTURE PERSPECTIVES OF MITOCHONDRIAL PHARMACEUTICS IN CARDIOVASCULAR DISEASES
The increase of mitochondrial concentrations of antioxidant drugs by selective targeting mitochondria should be a practical approach for a wide range of human diseases. Mitochondria-targeted antioxidants have been developed as pharmaceuticals and have been shown to be safe and effective in phase II clinical trials. Various antioxidant molecules targeting mitochondria in cardiomyocytes are given in Table 1. In general, attempts to achieve cell protection using antioxidants have been successfully undertaken with two free radical scavengers, such as 4-hydroxy-2,2,6,6-tetramethylpiperidin-N-oxide and Salen-Mn(III) complex of o-vanillin (EUK-134). Inorganic MnSOD mimetics, such as EUK-8 and EUK-134, possess antioxidant properties of both MnSOD and catalase and have been successfully synthesized and partially tested in terms of their antioxidant and anti-apoptotic properties that appear to be effective in the heart[49]. The mitochondrial-targeted version of vitamin E protected mitochondria from oxidative damage induced by iron/ascorbate far more effectively than vitamin E itself[50].
Table 1.
Mitochondria-targeted antioxidants
| Sl no. | Antioxidants |
| 1 | 4-hydroxy-2,2,6,6-tetramethylpiperidin-N-oxide (TEMPOL) |
| 2 | Salen-Mn(III) complex of o-vanillin (EUK-8, EUK-134) |
| 3 | Choline esters of glutathione and N-acetyl-l-cysteine |
| 4 | Triphenylphosphonium ligated vitamin E, lipoic acid, plastoquinone, Mito SOD and Mito CoQ10 |
| 5 | SS-peptides (SS-02 and SS-31) |
SS: Szeto-Schiller.
The GSH pool in mitochondria, approximately 15% of total cellular GSH, is found to be reduced during oxidative stress. Choline esters of GSH and N-acetyl-L-cysteine were prepared as mitochondria-targeted antioxidants[51]. However, in vivo data are not available to support their efficacy. Recently, many trials have been conducted in which cationic molecules are targeted using the negative membrane potential of the inner membrane as a promising approach in this field. Triphenylphosphonium (TPP) cation is one among such molecules that are conjugated to a range of antioxidants. Antioxidants ligated with TPP, such as vitamin E[50], LA[52], plastoquinone[53] and mitochondrial CoQ10 (MitoQ)[54], have been experimentally confirmed to be effective in ameliorating mitochondrial oxidative stress in CVD. TPP, like pentaaza macrocyclic Mn(II) superoxide dismutase (SOD) mimetic, MitoSOD, is found to be very effective in selectively protecting mitochondria from damage[55].
Adlam et al[54] reported that the myocardium of the rat administered with MitoQ can render protection against heart dysfunction, tissue damage and mitochondrial dysfunction induced from ischemia-reperfusion injury. It can be given either as iv or orally without toxicity. Graham et al[56] showed that MitoQ protects the development of hypertension, improves endothelial functions and reduces cardiac hypertrophy in young hypertensive rats. MitoQ is also a promising, novel strategy for preserving vascular endothelial function with advancing age and can prevent age-related CVD in mice[57]. However, MitoQ was not useful in protecting oxidative damage to cardiolipin, accumulation of protein carbonyls, activity of mitochondrial respiratory complexes, mtDNA copy number, or damage to mtDNA[58-60].
Another critical molecule in this field is a synthetic peptide called Szeto-Schiller (SS) - peptides, synthesized from basic and aromatic amino acids. SS-peptides comprised four alternating aromatic/basic D-amino acids in the first or second position with three positive charges at physiological pH. SS-02 and SS-31 were shown to be protective against cardiac ischemia-reperfusion injury when administered on reperfusion by iv, ip or subcutaneously[61]. Pre-ischemic intraperitoneal administration of these peptides to rats significantly reduced infarct size[62]. SS-02 has more efficacy as the free radical scavenger than SS-31. The uptake into tissues or metabolism of these peptides has not yet been thoroughly reported. However, studies with isolated mitochondria showed that despite the cationic nature, these peptides were found to predominantly target the IMM rather than the mitochondrial matrix[63]. SS-31 is currently in clinical trials for ischemia-reperfusion injury and protects mitochondrial cristae by interacting with cardiolipin on the IMM. SS peptides scavenge H2O2 and peroxynitrite inhibits lipid peroxidation. They can also inhibit the decline of MPT and cytochrome c release, thus preventing oxidant-induced cell death[64]. Although the delivery of antioxidants may protect mitochondria from oxidative stress caused by a variety of insults, the area of mitochondria-specific delivery of drugs is still in its infancy. Among the molecules studied in CVD, clinical trials on CoQ10 have found that it can be considered a safe adjunct to conventional therapies in CVD. Despite the beneficial effect of CoQ10, given alone or in addition to conventional therapies in hypertension and in HF, less extensive evidence in IHD has been found[65]. The present findings demonstrate that mitochondrial damage plays a prominent role in HF following MI and further research into the role of mitochondria-targeted agents to prevent the HF is compulsory.
CONCLUSION
Mitochondrial dysfunction plays a key role in the pathogenesis of ischemia and reperfusion injury and cardiomyopathy. Mutations in mt DNA and abnormalities in mitochondrial function are associated with common forms of cardiac diseases. Despite the promising mitochondria-targeted drugs that are emerging from the laboratory, very few have successfully completed clinical trials. Antioxidants ligated with TPP, such as vitamin E, lipoic acid, plastoquinone, MnSOD and mitochondrial CoQ10, have been experimentally determined as effective in ameliorating the mitochondrial oxidative stress associated with CVD. Among the molecules targeting mitochondria, MitoQ provides a novel approach to attenuate oxidative damage with the potential to become a new therapeutic intervention in humans. However, there are insufficient data from well designed randomized trials to issue a general recommendation for people to take antioxidant supplements in order to prevent heart disease. Since mitochondrial damage is central to the pathophysiology of HF, various approaches used to target antioxidant compounds at mitochondria should be explored in the development for the treatment of HF. A great deal of future research will be needed before mitochondria-directed therapies are made available for the prevention and treatment of CVD.
Footnotes
P- Reviewer: Alexeyev M, Fujiwara N, Julie NL, Song GB, Zeng LF S- Editor: Song XX L- Editor: Roemmele A E- Editor: Wu HL
References
- 1.McMurray JJ, Pfeffer MA. Heart failure. Lancet. 2005;365:1877–1889. doi: 10.1016/S0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- 2.He J, Ogden LG, Bazzano LA, Vupputuri S, Loria C, Whelton PK. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch Intern Med. 2001;161:996–1002. doi: 10.1001/archinte.161.7.996. [DOI] [PubMed] [Google Scholar]
- 3.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 2013;61:599–610. doi: 10.1016/j.jacc.2012.08.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballinger SW. Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med. 2005;38:1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 6.Fosslien E. Review: Mitochondrial medicine--cardiomyopathy caused by defective oxidative phosphorylation. Ann Clin Lab Sci. 2003;33:371–395. [PubMed] [Google Scholar]
- 7.Chevion M, Jiang Y, Har-El R, Berenshtein E, Uretzky G, Kitrossky N. Copper and iron are mobilized following myocardial ischemia: possible predictive criteria for tissue injury. Proc Natl Acad Sci USA. 1993;90:1102–1106. doi: 10.1073/pnas.90.3.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy BR, Kloner RA, Przyklenk K. Early treatment with deferoxamine limits myocardial ischemic/reperfusion injury. Free Radic Biol Med. 1989;7:45–52. doi: 10.1016/0891-5849(89)90099-3. [DOI] [PubMed] [Google Scholar]
- 9.Sudheesh NP, Ajith TA, Janardhanan KK. Ganoderma lucidum ameliorate mitochondrial damage in isoproterenol-induced myocardial infarction in rats by enhancing the activities of TCA cycle enzymes and respiratory chain complexes. Int J Cardiol. 2013;165:117–125. doi: 10.1016/j.ijcard.2011.07.103. [DOI] [PubMed] [Google Scholar]
- 10.Sudheesh NP, Ajith TA, Janardhanan KK, Krishnan CV. Effect of POLY-MVA, a palladium alpha-lipoic acid complex formulation against declined mitochondrial antioxidant status in the myocardium of aged rats. Food Chem Toxicol. 2010;48:1858–1862. doi: 10.1016/j.fct.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 11.McCommis KS, McGee AM, Laughlin MH, Bowles DK, Baines CP. Hypercholesterolemia increases mitochondrial oxidative stress and enhances the MPT response in the porcine myocardium: beneficial effects of chronic exercise. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1250–R1258. doi: 10.1152/ajpregu.00841.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marín-García J, Goldenthal MJ, Moe GW. Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc Res. 2001;52:103–10. doi: 10.1016/s0008-6363(01)00368-6. [DOI] [PubMed] [Google Scholar]
- 13.Tsutsui T, Tsutamoto T, Wada A, Maeda K, Mabuchi N, Hayashi M, Ohnishi M, Kinoshita M. Plasma oxidized low-density lipoprotein as a prognostic predictor in patients with chronic congestive heart failure. J Am Coll Cardiol. 2002;39:957–62. doi: 10.1016/s0735-1097(02)01721-7. [DOI] [PubMed] [Google Scholar]
- 14.Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep. 2013;3:1077. doi: 10.1038/srep01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davidson SM. Endothelial mitochondria and heart disease. Cardiovasc Res. 2010;88:58–66. doi: 10.1093/cvr/cvq195. [DOI] [PubMed] [Google Scholar]
- 16.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 17.Wei YH. Mitochondrial DNA alterations as ageing-associated molecular events. Mutat Res. 1992;275:145–55. doi: 10.1016/0921-8734(92)90019-l. [DOI] [PubMed] [Google Scholar]
- 18.Wei YH. Mitochondrial DNA mutations and oxidative damage in aging and diseases: an emerging paradigm of gerontology and medicine. Proc Natl Sci Counc Repub China B. 1998;22:55–67. [PubMed] [Google Scholar]
- 19.Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A, Magrini G, et al. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol. 1998;153:1501–10. doi: 10.1016/S0002-9440(10)65738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vasquez-Vivar J, Kalyanaraman B, Kennedy MC. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem. 2000;275:14064–14069. doi: 10.1074/jbc.275.19.14064. [DOI] [PubMed] [Google Scholar]
- 22.Batandier C, Leverve X, Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J Biol Chem. 2004;279:17197–17204. doi: 10.1074/jbc.M310329200. [DOI] [PubMed] [Google Scholar]
- 23.Grijalba MT, Vercesi AE, Schreier S. Ca2 -induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2 -stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry. 1999;38:13279–13287. doi: 10.1021/bi9828674. [DOI] [PubMed] [Google Scholar]
- 24.Wan B, LaNoue KF, Cheung JY, Scaduto RC. Regulation of citric acid cycle by calcium. J Biol Chem. 1989;264:13430–13439. [PubMed] [Google Scholar]
- 25.Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO () signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free Radic Biol Med. 2002;32:370–74. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 26.Brookes PS, Darley-Usmar VM. Role of calcium and superoxide dismutase in sensitizing mitochondria to peroxynitrite-induced permeability transition. Am J Physiol Heart Circ Physiol. 2004;286:H39–H46. doi: 10.1152/ajpheart.00742.2003. [DOI] [PubMed] [Google Scholar]
- 27.Subramanian S, Kalyanaraman B, Migrino RQ. Mitochondrially targeted antioxidants for the treatment of cardiovascular diseases. Recent Pat Cardiovasc Drug Discov. 2010;5:54–65. doi: 10.2174/157489010790192601. [DOI] [PubMed] [Google Scholar]
- 28.Xiang F, Huang YS, Shi XH, Zhang Q. Mitochondrial chaperone tumour necrosis factor receptor-associated protein 1 protects cardiomyocytes from hypoxic injury by regulating mitochondrial permeability transition pore opening. FEBS J. 2010;277:1929–1938. doi: 10.1111/j.1742-4658.2010.07615.x. [DOI] [PubMed] [Google Scholar]
- 29.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 30.Hagen TM, Moreau R, Suh JH, Visioli F. Mitochondrial decay in the aging rat heart: evidence for improvement by dietary supplementation with acetyl-L-carnitine and/or lipoic acid. Ann N Y Acad Sci. 2002;959:491–507. doi: 10.1111/j.1749-6632.2002.tb02119.x. [DOI] [PubMed] [Google Scholar]
- 31.Garrido-Maraver J, Cordero MD, Oropesa-Avila M, Vega AF, de la Mata M, Pavon AD, Alcocer-Gomez E, Calero CP, Paz MV, Alanis M, et al. Clinical applications of coenzyme Q10. Front Biosci. 2014;19:619–633. doi: 10.2741/4231. [DOI] [PubMed] [Google Scholar]
- 32.Ajith TA, Divya KR. An in vitro comparative study on the antibacterial and antioxidant activities of atorvastatin and simvastatin. Pharmaceutical Biol. 2007;45:1–5. [Google Scholar]
- 33.Ajith TA, Riji T, Anu V. In vitro antioxidant and DNA protective effects of a novel HMG.CoA reductase inhibitor, rosuvastatin. Clin Exp Physiol Pharmacol. 2008;35:625–29. doi: 10.1111/j.1440-1681.2007.04853.x. [DOI] [PubMed] [Google Scholar]
- 34.Littarru GP, Langsjoen P. Coenzyme Q10 and statins: biochemical and clinical implications. Mitochondrion. 2007;7 Suppl:S168–S174. doi: 10.1016/j.mito.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am J Cardiol. 2007;99:1409–1412. doi: 10.1016/j.amjcard.2006.12.063. [DOI] [PubMed] [Google Scholar]
- 36.Langsjoen PH, Langsjoen JO, Langsjoen AM, Lucas LA. Treatment of statin adverse effects with supplemental Coenzyme Q10 and statin drug discontinuation. Biofactors. 2005;25:147–152. doi: 10.1002/biof.5520250116. [DOI] [PubMed] [Google Scholar]
- 37.Rizos I. Three-year survival of patients with heart failure caused by dilated cardiomyopathy and L-carnitine administration. Am Heart J. 2000;139:S120–S123. doi: 10.1067/mhj.2000.103917. [DOI] [PubMed] [Google Scholar]
- 38.Serati AR, Motamedi MR, Emami S, Varedi P, Movahed MR. L-carnitine treatment in patients with mild diastolic heart failure is associated with improvement in diastolic function and symptoms. Cardiology. 2010;116:178–182. doi: 10.1159/000318810. [DOI] [PubMed] [Google Scholar]
- 39.Colonna P, Iliceto S. Myocardial infarction and left ventricular remodeling: results of the CEDIM trial. Carnitine Ecocardiografia Digitalizzata Infarto Miocardico. Am Heart J. 2000;139:S124–S130. doi: 10.1067/mhj.2000.103918. [DOI] [PubMed] [Google Scholar]
- 40.Tarantini G, Scrutinio D, Bruzzi P, Boni L, Rizzon P, Iliceto S. Metabolic treatment with L-Carnitine in acute anterior ST segment elevation myocardial infarction. A randomized controlled trial. Cardiology. 2006;106:215–223. doi: 10.1159/000093131. [DOI] [PubMed] [Google Scholar]
- 41.Study on propionyl-L-carnitine in chronic heart failure. Eur Heart J. 1999;20:70–76. doi: 10.1053/euhj.1998.1271. [DOI] [PubMed] [Google Scholar]
- 42.Cheng J, Kamiya K, Kodama I. Carvedilol: molecular and cellular basis for its multifaceted therapeutic potential. Cardiovasc Drug Rev. 2001;19:152–171. doi: 10.1111/j.1527-3466.2001.tb00061.x. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto K, Origasa H, Suzuki Y, Takahashi T, Shinozaki T, Watanabe T, Sakata Y, Izumi C, Taira K, Hori M. Relation of risk factors with response to carvedilol in heart failure with preserved ejection fraction - A report from the Japanese Diastolic Heart Failure Study (J-DHF) J Cardiol. 2013;S0914-5087:326–332. doi: 10.1016/j.jjcc.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 44.Yanagishita T, Tomita M, Itoh S, Mukae S, Arata H, Ishioka H, Geshi E, Konno N, Katagiri T. Protective effect of captopril on ischemic myocardium. Jpn Circ J. 1997;61:161–169. doi: 10.1253/jcj.61.161. [DOI] [PubMed] [Google Scholar]
- 45.Sanbe A, Tanonaka K, Kobayasi R, Takeo S. Effects of long-term therapy with ACE inhibitors, captopril, enalapril and trandolapril, on myocardial energy metabolism in rats with heart failure following myocardial infarction. J Mol Cell Cardiol. 1995;27:2209–2222. doi: 10.1016/s0022-2828(95)91551-6. [DOI] [PubMed] [Google Scholar]
- 46.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2008;(2):CD007176. doi: 10.1002/14651858.CD007176. [DOI] [PubMed] [Google Scholar]
- 47.Cochemé HM, Murphy MP. Can antioxidants be effective therapeutics? Curr Opin Investig Drugs. 2010;11:426–431. [PubMed] [Google Scholar]
- 48.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 49.Pucheu S, Boucher F, Sulpice T, Tresallet N, Bonhomme Y, Malfroy B, de Leiris J. EUK-8 a synthetic catalytic scavenger of reactive oxygen species protects isolated iron-overloaded rat heart from functional and structural damage induced by ischemia/reperfusion. Cardiovasc Drugs Ther. 1996;10:331–339. doi: 10.1007/BF02627957. [DOI] [PubMed] [Google Scholar]
- 50.Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263:709–716. doi: 10.1046/j.1432-1327.1999.00543.x. [DOI] [PubMed] [Google Scholar]
- 51.Sheu SS, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim Biophys Acta. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 52.Brown SE, Ross MF, Sanjuan-Pla A, Manas AR, Smith RA, Murphy MP. Targeting lipoic acid to mitochondria: synthesis and characterization of a triphenylphosphonium-conjugated alpha-lipoyl derivative. Free Radic Biol Med. 2007;42:1766–1780. doi: 10.1016/j.freeradbiomed.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 53.Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, Filenko OF, Kalinina NI, Kapelko VI, Kolosova NG, Kopnin BP, Korshunova GA, Lichinitser MR, Obukhova LA, Pasyukova EG, Pisarenko OI, Roginsky VA, Ruuge EK, Senin II, Severina II, Skulachev MV, Spivak IM, Tashlitsky VN, Tkachuk VA, Vyssokikh MY, Yaguzhinsky LS, Zorov DB. An attempt to prevent senescence: a mitochondrial approach. Biochim Biophys Acta. 2009;1787:437–461. doi: 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 55.Kelso GF, Maroz A, Cochemé HM, Logan A, Prime TA, Peskin AV, Winterbourn CC, James AM, Ross MF, Brooker S, et al. A mitochondria-targeted macrocyclic Mn(II) superoxide dismutase mimetic. Chem Biol. 2012;19:1237–1246. doi: 10.1016/j.chembiol.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cochemé HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 57.Gioscia-Ryan RA, Larocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014:(Accepted Article). doi: 10.1113/jphysiol.2013.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009;45:643–650. doi: 10.1016/j.ceca.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 59.Davies SM, Poljak A, Duncan MW, Smythe GA, Murphy MP. Measurements of protein carbonyls, ortho- and meta-tyrosine and oxidative phosphorylation complex activity in mitochondria from young and old rats. Free Radic Biol Med. 2001;31:181–190. doi: 10.1016/s0891-5849(01)00576-7. [DOI] [PubMed] [Google Scholar]
- 60.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 61.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 62.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 63.Zhao K, Zhao G-M, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 64.Rocha M, Hernandez-Mijares A, Garcia-Malpartida K, Bañuls C, Bellod L, Victor VM. Mitochondria-targeted antioxidant peptides. Curr Pharm Des. 2010;16:3124–31. doi: 10.2174/138161210793292519. [DOI] [PubMed] [Google Scholar]
- 65.Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion. 2007;7 Suppl:S154–S167. doi: 10.1016/j.mito.2007.02.005. [DOI] [PubMed] [Google Scholar]