

Abstract
The harmful use of alcohol is a worldwide problem. It has been estimated that alcohol abuse represents the world’s third largest risk factor for disease and disability; it is a causal factor of 60 types of diseases and injuries and a concurrent cause of at least 200 others. Liver is the main organ responsible for metabolizing ethanol, thus it has been considered for long time the major victim of the harmful use of alcohol. Ethanol and its bioactive products, acetaldehyde-acetate, fatty acid ethanol esters, ethanol-protein adducts, have been regarded as hepatotoxins that directly and indirectly exert their toxic effect on the liver. A similar mechanism has been postulated for the alcohol-related pancreatic damage. Alcohol and its metabolites directly injure acinar cells and elicit stellate cells to produce and deposit extracellular matrix thus triggering the “necrosis-fibrosis” sequence that finally leads to atrophy and fibrosis, morphological hallmarks of alcoholic chronic pancreatitis. Even if less attention has been paid to the upper and lower gastrointestinal tract, ethanol produces harmful effects by inducing: (1) direct damaging of the mucosa of the esophagus and stomach; (2) modification of the sphincterial pressure and impairment of motility; and (3) alteration of gastric acid output. In the intestine, ethanol can damage the intestinal mucosa directly or indirectly by altering the resident microflora and impairing the mucosal immune system. Notably, disruption of the intestinal mucosal barrier of the small and large intestine contribute to liver damage. This review summarizes the most clinically relevant alcohol-related diseases of the digestive tract focusing on the pathogenic mechanisms by which ethanol damages liver, pancreas and gastrointestinal tract.
Keywords: Alcoholic liver disease, Alcoholic pancreatitis, Alcohol and gastrointestinal tract
Core tip: Alcohol abuse represents the world’s third largest risk factor for disease and disability. According to a “hepatocentric” vision of the problem, liver has been considered for long time the main victim of the harmful use of alcohol. However, growing evidence suggests that alcoholic disease should not be considered limited to the liver but as a true systemic disease including damage to the digestive tract, the central and peripheral nervous systems, the heart and vascular system, the bone and skeletal muscle, the endocrine and immune systems and disruption of nutritional status and finally cancer.
BURDEN OF DISEASE
Alcohol is a major risk factor for chronic diseases and one of the leading causes of “preventable” morbidity and mortality worldwide. It has been estimated that the harmful use of alcohol results in approximately 2.5 million deaths each year with much of the burden depending on alcoholic liver disease (ALD)[1]. Even if the liver has been for long time considered the major victim of the harmful use of alcohol, the likelihood of systemic effects parallels the severity of liver damage dependent on the alcohol abuse. According to the World Health Organization Report on Alcohol and Health (2011), alcohol abuse is responsible for at least 60 major types of systemic diseases. Furthermore, alcohol consumption significantly increases the overall risk of developing cancer[1].
This review will focus on ethanol-dependent damages to liver, pancreas and digestive apparatus.
ALCOHOL AND LIVER
After C virus-related hepatitis, alcohol represents the most common cause of chronic liver disease in the majority of the industrialized countries[2]. Overall, about 25% of the cases of liver cirrhosis recognize as initial trigger the over-exposition to alcohol[2,3]. However, although virtually all individuals chronically exposed to alcohol develop fatty liver, the earliest response of the liver to alcohol consumption, only a minority progresses up to cirrhosis[4,5]. Duration and amount of alcohol ingested remain the most important risk factors for the development of a progressive form of alcohol-dependent liver disease even if there is not a definite cut-off of alcohol consumption able to predict a severe form of ALD[6,7]. It has been reported that a daily consumption of 60-80 g/d of alcohol for 10 years or longer in men, and 20 g/d in women leads to an advanced form of liver disease in < 40% of the cases[4,5]. A recent population-based study including 6917 northern Italian subjects demonstrated that only 13.5% developed ALD even when exposed to a very high daily alcohol intake (120 g/d)[8]. Female sex, obesity, non-sex-linked genetic factors and cigarette smoking can modulate the host susceptibility to develop ALD and contribute to overall risk of developing the severe form of the disease[9]. Furthermore, alcohol synergistically interacts with other well-known causative agents of liver damage such as hepatitis virus B or C and/or human immunodeficiency virus infection, nonalcoholic fatty liver disease, and disorders such as hemochromatosis to promote the progression of alcohol-related liver injury[10].
Many hypothesis have been advanced to explain the pathogenic mechanisms of ALD. Firstly, the liver is the main organ responsible for metabolizing ethanol, thus it is conceivable that ethanol and its metabolites [acetaldehyde-acetate, fatty acid ethanol esters (FAEEs), ethanol-protein adducts] can exert a direct cytotoxic effect acting as hepatotoxins[11]. Hepatic metabolism of the ethanol proceeds via oxidative and non-oxidative pathways (Figure 1). The main steps of the oxidative pathway are mediated by alcohol dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH) that transform ethanol to acetaldehyde and acetaldehyde to acetate, respectively[12]. The end products of this reaction are acetaldehyde, acetate and high levels of NADH. Acetaldehyde damages liver by directly triggering inflammation, extracellular matrix (ECM) remodeling and fibrogenesis[13]. Furthermore, it covalently binds to proteins and DNA leading to the production of immunogenic adducts (i.e., malondialdehyde) in the hepatocytes[14]. Finally, acetaldehyde stimulates transforming growth factor (TGF)-beta signaling in hepatic stellate cells that acquire a pro-fibrogenic and pro-inflammatory profile[15]. Electrons from alcohol are transferred to NADP+ by ADH. Changes in NADH/NAD+ ratio may affect biochemical reactions in the mitochondria and gene expression in nucleus. The burn of NADH requires additional oxygen amount in the mitochondria; the hepatocytes take up more than their normal share of oxygen from arterious blood but not enough to adequately supply all liver regions. Thus, alcohol consumption results in significant hypoxia of the perivenous hepatocytes that are the first ones to show evidence of damage from chronic alcohol consumption[16,17].
Figure 1.
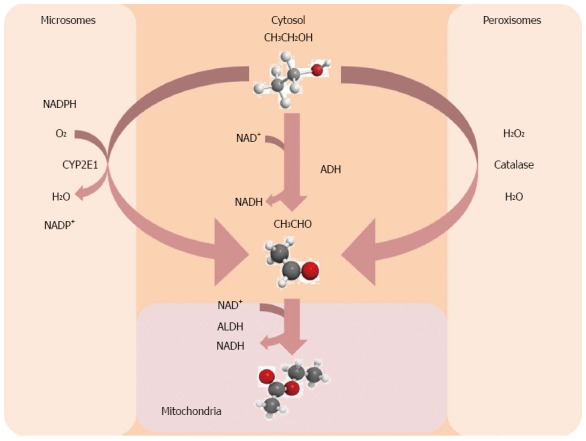
Main steps of alcohol metabolism in the liver. ADH: Alcohol dehydrogenase; ALDH: Acetaldehyde dehydrogenase.
Cytochrome P450s and especially cytochrome P450 2E1 (CYP2E1) is up-regulated in condition of chronic alcohol abuse and assists ADH in converting alcohol to acetaldehyde[11]. Reactive oxygen species (ROS), such as hydrogen peroxide and superoxide ions, generated by CYP2E1 are responsible for the pro-inflammatory profile of the alcohol-related liver injury by: (1) activating redox-sensitive transcription factors [i.e., nuclear factor kappa B (NF-κB)][18]; (2) recruiting neutrophils and other immune cells; (3) increasing the level of circulating pro-inflammatory cytokines; and (4) contributing to the lipid peroxidation associated with alcoholic liver injury[11].
Catalase, a peroxisomal enzyme, is the master regulator of non-oxidative metabolism of alcohol the final product of which is FAEE responsible for alcoholic steatosis[9,11] and useful as biomarker of chronic alcohol consumption[19,20].
Gut-derived lipopolysaccharide (LPS) is another critical trigger of liver steatosis, inflammation, and fibrosis. Gut houses billions of microorganisms but, under physiological condition, LPS derived from Gram-negative bacteria enters the portal circulation only in trace to be cleared by Kupffer cells (resident macrophages) and hepatocytes that possess different LPS recognition systems[21-23]. Alcohol impairs intestinal barrier leading to increased circulating endotoxin levels which binds to the surface receptor CD14 on hepatic Kupffer cells via LPS-binding protein (LBP). The CD14-LPBLPS complex, via NADPH oxidase, produces ROS and stimulates the toll-like receptor 4 (TLR4) signaling cascade with the end result of the activation of NF-κB and the release of inflammatory cytokines, notably tumor necrosis factor (TNF)-α[24,25]. TNF-α, in turn, sustains liver injury by worsening the gut permeability on one side, and upholding the necro-inflammatory hepatic damage, on the other side[26].
ALCOHOL AND PANCREAS
Since the initial observation by Friedreich in 1878, growing evidence linked alcohol misuse and pancreatic damage. Because of difficulties in accurately identifying alcohol abuse, differences in the populations studied and classification of pancreatitis, based on morphology rather than on etiology, the proportion of cases of alcohol-related pancreatitis widely varies between and even in the same country. Overall, it has been estimated that prevalence of pancreatitis in alcoholics increased approximately 4-fold when compared with teetotalers[27]. Although a dose-related toxic effect of the alcohol on the pancreas has been described, the alcohol consumption and risk of developing pancreatitis are not linearly linked[28]. In fact, the risk curve remains relatively flat up to the threshold of approximately 4-5 drinks/d and markedly increases as levels of consumption increase[29-31]. Notably, after a first acute episode of pancreatitis, alcoholics have a risk of developing chronic disease approximately of 14% with complete abstinence or occasional drinking and 41% if alcohol intake persists[32].
The pancreas directly metabolizes ethanol producing metabolites and byproducts responsible for injury of acinar cells and stimulation of stellate cells to produce and deposit ECM[33]. Acetaldehyde and ROS induce microtubular dysfunction and disruption of the actin cytoskeleton, alteration in the intracellular redox state and destabilization of zymogen granules and lysosomes. Even if FAEE tissue levels are lower than those of acetate (product of alcohol oxidation), they have been shown to be sufficient to produce pancreatic injury[33]. FAAEs exert their toxic effects by directly interacting with cellular membranes, stimulating cholesteryl ester synthesis and free fatty acids release with consequent mitochondrial damage. Furthermore, FAAEs can induce activation of NF-κB and AP-1 transcription factors[33] and alteration of intracellular calcium metabolism[34,35]. Damaging of microtubular function and instability of zymogen granules and lysosomes results in an inappropriate activation of the digestive enzymes and autodigestion of the pancreatic glands. Acinar cell death (both by necrosis or apoptosis), activated by proliferative and profibrinogenic growth factors such as platelet-derived growth factor (PDGF), TGF-α and connective growth factor, drives pancreatic stellate cells (PSCs) into a highly proliferative state triggering their myofibroblast-like transformation and subsequent tissue fibrosis[36,37]. However, exposure to alcohol or bacterial endotoxin-like LPS may also directly activate PSCs. In a rat model, the administration of alcohol and LPS was effective in inducing pancreatic injury while alcohol alone failed to trigger inflammatory process suggesting that alcohol and LPS exert synergistic effects on PSCs activation[37]. The “necrosis-fibrosis” sequence leads to acinar atrophy and fibrosis, morphological hallmarks of chronic pancreatitis, resulting in exocrine and endocrine glandular dysfunction.
ALCOHOL AND UPPER GASTROINTESTINAL TRACT
Despite the association between excessive drinking and risk of gastric bleeding dates back 170 years, the effects of alcohol on upper gastrointestinal (GI) tract have systematically been investigated only in the last 15 years. Both acute and chronic alcohol consumption affects upper GI tract by multiple and complex mechanisms depending either by direct contact of ethanol and/or its metabolite acetaldehyde with the mucosa as well as by non-alcoholic components of alcoholic beverages (i.e., fermentation products). These mechanisms result in: (1) inflammation of the esophageal and gastric mucosa; (2) modification of sphincterial pressure and impairment of motility; and (3) alteration of gastric acid output. All these effects are dose-dependent and reversible following abstinence[38].
Increased prevalence of heartburn and increased risk of gastro-esophageal reflux disease (GERD) or erosive esophagitis have been reported in alcoholics[39-42] as well as GERD patients are more likely to consume alcohol than controls[43]. Ethanol-induced inflammation of the esophageal mucosa depends on the direct damage of the mucosal barrier that, in turn, predisposes tissue to acid injury. In rabbit, the exposition of esophageal epithelium to chloride acid (HCl) alone induced little or no morphological alterations or functional changes as measured by means of mucosal potential difference (PD) and electrical resistance of the mucosa. However, the simultaneous exposure to both ethanol and HCl resulted in morphologic damage and significantly greater declines in PD[44].
Both acute and chronic alcohol consumption affect esophageal motility. Acute ethanol administration in vivo, in man as well as in cats, transiently decreased lower esophageal sphinteric (LES) pressure, degree of contraction of the smooth muscle layer of the lower esophagus and mucosal clearance due to a primary and secondary peristalsis reduction[45]. In a cat model, neural decentralization (cervical vagotomy) or nerve block did not prevent the effects of acute ethanol administration suggesting a direct inhibitory effect of alcohol on esophageal muscle cells[46,47]. In an in vitro model, the pre-exposition of esophageal smooth muscle cells to ethanol significantly decreased carbachol-dependent shortening of the cells, thus confirming that ethanol directly inhibits the contractile activity of the esophageal muscle cells[46]. Paradoxically, chronic effect of alcohol on esophageal motility consisted in an increased tonus of the LES and reduced esophageal clearance[45]. In an alcohol-fed rat model, Yazir et al[48] recently demonstrated that chronic alcohol consumption impaired relaxant and contractile responses of both LES and tunica muscularis mucosae of the esophagus.
More than 40 years ago Roberts[49] already reported that gastritis and gastroduodenal ulcer were common in alcoholics. However, the mechanisms by which alcohol damages gastric mucosa are still largely unknown. Ethanol can cause mucosal edema, erosion, hemorrhage and necrosis by directly damaging the gastric mucosal barrier and thus affecting the ability of the gastric mucosa to defend itself by gastric acid, bile and digestive enzymes[49]. Recent studies demonstrated that an impaired gastric microcirculation, accompanied by increased blood levels of endothelin-1 (ET-1) and decreased levels of nitric oxide (NO) and prostaglandin (PG)E2 can critically contribute to mucosal barrier damage in a rat model[50]. NO and PGE2 accelerate the flow of the gastric mucosal microcirculation, promote the secretion of bicarbonate, mediate the adaptive immune protective function, increase protein synthesis and cell renewal, and finally enhance the repair ability of the damaged gastric mucosa[50]. In contrast, ET-1 is the strongest vasoconstrictor of gastric mucosa microcirculation. Pre-injection of an endothelin receptor antagonist significantly reduced ET-1-dependent gastric mucosal damage in rat. In addition, high ethanol intake rats had ET-1 plasma levels significantly higher and NO and PGE2 decreased in respect to the normal control group[51]. Taken together these data suggests that ethanol damages gastric mucosa and weakens its ability to repair by stimulating ET-1 secretion and inhibiting NO and PGE2 synthesis and secretion[51].
Contradictory results exist on the effect of alcohol upon gastric emptying, depending on dose and type of beverage involved. In fact gastric emptying seems to be accelerated after a low alcohol dose while high doses of ethanol delay emptying and reduce motility[52]. Several reports emphasized the potential role of NO in alcohol-seeking behavior as a main target of alcohol-related GI motility disorders[53,54]. Studies in NO sintase knockout animals demonstrated that NO is the main inhibitory nonadrenergic noncholinergic neurotransmitter in the enteric neuronal system and that NOS inhibitors delay gastric emptying and colonic transit[53]. Recently, it has been shown that chronic ethanol administration in rats markedly increases NOS activity, the amount of NOS protein and the number of nitrergic neurons prompting to conclude that the overproduction of NO may be a direct cause of gastrointestinal motility disturbances in alcoholics[54].
Finally, alcohol can differently affect gastric acid output. Low doses of ethanol stimulate gastric acid secretion while high doses either exert or not inhibitory effect[38]. Similarly contradictory results have been reported on the effect of alcohol on endocrine cells regulating acid secretion (G cells). According to one group of authors, chronic alcohol consumption reduced the number of G cells while increased gastrin plasma level in experimental animals[55]. On the other hand, another study found that neither acute nor chronic ethanol consumption significantly changed the number of G cells or gastrin serum level[56]. Finally, a recent study demonstrated that ethanol consumption over a 4-mo period, resulted in a non-significant decrease of antral and plasma levels of gastrin and an increased volume density and diameter of G cells suggesting that a more complex network of endocrine mechanisms (i.e., simultaneous reduction of D cell and somatostatin levels) can be involved in ethanol acid output modifications[57].
ALCOHOL AND LOWER GASTROINTESTINAL TRACT
Alcohol can affect lower gastrointestinal tract directly by damaging the intestinal mucosa or indirectly by altering the resident microflora and impairing the mucosal immune system[58].
In spite the “first-pass” metabolism of ethanol by gastric ADH, high concentration of ethanol can reach duodenum and upper jejunum where direct effects on the mucosa are plausible. In experimental animal models (rodents or dogs), even low concentration of ethanol (≥ 4%, vol/vol) damages the intestinal mucosa[59]. The histological hallmarks of alcohol exposition that are loss of epithelium at the tips of the villi, hemorrhagic erosions of the mucosa and hemorrhage in the lamina propria have been also demonstrated in volunteers with previously normal upper gastrointestinal endoscopy 3 h after the assumption of 1 g/kg body weight of ethanol[60]. According to these findings, a case-control study conducted in United States, Sweden and Hungary demonstrated that the risk of major duodenal bleeding was significantly higher in non-predisposed drinkers than in non-drinkers, particularly in the heaviest consumers[61]. The mechanisms by which alcohol causes the structural changes of the intestinal mucosa have not been completely clarified. It is likely that alcohol directly causes villus contraction with consequent villus tip bleb formation, lymphatic obstruction and exfoliation of the tips of the villi[62]. In addition, ethanol can indirectly contribute to the mucosal damage by triggering an “inflammatory state” of the intestinal mucosa with invasion of leukocytes and release of noxious mediators, such as ROS[63], leukotrienes[64] and release of histamine by mast cells[65]. Finally, alcohol can increase the permeability of the mucosal micro-vessels with enhanced transcapillary fluid filtration and disruption of the epithelial continuity[58]. If the damaging effect of acute alcohol exposure is clear, whether or not chronic alcohol ingestion damages the mucosa of the small intestine remains controversial. Duodenal biopsies of chronic alcohol abusers either had normal histology or showed reduced villus height, increase in the number of intra-epithelial mononuclear cells, goblet cell hyperplasia and gastric metaplasia[58,66]. Furthermore, chronic alcohol abuse can induce fibrosis of the intestinal mucosa by increasing number of myofibroblast-like cells in the duodenal mucosa[67]. The loss of the anatomical integrity of the mucosa can, at least in part, account for the alcohol-related increased intestinal permeability. In animal models it has been reported that alcohol exposition can induce increased permeability for hemoglobin, horseradish peroxidase and polyethyleneglycol (PEG) 1500 Mr[58]. In human, a significantly increased intestinal permeability for macromolecules such as PEG 4.000 Mr and 10000 Mr has been reported in actively drinking alcoholics[24]. The increased intestinal permeability to macromolecules may account for the transient endotoxaemia described in healthy volunteers after acute alcohol consumption and in alcoholics with fatty liver[24,68]. The increased permeability of the gut mucosa reported in alcoholics since the early stage of liver disease, further supports the hypothesis that it is caused by ethanol itself and is not a consequence of advanced ALD.
Alcohol can also affect the intestinal microenvironment by inducing qualitative and quantitative alterations of the jejunal microflora[69]. Small intestinal bacterial overgrowth, defined as 105 colony forming units/mL of jejunal juice, has been demonstrated in about half of individuals chronically exposed to alcohol[58]. Similar results were obtained by using glucose or lactulose H2-breath test[69]. Small intestinal bacterial overgrowth not only contributes to the direct damage of the intestinal mucosa but it also leads to an increased production of bacterial toxins, especially those derived from Gram-negative bacteria. The combination of mucosal injury and the increased bacterial-derived products results in the enhanced endotoxin translocation from the luminal side into the portal blood[70].
Both specific and unspecific immune system is affected by alcohol ingestion[71]. In animal models, perfusion of jejunum with 6% ethanol triggered inflammatory reactions characterized by leukocyte infiltration and mast cells release of histamine and ROS[63,72]. The abstention from alcohol for up to 5 d increased the number of B-lymphocytes and halved the number of mononuclear macrophages in the lamina propria of duodenal mucosa[73]. These changes were no longer observed in subjects abstaining for 5-10 d.
Finally, both acute and chronic exposure of the small intestine to alcohol can impair the absorption of monosaccharides, several L-amino acid residues and lipids (fatty acids, monoglycerides) as well as some vitamins even in absence of advanced liver disease or pancreatic insufficiency[58]. The functional interference of ethanol with the physiological absorption of nutrients can contribute to qualitative and quantitative malnutrition frequently observed in alcoholics.
In contrast to duodenum and upper jejunum, alcohol does not directly reach the large bowel. Indeed, the concentration of ethanol in the colon, even when assumed in large quantity, is similar to that observed in the blood, thus suggesting that ethanol enters this organ from the vascular space. The oxidation of alcohol to acetaldehyde by colonic bacteria via ADH largely exceeds the capability to metabolize acetaldehyde to acetate, thus leading to a significant increase of the former[74]. The increased level of acetaldehyde not only damages the colonic mucosa but, after absorption into the portal blood, contributes to liver injury.
Finally, acetaldehyde can exert a local carcinogenic by stimulating the proliferation rate of the epithelial cells in the rectum of both animal and humans[75,76].
CONCLUSION
Alcohol abuse represents a serious public health concern in many countries. According to a “hepatocentric” vision of the problem, liver has been considered for long time the main victim of the harmful use of alcohol. However, growing evidence suggest that ALD have to be considered a true systemic disease. The “multisystemic scenario” of the alcohol-related diseases underlies the urgent need to promote research oriented to pathophysiology-targeted therapies as well as preventive policy strategies to reduce the clinical and economic burden of alcohol abuse.
Footnotes
P- Reviewer: Gong NQ, Zerem E S- Editor: Wen LL L- Editor: A E- Editor: Zhang DN
References
- 1.World Health Organization. Global Status Report on Alcohol and Health 2011. Available from: http://www.who.int/substance_abuse/publications/global_alcohol_report/msbgsruprofiles.pdf.
- 2.Singal AK, Anand BS. Recent trends in the epidemiology of alcoholic liver disease. Clin Liv Dis. 2013;2:53–56. doi: 10.1002/cld.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mann RE, Smart RG, Govoni R. The epidemiology of alcoholic liver disease. Alcohol Res Health. 2003;27:209–219. [PMC free article] [PubMed] [Google Scholar]
- 4.Mendenhall CL. Anabolic steroid therapy as an adjunct to diet in alcoholic hepatic steatosis. Am J Dig Dis. 1968;13:783–791. doi: 10.1007/BF02233094. [DOI] [PubMed] [Google Scholar]
- 5.Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis. 2004;24:217–232. doi: 10.1055/s-2004-832936. [DOI] [PubMed] [Google Scholar]
- 6.Kamper-Jørgensen M, Grønbaek M, Tolstrup J, Becker U. Alcohol and cirrhosis: dose--response or threshold effect? J Hepatol. 2004;41:25–30. doi: 10.1016/j.jhep.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Ramstedt M. Per capita alcohol consumption and liver cirrhosis mortality in 14 European countries. Addiction. 2001;96 Suppl 1:S19–S33. doi: 10.1080/09652140020021152. [DOI] [PubMed] [Google Scholar]
- 8.Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, Saveria Crocè L, Sasso F, Pozzato G, Cristianini G, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut. 1997;41:845–850. doi: 10.1136/gut.41.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day CP. Who gets alcoholic liver disease: nature or nurture? J R Coll Physicians Lond. 2000;34:557–562. [PMC free article] [PubMed] [Google Scholar]
- 11.Seth D, Haber PS, Syn WK, Diehl AM, Day CP. Pathogenesis of alcohol-induced liver disease: classical concepts and recent advances. J Gastroenterol Hepatol. 2011;26:1089–1105. doi: 10.1111/j.1440-1746.2011.06756.x. [DOI] [PubMed] [Google Scholar]
- 12.Lieber CS. Role of oxidative stress and antioxidant therapy in alcoholic and nonalcoholic liver diseases. Adv Pharmacol. 1997;38:601–628. doi: 10.1016/s1054-3589(08)61001-7. [DOI] [PubMed] [Google Scholar]
- 13.Mello T, Ceni E, Surrenti C, Galli A. Alcohol induced hepatic fibrosis: role of acetaldehyde. Mol Aspects Med. 2008;29:17–21. doi: 10.1016/j.mam.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Niemelä O. Distribution of ethanol-induced protein adducts in vivo: relationship to tissue injury. Free Radic Biol Med. 2001;31:1533–1538. doi: 10.1016/s0891-5849(01)00744-4. [DOI] [PubMed] [Google Scholar]
- 15.Chen A. Acetaldehyde stimulates the activation of latent transforming growth factor-beta1 and induces expression of the type II receptor of the cytokine in rat cultured hepatic stellate cells. Biochem J. 2002;368:683–693. doi: 10.1042/BJ20020949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishak KG, Zimmerman HJ, Ray MB. Alcoholic liver disease: pathologic, pathogenetic and clinical aspects. Alcohol Clin Exp Res. 1991;15:45–66. doi: 10.1111/j.1530-0277.1991.tb00518.x. [DOI] [PubMed] [Google Scholar]
- 17.Beier JI, McClain CJ. Mechanisms and cell signaling in alcoholic liver disease. Biol Chem. 2010;391:1249–1264. doi: 10.1515/BC.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 19.Soderberg BL, Salem RO, Best CA, Cluette-Brown JE, Laposata M. Fatty acid ethyl esters. Ethanol metabolites that reflect ethanol intake. Am J Clin Pathol. 2003;119 Suppl:S94–S99. doi: 10.1309/6F39-EAR2-L4GY-X5G6. [DOI] [PubMed] [Google Scholar]
- 20.Kaphalia BS, Cai P, Khan MF, Okorodudu AO, Ansari GA. Fatty acid ethyl esters: markers of alcohol abuse and alcoholism. Alcohol. 2004;34:151–158. doi: 10.1016/j.alcohol.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 21.Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity. 2009;31:368–376. doi: 10.1016/j.immuni.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis. 2009;29:141–154. doi: 10.1055/s-0029-1214370. [DOI] [PubMed] [Google Scholar]
- 24.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 25.Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, Remick DG, Wang SC. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology. 2000;31:932–936. doi: 10.1053/he.2000.5634. [DOI] [PubMed] [Google Scholar]
- 26.Bradham CA, Plümpe J, Manns MP, Brenner DA, Trautwein C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am J Physiol. 1998;275:G387–G392. doi: 10.1152/ajpgi.1998.275.3.G387. [DOI] [PubMed] [Google Scholar]
- 27.Wilson JS, Korsten MA, Pirola RC. Alcohol-induced pancreatic injury (Part I). Unexplained features and ductular theories of pathogenesis. Int J Pancreatol. 1989;4:109–125. [PubMed] [Google Scholar]
- 28.Kristiansen L, Grønbaek M, Becker U, Tolstrup JS. Risk of pancreatitis according to alcohol drinking habits: a population-based cohort study. Am J Epidemiol. 2008;168:932–937. doi: 10.1093/aje/kwn222. [DOI] [PubMed] [Google Scholar]
- 29.Corrao G, Bagnardi V, Zambon A, La Vecchia C. A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev Med. 2004;38:613–619. doi: 10.1016/j.ypmed.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 30.Irving HM, Samokhvalov AV, Rehm J. Alcohol as a risk factor for pancreatitis. A systematic review and meta-analysis. JOP. 2009;10:387–392. [PMC free article] [PubMed] [Google Scholar]
- 31.Yadav D, Hawes RH, Brand RE, Anderson MA, Money ME, Banks PA, Bishop MD, Baillie J, Sherman S, DiSario J, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. 2009;169:1035–1045. doi: 10.1001/archinternmed.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeyama Y. Long-term prognosis of acute pancreatitis in Japan. Clin Gastroenterol Hepatol. 2009;7:S15–S17. doi: 10.1016/j.cgh.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 33.Gukovskaya AS, Mouria M, Gukovsky I, Reyes CN, Kasho VN, Faller LD, Pandol SJ. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology. 2002;122:106–118. doi: 10.1053/gast.2002.30302. [DOI] [PubMed] [Google Scholar]
- 34.Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, Petersen OH. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;130:781–793. doi: 10.1053/j.gastro.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 35.Del Castillo-Vaquero A, Salido GM, González A. Increased calcium influx in the presence of ethanol in mouse pancreatic acinar cells. Int J Exp Pathol. 2010;91:114–124. doi: 10.1111/j.1365-2613.2009.00691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Apte MV, Phillips PA, Fahmy RG, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Naidoo D, Wilson JS. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology. 2000;118:780–794. doi: 10.1016/s0016-5085(00)70148-x. [DOI] [PubMed] [Google Scholar]
- 37.Vonlaufen A, Xu Z, Daniel B, Kumar RK, Pirola R, Wilson J, Apte MV. Bacterial endotoxin: a trigger factor for alcoholic pancreatitis? Evidence from a novel, physiologically relevant animal model. Gastroenterology. 2007;133:1293–1303. doi: 10.1053/j.gastro.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 38.Teyssen S, Singer MV. Alcohol-related diseases of the oesophagus and stomach. Best Pract Res Clin Gastroenterol. 2003;17:557–573. doi: 10.1016/s1521-6918(03)00049-0. [DOI] [PubMed] [Google Scholar]
- 39.Minatsuki C, Yamamichi N, Shimamoto T, Kakimoto H, Takahashi Y, Fujishiro M, Sakaguchi Y, Nakayama C, Konno-Shimizu M, Matsuda R, et al. Background factors of reflux esophagitis and non-erosive reflux disease: a cross-sectional study of 10,837 subjects in Japan. PLoS One. 2013;8:e69891. doi: 10.1371/journal.pone.0069891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akiyama T, Inamori M, Iida H, Mawatari H, Endo H, Hosono K, Yoneda K, Fujita K, Yoneda M, Takahashi H, et al. Alcohol consumption is associated with an increased risk of erosive esophagitis and Barrett’s epithelium in Japanese men. BMC Gastroenterol. 2008;8:58. doi: 10.1186/1471-230X-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson LA, Cantwell MM, Watson RG, Johnston BT, Murphy SJ, Ferguson HR, McGuigan J, Comber H, Reynolds JV, Murray LJ. The association between alcohol and reflux esophagitis, Barrett’s esophagus, and esophageal adenocarcinoma. Gastroenterology. 2009;136:799–805. doi: 10.1053/j.gastro.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Gunji T, Sato H, Iijima K, Fujibayashi K, Okumura M, Sasabe N, Urabe A, Matsuhashi N. Risk factors for erosive esophagitis: a cross-sectional study of a large number of Japanese males. J Gastroenterol. 2011;46:448–455. doi: 10.1007/s00535-010-0359-5. [DOI] [PubMed] [Google Scholar]
- 43.Conio M, Filiberti R, Blanchi S, Ferraris R, Marchi S, Ravelli P, Lapertosa G, Iaquinto G, Sablich R, Gusmaroli R, et al. Risk factors for Barrett’s esophagus: a case-control study. Int J Cancer. 2002;97:225–229. doi: 10.1002/ijc.1583. [DOI] [PubMed] [Google Scholar]
- 44.Bor S, Bor-Caymaz C, Tobey NA, Abdulnour-Nakhoul S, Orlando RC. Esophageal exposure to ethanol increases risk of acid damage in rabbit esophagus. Dig Dis Sci. 1999;44:290–300. doi: 10.1023/a:1026646215879. [DOI] [PubMed] [Google Scholar]
- 45.Franke A, Teyssen S, Singer MV. Alcohol-related diseases of the esophagus and stomach. Dig Dis. 2005;23:204–213. doi: 10.1159/000090167. [DOI] [PubMed] [Google Scholar]
- 46.Fields JZ, Jacyno M, Wasyliw R, Winship D, Keshavarzian A. Ethanol inhibits contractility of esophageal smooth muscle strips. Alcohol Clin Exp Res. 1995;19:1403–1413. doi: 10.1111/j.1530-0277.1995.tb00999.x. [DOI] [PubMed] [Google Scholar]
- 47.Keshavarzian A, Muska B, Sundaresan R, Urban G, Fields J. Ethanol at pharmacologically relevant concentrations inhibits contractility of isolated smooth muscle cells of cat esophagus. Alcohol Clin Exp Res. 1996;20:180–184. doi: 10.1111/j.1530-0277.1996.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 48.Yazir Y, Tugay M, Utkan Z, Utkan T. Effects of chronic ethanol consumption on rat upper gastrointestinal system: functional and histologic findings. Alcohol. 2012;46:649–655. doi: 10.1016/j.alcohol.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 49.Roberts DM. Chronic gastritis, alcohol, and non-ulcer dyspepsia. Gut. 1972;13:768–774. doi: 10.1136/gut.13.10.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ning JW, Lin GB, Ji F, Xu J, Sharify N. Preventive effects of geranylgeranylacetone on rat ethanol-induced gastritis. World J Gastroenterol. 2012;18:2262–2269. doi: 10.3748/wjg.v18.i18.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lazaratos S, Irukayama-Tomobe Y, Miyauchi T, Goto K, Nakahara A. Oxygen radicals mediate the final exacerbation of endothelin-1-induced gastric ulcer in rat. Eur J Pharmacol. 2001;413:121–129. doi: 10.1016/s0014-2999(01)00752-x. [DOI] [PubMed] [Google Scholar]
- 52.Bujanda L. The effects of alcohol consumption upon the gastrointestinal tract. Am J Gastroenterol. 2000;95:3374–3382. doi: 10.1111/j.1572-0241.2000.03347.x. [DOI] [PubMed] [Google Scholar]
- 53.Mashimo H, He XD, Huang PL, Fishman MC, Goyal RK. Neuronal constitutive nitric oxide synthase is involved in murine enteric inhibitory neurotransmission. J Clin Invest. 1996;98:8–13. doi: 10.1172/JCI118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bagyánszki M, Torfs P, Krecsmarik M, Fekete E, Adriaensen D, Van Nassauw L, Timmermans JP, Kroese AB. Chronic alcohol consumption induces an overproduction of NO by nNOS- and iNOS-expressing myenteric neurons in the murine small intestine. Neurogastroenterol Motil. 2011;23:e237–e248. doi: 10.1111/j.1365-2982.2011.01707.x. [DOI] [PubMed] [Google Scholar]
- 55.Yamada H, Kimura K, Chen D. Effects of daily fat or ethanol ingestion on the cholecystokinin- pancreas and the gastrin- enterochromaffin-like cell axes in rats. Digestion. 1998;59:331–334. doi: 10.1159/000007511. [DOI] [PubMed] [Google Scholar]
- 56.Koko V, Todorović V, Varagić J, Micev M, Korać A, Bajcetić M, Cakić-Milosević M, Nedeljković M, Drndarević N. Gastrin producing G-cells after chronic ethanol and low protein nutrition. Indian J Exp Biol. 1998;36:1093–1101. [PubMed] [Google Scholar]
- 57.Todorović V, Koko V, Budec M, Mićić M, Micev M, Pavlović M, Vignjević S, Drndarević N, Mitrović O. G cells and gastrin in chronic alcohol-treated rats. Alcohol. 2008;42:37–45. doi: 10.1016/j.alcohol.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Bode C, Bode JC. Effect of alcohol consumption on the gut. Best Pract Res Clin Gastroenterol. 2003;17:575–592. doi: 10.1016/s1521-6918(03)00034-9. [DOI] [PubMed] [Google Scholar]
- 59.Beck IT, Dinda PK. Acute exposure of small intestine to ethanol: effects on morphology and function. Dig Dis Sci. 1981;26:817–838. doi: 10.1007/BF01309614. [DOI] [PubMed] [Google Scholar]
- 60.Gottfried EB, Korsten MA, Lieber CS. Alcohol-induced gastric and duodenal lesions in man. Am J Gastroenterol. 1978;70:587–592. [PubMed] [Google Scholar]
- 61.Kelly JP, Kaufman DW, Koff RS, Laszlo A, Wiholm BE, Shapiro S. Alcohol consumption and the risk of major upper gastrointestinal bleeding. Am J Gastroenterol. 1995;90:1058–1064. [PubMed] [Google Scholar]
- 62.Ray M, Dinda PK, Beck IT. Mechanism of ethanol-induced jejunal microvascular and morphologic changes in the dog. Gastroenterology. 1989;96:345–354. doi: 10.1016/0016-5085(89)91558-8. [DOI] [PubMed] [Google Scholar]
- 63.Dinda PK, Kossev P, Beck IT, Buell MG. Role of xanthine oxidase-derived oxidants and leukocytes in ethanol-induced jejunal mucosal injury. Dig Dis Sci. 1996;41:2461–2470. doi: 10.1007/BF02100144. [DOI] [PubMed] [Google Scholar]
- 64.Beck IT, Boyd AJ, Dinda PK. Evidence for the involvement of 5-lipoxygenase products in ethanol-induced intestinal plasma protein loss. Am J Physiol. 1988;254:G483–G488. doi: 10.1152/ajpgi.1988.254.4.G483. [DOI] [PubMed] [Google Scholar]
- 65.Dinda PK, Leddin DJ, Beck IT. Histamine is involved in ethanol-induced jejunal microvascular injury in rabbits. Gastroenterology. 1988;95:1227–1233. doi: 10.1016/0016-5085(88)90355-1. [DOI] [PubMed] [Google Scholar]
- 66.Persson J. Alcohol and the small intestine. Scand J Gastroenterol. 1991;26:3–15. doi: 10.3109/00365529108996478. [DOI] [PubMed] [Google Scholar]
- 67.Casini A, Galli A, Calabro’ A, Di Lollo S, Orsini B, Arganini L, Jezequel AM, Surrenti C. Ethanol-induced alterations of matrix network in the duodenal mucosa of chronic alcohol abusers. Virchows Arch. 1999;434:127–135. doi: 10.1007/s004280050316. [DOI] [PubMed] [Google Scholar]
- 68.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94:200–207. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 69.Casafont Morencos F, de las Heras Castaño G, Martín Ramos L, López Arias MJ, Ledesma F, Pons Romero F. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig Dis Sci. 1996;41:552–556. doi: 10.1007/BF02282340. [DOI] [PubMed] [Google Scholar]
- 70.Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, Kang YJ, Keshavarzian A, Rao R, Sartor RB, et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol. 2008;42:349–361. doi: 10.1016/j.alcohol.2008.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33:220–232. doi: 10.1111/j.1530-0277.2008.00842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaquera J, Vaquera A, Girbes T. Effects of chronic administration of either ethanol or pentanol on rat duodenum morphology. Histol Histopathol. 2002;17:199–203. doi: 10.14670/HH-17.199. [DOI] [PubMed] [Google Scholar]
- 73.Maier A, Bode C, Fritz P, Bode JC. Effects of chronic alcohol abuse on duodenal mononuclear cells in man. Dig Dis Sci. 1999;44:691–696. doi: 10.1023/a:1026697305769. [DOI] [PubMed] [Google Scholar]
- 74.Salaspuro M. Bacteriocolonic pathway for ethanol oxidation: characteristics and implications. Ann Med. 1996;28:195–200. doi: 10.3109/07853899609033120. [DOI] [PubMed] [Google Scholar]
- 75.Simanowski UA, Seitz HK, Baier B, Kommerell B, Schmidt-Gayk H, Wright NA. Chronic ethanol consumption selectively stimulates rectal cell proliferation in the rat. Gut. 1986;27:278–282. doi: 10.1136/gut.27.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simanowski UA, Suter P, Russell RM, Heller M, Waldherr R, Ward R, Peters TJ, Smith D, Seitz HK. Enhancement of ethanol induced rectal mucosal hyper regeneration with age in F344 rats. Gut. 1994;35:1102–1106. doi: 10.1136/gut.35.8.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]