

Abstract
Alcoholic fatty liver disease (AFLD), a potentially pathologic condition, can progress to steatohepatitis, fibrosis, and cirrhosis, leading to an increased probability of hepatic failure and death. Alcohol induces fatty liver by increasing the ratio of reduced form of nicotinamide adenine dinucleotide to oxidized form of nicotinamide adenine dinucleotide in hepatocytes; increasing hepatic sterol regulatory element-binding protein (SREBP)-1, plasminogen activator inhibitor (PAI)-1, and early growth response-1 activity; and decreasing hepatic peroxisome proliferator-activated receptor-α activity. Alcohol activates the innate immune system and induces an imbalance of the immune response, which is followed by activated Kupffer cell-derived tumor necrosis factor (TNF)-α overproduction, which is in turn responsible for the changes in the hepatic SREBP-1 and PAI-1 activity. Alcohol abuse promotes the migration of bone marrow-derived cells (BMDCs) to the liver and then reprograms TNF-α expression from BMDCs. Chronic alcohol intake triggers the sympathetic hyperactivity-activated hepatic stellate cell (HSC) feedback loop that in turn activates the HSCs, resulting in HSC-derived TNF-α overproduction. Carvedilol may block this feedback loop by suppressing sympathetic activity, which attenuates the progression of AFLD. Clinical studies evaluating combination therapy of carvedilol with a TNF-α inhibitor to treat patients with AFLD are warranted to prevent the development of alcoholic liver disease.
Keywords: Alcohol, Fatty liver, Tumor necrosis factor-α, Hepatic stellate cell, Bone marrow-derived cell, Alcoholic liver disease
Core tip: Alcohol induces fatty liver by increasing the nicotinamide adenine dinucleotide/nicotinamide adenine dinucleotide ratio; increasing the activity of sterol regulatory element-binding protein (SREBP)-1, plasminogen activator inhibitor (PAI)-1, and early growth response-1; and decreasing peroxisome proliferator-activated receptor-α activity in liver. Alcohol activates the innate immune system and induces an imbalance in the immune response followed by the activation of Kupffer cell-derived tumor necrosis factor (TNF)-α overproduction, which is responsible for the dysregulated SREBP-1 and PAI-1 activity. Bone marrow-derived cells and sympathetic hyperactivity-activated hepatic stellate cells are also responsible for TNF-α overproduction in ethanol-induced hepatosteatosis. Carvedilol may attenuate the progression of ethanol-induced hepatosteatosis by suppressing sympathetic activity.
INTRODUCTION
Alcohol consumption is a major risk factor for chronic disease. Based on 58 studies from 17 Global Burden of Diseases (GBD) regions, alcohol use disorders accounted for 9.6% (7.7%-11.18%) of age-standardized disability-adjusted life years (DALYs) worldwide in 2010[1]. Alcohol-induced liver cirrhosis was responsible for 0.9% of all global deaths and 47.9% of all liver cirrhosis deaths in 2010[2]. In addition, alcohol accelerates the progression of other liver diseases, such as hepatitis C virus infection[3], hepatocellular carcinoma[4], and graft dysfunction in patients with liver transplantation[5]. Alcoholic liver disease (ALD) is a potentially avoidable disease because excess alcohol consumption is required for its development. However, alcohol consumption is not sufficient to elicit ALD because only a minority of heavy drinkers progress from alcoholic fatty liver disease (AFLD) to steatohepatitis, fibrosis, and cirrhosis[6-8].
Lieber et al[9] demonstrated that, as in humans, alcohol alone can induce hepatosteatosis in rats. Alcohol, as a hepatotoxin, causes hepatocellular damage via ethanol metabolism-induced oxidative stress and the inflammatory response in the liver[9,10]. Changes in the fibronectin levels in both plasma and hepatic cells are an early response to liver damage in mice with carbon tetrachloride-induced liver injury[11]. Our recent study also showed that fatty liver is associated with zone 3 (perivenular) fibrinogenesis in AFLD rats that have mildly elevated serum alanine aminotransferase levels, a marker of liver injury[12] (Figure 1). Other studies have illustrated that fatty liver is especially susceptible to endotoxins and that it progresses to steatohepatitis, fibrosis, cirrhosis and even hepatocellular carcinoma, especially when accompanied with other co-morbidity factors[13], such as hepatitis C virus infection[3,14], diabetes[15], and smoking[16]. This review first summarizes the classical concepts on the pathogenesis of AFLD and the role of tumor necrosis factor (TNF)-α, the major pro-inflammatory cytokine in ALD, in the induction of fatty liver, and then focuses on the roles of lipid metabolism-associated transcription factors [sterol regulatory element-binding protein (SREBP)-1 and peroxisome proliferator-activated receptor (PPAR)-α], plasminogen activator inhibitor (PAI)-1, and early growth response (Egr)-1 in the pathogenesis of AFLD. This report also describes the recent studies that have characterized the alcohol-mediated changes in bone marrow-derived cell (BMDC) mobilization and recruitment in the liver, sympathetic nervous system (SNS) activity, and TNF-α overproduction from BMDCs and SNS-activated hepatic stellate cells (HSCs). In addition, our recent research suggests that carvedilol, which blocks the SNS via β1, β2, and α1 adrenergic receptors, can block the sympathetic hyperactivity-activated HSC feedback loop to down-regulate TNF-α overproduction and, thereby, attenuate the progression of AFLD in rats. Further understanding of these underlying mechanisms could generate therapeutic interventions to reduce the progression of ALD from the benign condition (fatty liver) to severe forms of liver injury (steatohepatitis, fibrosis, and cirrhosis).
Figure 1.
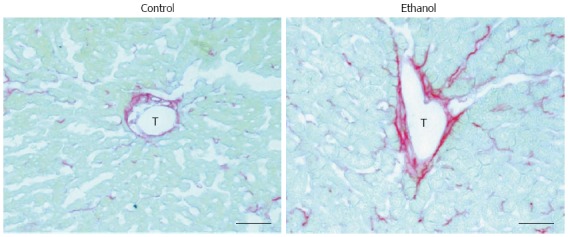
Fatty liver is associated with perivenular fibrinogenesis in rats. Control: 7-wk control liquid diet-fed rat; Ethanol: 7-wk 5 g/dL ethanol liquid diet-fed rat. T: Terminal hepatic venule, scale bars = 50 μm.
SPECTRUM AND RISK FACTORS
Chronic alcohol abuse leads to liver injury, which presents as a broad spectrum of disorders. Fatty liver, also known as AFLD, is the earliest sign of alcohol-induced liver injury. AFLD occurs in 80% of unselected heavy drinkers who consume an excess of 80 g of alcohol a day[17]. Approximately 20%-40% of alcohol abusers will progress to the next stage, alcoholic steatohepatitis, which is characterized by inflammation and hepatocyte death[17]. Thirty to 60% of alcoholic steatohepatitis results in severe complications (liver failure and portal hypertension) with high short-term mortality[17,18]. Approximately 40% of alcoholic steatohepatitis develops to necroinflammation and fibrosis[17]. Approximately 10% of heavy drinkers progress to cirrhosis[17,19,20]. Among alcoholic-related cirrhosis cases, 1%-2% of cases per year develop to hepatocellular carcinoma[21].
The major ALD risk factors include sex, obesity, drinking patterns, co-existing viral infection, and genetic factors[22,23]. Being female is a risk factor for ALD due to lower first-pass metabolism and gastric alcohol dehydrogenase (ADH) activity[24]. Obesity exacerbates the abnormalities in hepatic lipid oxidation[25] and accelerates fibrosis and cirrhosis progression in ALD[26]. Experimental studies indicate that ethanol feeding augments the impairment of hepatic sirtuin1-adenosine monophosphate-activated protein kinase signaling in obese mice[25]. Certain patterns of drinking, such as commencing drinking at an early age and frequent drinking, as well as dietary compositions, increase the risk that severe forms of ALD will develop from fatty liver disease[19,27]. However, cumulative alcohol consumption is the most strongly correlated factor with the progression of AFLD[28]. Co-existing viral infection amplifies alcohol-related hepatotoxicity and then enhances the development of cirrhosis due to the correlation between increased iron deposition in hepatocytes and Kupffer cells, resulting in increased oxidative stress, cellular injury, and fibrogenesis in ALD[29,30].
Genetic factors are also responsible for the susceptibility to and death rate from ALD[31,32]. The gene for patatin-like phospholipase domain-containing protein 3 (PNPLA3), a genetic risk factor for increased fat accumulation in patients with non-alcoholic fatty liver disease (NAFLD)[33], has also been analyzed in patients with AFLD[34,35]. The data for AFLD indicate that PNPLA3 carriers are at a high risk for developing alcoholic liver injury[36].
Taken together, the previous studies support the possibility that AFLD is a complex disease where subtle interpatient genetic variations and the environment interact to produce the disease phenotype and determine disease progression[36]. This may partly explain why some heavy drinkers do not progress to alcoholic steatohepatitis, while some mild alcoholics develop steatohepatitis.
FATTY LIVER AND ALCOHOL METABOLISM
Fatty liver is characterized by the accumulation of fat (mainly triglycerides, phospholipids, and cholesterol esters) in zone 3 (perivenular) hepatocytes; in steatosis, the fat is diffused into zone 2 and zone 1 (periportal) hepatocytes during the development of AFLD (Figure 2).
Figure 2.
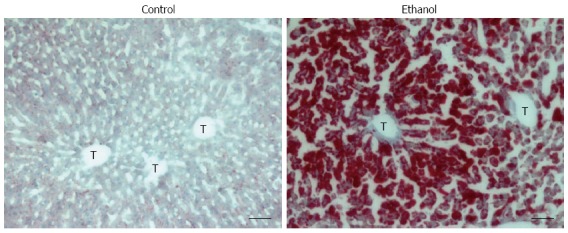
Alcohol-induced hepatosteatosis in rats. Oil red O staining of liver sections; Control: 7-wk control liquid diet-fed rat; Ethanol: 7-wk 5 g/dL ethanol liquid diet-fed. T: Terminal hepatic venule. Scale bars = 50 μm.
Alcohol is absorbed from the jejunum (the major site), and small amounts of fat are also absorbed from the mouth, esophageal, gastric, and large intestine mucosal membranes. Approximately 2% (at low blood-alcohol concentration) and 10% (at high blood-alcohol concentration) of alcohol is excreted directly through the lungs, urine, or sweat. Approximately 90% of ingested alcohol is metabolized in the liver[37]. Alcohol is primarily oxidized to acetaldehyde by ADH in the cytosol of hepatocytes, and it is partly metabolized by cytochrome P-450 and catalase in the hepatocyte microsomes and hepatocyte peroxisomes, respectively. Acetaldehyde dehydrogenase converts acetaldehyde to acetate primarily in the hepatocyte mitochondria (Figure 3).
Figure 3.
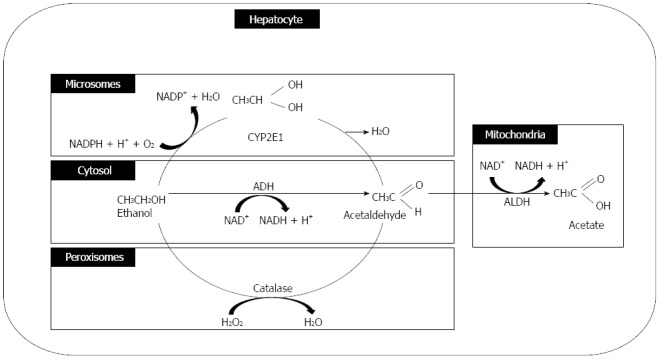
Ethanol metabolism. ADH: Alcohol dehydrogenase; ALDH: Aldehyde dehydrogenase.
Acetaldehyde is the key toxin in alcohol-induced liver injury, causing cellular damage, inflammation, extracellular matrix remodeling, and fibrogenesis[38]. Acetaldehyde increases the ratio of the reduced form of nicotinamide adenine dinucleotide (NADH) to the oxidized form of nicotinamide adenine dinucleotide (NAD+) in the hepatocytes to decrease the β-oxidation of fatty acids by the mitochondria, resulting in fatty liver[39-41]. Acetaldehyde also forms an adduct with tubulin that induces microtubule dysfunction, resulting in decreased lipoprotein transportation from the liver[39].
Acetate, which is largely present in other tissues, can be incorporated into acetyl-CoA, a mitochondrial fuel, for use in Krebs cycle oxidation and fatty acid synthesis[42]. This conversion is catalyzed by the acyl-CoA synthetase short-chain family member and contributes to lipid synthesis and energy generation[43,44]. Acetate can affect histone modification to up-regulate acetyl-CoA and enhance the inflammatory response in ethanol-exposed macrophages by reducing histone deacetylase activity[45]. Moreover, a recent study demonstrated that the formation of acetate from alcohol is key to the process of alcohol-induced inflammatory gene expression by promoter histone acetylation in acute alcoholic steatohepatitis[46]. However, the effect of acetate on the development of fatty liver via histone modification is not well understood.
Under chronic and heavy alcohol intake conditions, alcohol oxidation also occurs via cytochrome P450s, resulting in increased levels of cytochrome P450 2E1, which in turn causes oxidative stress through the generation of reactive oxygen species (ROS)[47,48]. ROS are responsible for lipid peroxidation and alcoholic liver injury[49]. The non-oxidative metabolism of alcohol, mediated by catalase, is responsible for AFLD via the production of fatty acid ethyl ester[50]. Increased blood alcohol concentrations increase the levels of fatty acid ethyl esterase, which can be used as a marker for chronic alcohol consumption[51,52].
Changes in the degree of fatty liver do not parallel the changes due to the chronic consumption of alcohol in animal models[53], and antioxidant treatment was not as successful as expected for treating ALD[54]. Other aspects of ethanol-induced liver injury besides the alcohol-metabolism-related mechanism underlying ALD should be examined.
FATTY LIVER AND TNF-α OVERPRODUCTION
The production of TNF-α is one of the earliest liver responses to injury[55]. TNF-α, a mediator of the mammalian inflammatory response, transduces differential signals to regulate cellular activation and proliferation, cytotoxicity, and apoptosis[56].
TNF-α overproduction due to the ethanol-induced imbalance of immune responses
ALD is associated with the imbalanced immune responses that result in the increased production of pro-inflammatory cytokines[57,58]. Cytokines, the low-molecular-weight polypeptide mediators of cellular communication, are produced and released by different cell types in the liver[58]. TNF-α, the major pro-inflammatory cytokine in ALD, is involved in inflammatory responses, steatosis and cell death[58,59]. Alcohol abuse increases gut permeability and the translocation of bacteria-derived lipopolysaccharide (LPS) from the gut to the liver. In the liver, LPS activates Kupffer cells through the LPS/Toll-like receptor-4 pathway, leading to TNF-α production after nuclear factor-κB activation[59]. Normally, only an occasional particle of LPS, derived from Gram-negative bacteria in the intestinal microflora, penetrates the mucosa and enters the portal circulation, resulting in clearance of LPS without significant inflammatory cell activation in the liver. However, alcoholics have increased circulating endotoxin levels, and patients with ALD have a high frequency of endotoxemia[60,61]. Studies using macromolecular markers have demonstrated a correlation between intestinal permeability and alcoholic liver damage[61,62]. LPS-binding protein and CD14[63,64] are LPS receptors that trigger different downstream signaling pathways and induce nuclear factor-κB activation, resulting in TNF-α production and liver injury. TNF-α has been shown to increase hepatic fatty acid synthesis by increasing hepatic acetyl CoA carboxylase (ACC) and fatty acid synthase (FAS) activities[65], decreasing lipoprotein lipase activity[66], and inhibiting fatty acid oxidation in hepatocytes[67]. Studies of transgenic mice lacking the TNF receptor[57] and studies that involve treating mice and rats with antibodies against TNF-α during chronic ethanol exposure[55] have shown that TNF-α overproduction plays an important role in the progression of ALD.
TNF-α overproduction due to ethanol-induced sympathetic hyperactivity
Our recent studies have indicated that alcohol-induced lipogenesis triggers sympathetic hyperactivity, activating HSCs in AFLD rats and leading to TNF-α overproduction[12] (Figure 4). Using the same rat model of AFLD, carvedilol, which can block the SNS via β1, β2, and α1 adrenergic receptors, blocked the SNS-activated HSC feedback loop and attenuated the development of fatty liver in rats[12] (Figure 5). The high fatty acids levels in the peripheral circulation enhance reflex vasoconstrictor responses[68] and activate the SNS indirectly through pathways originating in the liver[69]. Chronic alcohol administration is associated with observable sympathetic hyperactivity, as evidenced by a high level of 3-methoxy-4-hydroxyphenylglycol (noradrenalin metabolite) in the peripheral circulation[70] and a high level of tyrosine hydroxylase (the rate-limiting enzyme in the synthesis of catecholamine) in the liver[12]. Spontaneously hypertensive rats, which possess high sympathetic tone[71], develop severe liver injury when given hepatoxins[72]. Epinephrine pre-exposure enhances LPS treatment-induced liver damage[73]. Chronic alcohol exposure up-regulates the Kupffer cell α2A-adrenoreceptor to release TNF-α, resulting in liver injury[74].
Figure 4.
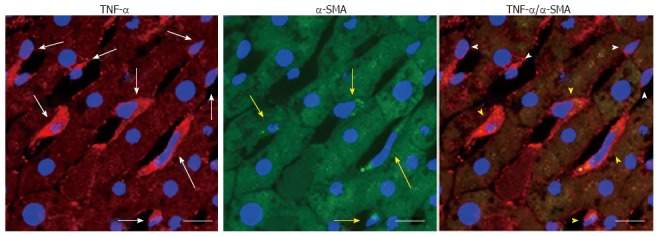
Ethanol-activated hepatic stellate cells, which can product tumor necrosis factor-α in rats fed with a 7-wk 5 g/dL ethanol liquid diet. The white arrows indicate tumor necrosis factor (TNF)-α positive cells; yellow arrows indicate cells positive for α-smooth muscle actin (α-SMA), a marker of activated hepatic stellate cells; white arrowheads indicate TNF-α positive cells that did not overlap with α-SMA positive cells; yellow arrowheads indicate cells that are both TNF-α- and α-SMA-positive; scale bar = 10 μm.
Figure 5.
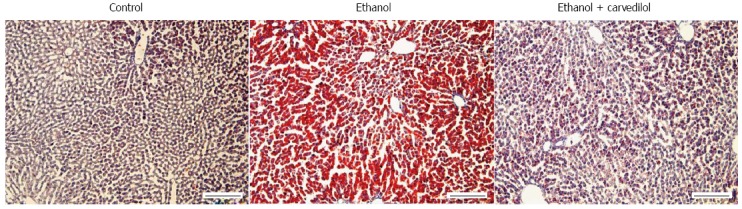
Carvedilol attenuates the development of ethanol-induced hepatosteatosis. Oil red O staining of liver sections; Control: 7-wk control liquid diet-fed rat; Ethanol: 7-wk 5 g/dL ethanol liquid diet-fed rat; Ethanol + carvedilol: 7-wk 5 g/dL ethanol liquid diet-fed rat with one-week carvedilol pretreatment (10 mg/kg body weight/d) before the end of the study. Scale bars = 200 μm.
TNF-α overproduction from the BMDCs in the liver
BMDCs are known to play important roles in parenchymal regeneration and liver injury[75-78]. Our recent study showed that BMDCs in the liver increase in a time-dependent manner after ethanol treatment in a mouse model of AFLD[79] (Figure 6). Furthermore, the BMDCs produce TNF-α in the same AFLD mouse model[79] (Figure 7), indicating that alcohol abuse may promote the migration of BMDCs to the liver and then reprogram TNF-α expression from the BMDCs to promote the development of AFLD in mice. Our results have given new insight into the mechanism of ALD; however, further in vitro studies with cultured cells are essential to understanding the changes in bone marrow cells and endogenous cells during ethanol exposure as well as to understand the recruitment of BMDCs from the bone marrow during the ALD progression.
Figure 6.
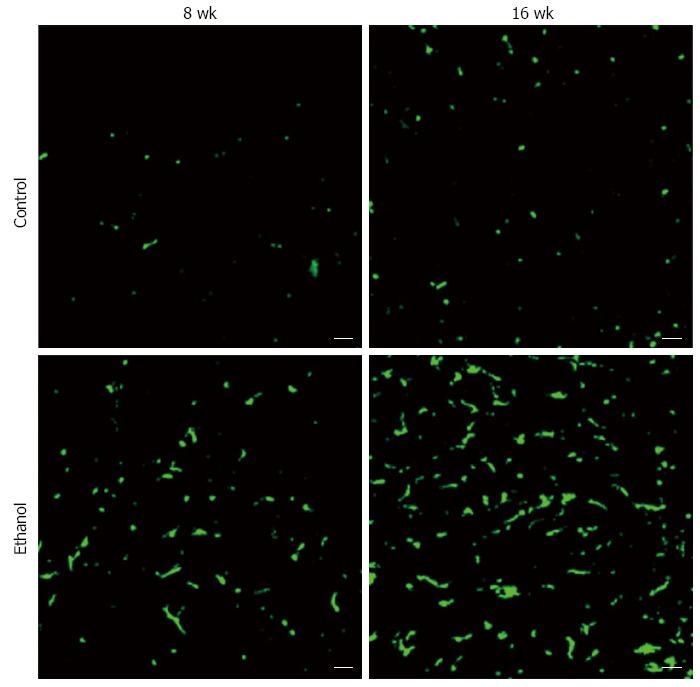
Bone marrow-derived cells increase in a time-dependent manner in the alcoholic fatty liver disease mouse liver. Control: 4 wk after the bone marrow transplantation [from male transgenic mice expressing green fluorescence protein (GFP) to female wild-type mice]. The mice were fed water and standard mouse pellet chow for 8 or 16 wk; Ethanol: 4 wk after the bone marrow transplantation (from male transgenic mice expressing GFP to female wild-type mice), the mice were fed 10 g/dL ethanol and standard mouse pellet chow for 8 or 16 wk; scale bar = 20 μm.
Figure 7.
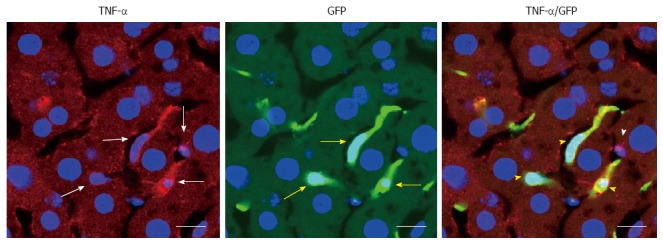
Tumor necrosis factor-α is produced by the bone marrow derived-cells in the alcoholic fatty liver disease mice. The white arrows indicate tumor necrosis factor (TNF)-α-positive cells; yellow arrows indicate cells positive for green fluorescence protein (GFP), a marker of bone marrow-derived cells; white arrowheads indicate TNF-α positive cells that did not overlap GFP positive cells; yellow arrowheads indicate cells that are both TNF-α- and GFP-positive cells; scale bar = 10 μm.
TNF-α overproduction and lipid metabolism-associated regulators
Administering TNF-α to mice increased the rates of fatty acid synthesis and the activation of SREBP-1, resulting in fatty liver development[80-82]. TNF-α up-regulates the expression of SREBP-1 mRNA in the livers of rats[12] and stimulates the maturation of the SREBP-1 protein in human hepatocytes[80]. An endotoxin-induced systemic inflammatory state can reduce PPAR-α expression[83]. The effects of ethanol on SREBP-1 and PPAR-α are mediated by increased portal endotoxin and hepatic TNF-α overproduction. Moreover, recent studies suggest that Egr-1[84,85] and PAI-1[86-88] are also responsible for AFLD via TNF-α overproduction.
Collectively, the previous studies have shown that TNF-α overproduction is closely coupled with alcoholic liver injury. However, the role of TNF-α in AFLD induction remains uncertain. It seems more likely that ethanol-induced TNF-α overproduction regulates lipid metabolism-associated transcription factor gene expression (SREBP and PPAR-α) as well as induces PAI-1 and Egr-1 gene expression, promoting the AFLD progression.
FATTY LIVER AND LIPID METABOLISM-ASSOCIATED TRANSCRIPTION FACTORS
Fatty liver and SREBP
SREBP is a family of transcription factors that regulates the enzymes responsible for the synthesis of cholesterol, fatty acids, and triglycerides in the liver and other tissues. The following are 3 isoforms of SREBP: SREBP-1a, SREBP-1c, and SREBP-2. SREBP-1a is the major form in most cultured cell lines[89], while SREBP-1c is the predominant form in most animal tissues including the liver[90]. SREBP-1 plays an important role in regulating the transcription of genes involved in hepatic triglyceride synthesis (such as fatty acid synthase, stearoyl-CoA desaturase, and ATP citrate lyase)[91]. However, SREBP-2 is responsible for regulating genes related to cholesterol metabolism[92].
Alcohol consumption directly up-regulates SREBP-1c gene expression via its metabolite acetaldehyde[91] and indirectly up-regulates SREBP-1c expression by activating the endoplasmic reticulum response to cell stress[93,94], gut-derived LPS[59], and SREBP downstream proteins, such as Egr-1[84,85] and TNF-α[80]. Acetaldehyde treatment increases the levels of SREBP-1 in HepG2 cells in a dose-dependent manner[94]. Elevation of the hepatic SREBP-1 level is associated with increased gene expression of fatty acid synthase and the accumulation of triglycerides in the mouse liver after liquid ethanol consumption[91]. The connection between SREBP and fatty liver has also been recognized in transgenic mice[95,96]. These results suggest that alcohol and the acetaldehyde produced from alcohol metabolism increase the synthesis of the SREBP-1 protein and enhance hepatic lipogenesis, resulting in ALD progression.
Alcohol can also modify SREBP expression via down-regulation of AMP-activated protein kinase (AMPK), promoting the AFLD progression[97,98]. AMPK, a lipid regulator, regulates the hepatic triglyceride and cholesterol synthesis pathways by phosphorylating and inhibiting enzymes related to lipid metabolism, such as 3-hydroxy-3-methyl glutamate-CoA reductase and ACC[99]. ACC, a rate-limiting enzyme in hepatic fatty acid biosynthesis, catalyzes the conversion of acetyl-CoA to malonyl-CoA. Malonyl-CoA is known as a precursor for the synthesis of fatty acids and an inhibitor of carnitine palmitoyltransferase-1 (CPT-1). AMPK can also activate malonyl-CoA decarboxylase (MCD) to reduce the malonyl-CoA levels, increasing fatty acid oxidation[99]. Fatty acids are transported from the cytoplasm into the mitochondria via CPT-1 and are metabolized through the mitochondrial β-oxidation pathway. Thus, the AMPK-related inhibition of ACC and activation of MCD can lead to decreased synthesis and increased degradation of malonyl-CoA and can then reduce the inhibition of mitochondrial CPT-1, resulting in increased fatty acid transportation into the mitochondria for oxidization.
Alcohol can reduce the blood level of adiponectin, which is a hormone produced from the adipose tissue that activates PPAR-α and AMPK as well as inhibits SREBP-1[100]. AMPK activation can decrease the stability of mature SREBP-1 protein in hepatocytes by accelerating its proteasomal degradation[101]. The alcohol-mediated reduction in AMPK activity is responsible for the decreased activity of MCD and the increased ACC activity by change the phosphorylation state of these enzymes, and then reduces the fatty acid oxidation via the increased malonyl-CoA levels and the decreased CPT-1 activity, all of which contribute to the induction of AFLD[98,102,103]. These results suggest that AMPK may become a therapeutic target for AFLD.
Fatty liver and PPAR-α
PPAR-α, a member of the nuclear hormone receptor superfamily, can be activated by binding free fatty acids to regulate the transcription of the genes involved in the oxidation, transport, and export of free fatty acids[104]. A PPAR-α agonist can negatively regulate ACC[105]; however, PPAR-α positively controls MCD[106]. PPAR-α-null mice that chronically receive a high-fat diet have severe fatty liver with elevated plasma free fatty acid levels even after a 24-h fast[107].
Alcohol consumption can inhibit fatty acid oxidation via suppression of PPAR-α in hepatocytes[108]. Acetaldehyde directly inhibits the gene transcription activity and DNA-binding ability of PPAR-α in hepatocytes[109]. Ethanol can also indirectly inhibit PPAR-α via up-regulation of cytochrome P450 2E1-mediated oxidative stress[110]. Wy14643 and clofibrate, the PPAR-α activator, reverse the ethanol-induced PPAR-α dysfunction and abnormalities in hepatic lipid metabolism in mice[111]. PPAR-α agonist treatment prevents alcohol-induced fatty liver, hepatic inflammation, and hepatic insulin resistance in mice with AFLD[111,112]. These results confirm the critical role of PPAR-α in lipid homeostasis and the progression of AFLD.
FATTY LIVER AND PAI-1
PAI-1 plays an important role in the development of ALD[86,113]. Increased fibrinolysis is common in ALD, and PAI-1 is the major factor influencing fibrinolysis via inhibiting plasminogen activators[86]. PAI-1 is normally expressed in adipocytes and endothelial cells. However, PAI-1 can be induced to high levels in multiple cell types under injury and/or inflammation conditions[113,114]. PAI-1 is a major inhibitor of both the tissue-type plasminogen activator and urokinase-type plasminogen activator, regulating fibrinolysis by inhibiting plasminogen activation.
Alcohol up-regulates PAI-1[115,116], and its level can be used as an index for the severity of the disease[116]. Recently, a human study showed that even patients with simple NAFLD have increased PAI-1 levels, and the increase in hepatic PAI-1 mRNA expression is related to the histological severity of the steatosis and the steatohepatitis[117]. Furthermore, the increased levels of PAI-1 are accompanied with increased grades of inflammation or severity of steatohepatitis, which is expressed by the non-alcoholic steatohepatitis (NASH) activity score[117]. This effect is most likely not restricted to NAFLD and NASH because PAI-1, as an acute-phase reactant, is increased in different types of both acute and chronic hepatic inflammation[118]. Acute ethanol treatment rapidly induces hepatic PAI-1 expression, and the development of a fatty liver was prevented under these conditions by genetic (PAI-1-/- mice) or pharmacologic inhibition of hepatic PAI-1 expression in mice[88]. Taken together, these data indicate that PAI-1 plays an important role in AFLD, which is similar to the previous findings in experimental NAFLD[119].
Various mechanisms could be responsible for the link between fatty liver and increased PAI-1 levels. One mechanism by which TNF-α could cause fatty liver is through the induction of PAI-1 expression. The ethanol-induced up-regulation of PAI-1 expression was blunted in TNFR1-/- mice[88], suggesting that TNF-α is a potent inducer of PAI-1 expression, most likely via the mitogen-activated protein kinases pathway[87]. Additionally, preventing the induction of PAI-1 expression blunts AFLD, which is likely mediated by an increase in very low-density lipoprotein synthesis in the genetic absence of this acute-phase protein[86,88]. Another possible mechanism is that increased liver fat could directly stimulate hepatocytes to secrete PAI-1, as evidenced by the fact that higher circulating levels of PAI-1 are associated with higher hepatic PAI-1 mRNA expression in patients with NAFLD[120].
FATTY LIVER AND EGR-1
Egr-1, known as nerve growth factor, is a zinc-finger transcription factor discovered to have a role in the regulation of cell growth and proliferation[121]. Egr-1 expression is rapidly and transiently induced in response to a variety of stimuli, such as cytokines, growth factors, environmental stress, ischemic injury, and tissue damage[121,122].
Egr-1 is an important contributor to increased LPS-stimulated TNF-α secretion from Kupffer cells after chronic ethanol exposure[123]. Enhanced sensitivity to LPS after chronic ethanol exposure is caused by enhanced expression and DNA-binding activity of the transcription factor Egr-1 in Kupffer and RAW 264.7 cells (a macrophage-like cell line)[124]. Overexpression of a dominant-negative form of Egr-1 in RAW 264.7 macrophages prevents the LPS-induced up-regulation of hepatic TNF-α mRNA expression after chronic ethanol treatment[124]. Collectively, these data suggest that Egr-1 may contribute to the increased sensitivity of macrophages to LPS-stimulated TNF-α production after chronic ethanol exposure.
ROLE OF OTHER REGULATORY FACTORS
The present review has summarized a few key factors responsible for the development of AFLD. However, the following factors may also contribute to the progression of AFLD. Adipocytes, which can secrete leptin and resistin, may be responsible for the development of AFLD by modulating insulin sensitivity and insulin resistance[125]. Mitochondria are a primary source of ROS production, and subsequently become a primary target of ethanol-induced oxidative stress. Mitochondrial functions are suppressed after exposure to toxic compounds (including ethanol)[126]. Alcohol abuse increases oxidative stress[10], which then leads to the modification and degradation of mitochondrial proteins, resulting in inhibition of their functions and/or down-regulation of their protein expression[127]. Alcohol-induced oxidative stress may activate the AMPK signaling system, which controls mitochondrial function[128]. Ethanol-induced ROS overproduction results in the phosphorylation of stress-activated protein kinases, including c-Jun N-terminal protein kinase, and inhibits insulin receptor-1[129]. Activation of the insulin signaling pathway by endogenous substances, such as the fat-derived hormone adiponectin[103], may contribute to decreased fat accumulation in AFLD livers. In addition to activating pro-inflammatory cytokines, the activation of the innate immune system also stimulates Kupffer cells to produce the hepatoprotective cytokine interleukin (IL)-6 and the anti-inflammatory cytokine IL-10 during the development of ALD[130,131]. IL-6-deficient mice are more susceptible to AFLD and liver injury[132,133]. IL-6 treatment ameliorates AFLD and prevents mortality associated with fatty liver transplants in rats[134]. The hepatoprotective function of IL-6 is mediated via signal transducer and activator of transcription (STAT) 3 activation[130,133,135]. Studies on cell-type-specific STAT3-knockout mice suggest that STAT3 in hepatocytes plays an important role in inhibiting fatty acid synthesis while promoting inflammation; on the other hand, STAT3 in macrophages/neutrophils inhibits inflammation during alcoholic liver injury[133,136]. In the liver, IL-10 inhibits the release of pro-inflammatory cytokines, such as TNF-α and IL-6, from macrophages/monocytes to attenuate the progression of fatty liver disease and liver injury[137]; however, IL-10 also inhibits hepatoprotective cytokines, such as IL-6, and promotes fatty liver disease[138]. The overall effect of IL-10 on fatty liver and liver injury depends on the balance of the pro- and anti-inflammatory factors in the system[59,130].
CONCLUSION
AFLD, a frequently observed and potentially pathological condition, plays an important role in the development of advanced liver disease. AFLD is induced through the complex interactions between alcohol doses, alcoholic metabolites, cytokines, transcriptional factors, SNS activity, BMDC mobilization, oxidative stress, and mitochondrial dysfunction. In addition to the major mechanism indicated in this review, ethanol consumption can also promote fat transport from peripheral adipose tissues into the liver and inhibit fat export from the liver[139]. Most studies in this review were conducted in cultured hepatocytes or animal models. The mechanisms summarized in this review could be used to understand the etiologic mechanisms of AFLD in humans. However, caution should always be taken in extrapolating data obtained from in vitro and in vivo animal studies. The mechanisms for AFLD and liver injury in humans are more complicated due to differences in food intake, genetic makeup, race, gender, age, and environmental factors. Lipid metabolism-associated regulators, such as SREBR-1, PPAR-α, PAI-1, and Egr-1, potentiate AFLD, and TNF-α is responsible for the changes in these lipid metabolism-associated regulators. Thus, inhibition of TNF-α overproduction by Kupffer cells, HSCs, and BMDCs has therapeutic potential in the treatment of AFLD. Furthermore, alcohol metabolism triggers the sympathetic hyperactivity-activated HSC feedback loop, leading to TNF-α production. Carvedilol can block this feedback loop and attenuate the development of fatty liver in rats. Clinical studies evaluating combination therapy of carvedilol with a TNF-α inhibitor to treat AFLD patients are warranted.
Other therapy targets, such as CXC chemokines[140]; pentoxifylline, a phosphodiesterase inhibitor[141]; oxidative stress[141]; and TNF-α[141], for alcoholic hepatitis were reviewed by Orman et al[140] and Dhanda et al[141]. Pentoxifylline also reduces the complications in patients with advanced cirrhosis[142]. There have been some advances in our understanding of the pathogenesis and clinical characteristics of alcoholic liver disease. However, standardized nomenclature and histologic classifications are lacking; the animal models do not accurately mimic advanced alcoholic liver disease; and the pathophysiologic significance of the serum levels of biomarkers is unclear (due to impaired liver clearance and ongoing bacterial infections). Additional detailed studies on these potential targets in humans and animal models are urgently needed.
The pathophysiological significance of hepatic lipid accumulation in the absence of significant alcohol consumption, defined as NAFLD, is also increasingly recognized and regarded as the hepatic manifestation of the metabolic syndrome (substantially reviewed by Hellerbrand[143] and Miyake et al[144]). Both AFLD and NAFLD encompass mild fatty liver to steatohepatitis with significant necroinflammation and progressive fibrosis. However, the interaction between alcohol and obesity is poorly understood, and it is unknown whether the combined effects of alcohol and obesity on the progression of liver injury progression are additive or synergistic. It is important to describe the single individual and combined effects of alcohol and the metabolic syndrome on both hepatic steatosis and other organs to understand the differences between AFLD and NAFLD.
TNF-α has also been found to have a crucial role in alcoholic hepatitis, and small preliminary studies have evaluated the effect of anti-TNF therapy in this condition[145]. However, the use of anti-TNF-α drugs in alcoholic hepatitis is still controversial and needs to be investigated further. TNF-α overproduction also occurs in juvenile idiopathic arthritis[146] and ulcerative colitis[147], and a TNF-α inhibitor has been used to treat both conditions. However, neither prolonged nor tapering treatment seems to influence the risk of relapse[146].
Footnotes
P- Reviewer: Daltro C, Kayadibi H, Lu HY, Qiu LX S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, Charlson FJ, Norman RE, Flaxman AD, Johns N, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382:1575–1586. doi: 10.1016/S0140-6736(13)61611-6. [DOI] [PubMed] [Google Scholar]
- 2.Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol. 2013;59:160–168. doi: 10.1016/j.jhep.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Missiha SB, Ostrowski M, Heathcote EJ. Disease progression in chronic hepatitis C: modifiable and nonmodifiable factors. Gastroenterology. 2008;134:1699–1714. doi: 10.1053/j.gastro.2008.02.069. [DOI] [PubMed] [Google Scholar]
- 4.Seitz HK, Becker P. Alcohol metabolism and cancer risk. Alcohol Res Health. 2007;30:38–41, 44-7. [PMC free article] [PubMed] [Google Scholar]
- 5.Ong J, Younossi ZM. Non-alcoholic fatty liver disease after liver transplantation: a case of nurture and nature. Am J Gastroenterol. 2010;105:621–623. doi: 10.1038/ajg.2009.720. [DOI] [PubMed] [Google Scholar]
- 6.Nelson S, Kolls JK. Alcohol, host defence and society. Nat Rev Immunol. 2002;2:205–209. doi: 10.1038/nri744. [DOI] [PubMed] [Google Scholar]
- 7.Haber PS, Warner R, Seth D, Gorrell MD, McCaughan GW. Pathogenesis and management of alcoholic hepatitis. J Gastroenterol Hepatol. 2003;18:1332–1344. doi: 10.1046/j.1440-1746.2003.03217.x. [DOI] [PubMed] [Google Scholar]
- 8.Chedid A, Mendenhall CL, Gartside P, French SW, Chen T, Rabin L. Prognostic factors in alcoholic liver disease. VA Cooperative Study Group. Am J Gastroenterol. 1991;86:210–216. [PubMed] [Google Scholar]
- 9.Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Hakucho A, Liu J, Liu X, Fujimiya T. Carvedilol improves ethanol-induced liver injury via modifying the interaction between oxidative stress and sympathetic hyperactivity in rats. Hepatol Res. 2014;44:560–570. doi: 10.1111/hepr.12143. [DOI] [PubMed] [Google Scholar]
- 11.Tsujimoto I, Moriya K, Sakai K, Dickneite G, Sakai T. Critical role of factor XIII in the initial stages of carbon tetrachloride-induced adult liver remodeling. Am J Pathol. 2011;179:3011–3019. doi: 10.1016/j.ajpath.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Takase I, Hakucho A, Okamura N, Fujimiya T. Carvedilol attenuates the progression of alcohol fatty liver disease in rats. Alcohol Clin Exp Res. 2012;36:1587–1599. doi: 10.1111/j.1530-0277.2012.01773.x. [DOI] [PubMed] [Google Scholar]
- 13.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otani K, Korenaga M, Beard MR, Li K, Qian T, Showalter LA, Singh AK, Wang T, Weinman SA. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology. 2005;128:96–107. doi: 10.1053/j.gastro.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 15.Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, Beasley P, Patt YZ. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206–1213. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- 16.Kuper H, Tzonou A, Kaklamani E, Hsieh CC, Lagiou P, Adami HO, Trichopoulos D, Stuver SO. Tobacco smoking, alcohol consumption and their interaction in the causation of hepatocellular carcinoma. Int J Cancer. 2000;85:498–502. [PubMed] [Google Scholar]
- 17.Levene AP, Goldin RD. The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology. 2012;61:141–152. doi: 10.1111/j.1365-2559.2011.04145.x. [DOI] [PubMed] [Google Scholar]
- 18.Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:663–678. doi: 10.1053/bega.2002.0333. [DOI] [PubMed] [Google Scholar]
- 19.Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, Saveria Crocè L, Sasso F, Pozzato G, Cristianini G, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut. 1997;41:845–850. doi: 10.1136/gut.41.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 21.Seitz HK, Stickel F. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes Nutr. 2010;5:121–128. doi: 10.1007/s12263-009-0154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 23.Wilfred de Alwis NM, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin Liver Dis. 2007;27:44–54. doi: 10.1055/s-2006-960170. [DOI] [PubMed] [Google Scholar]
- 24.Baraona E, Abittan CS, Dohmen K, Moretti M, Pozzato G, Chayes ZW, Schaefer C, Lieber CS. Gender differences in pharmacokinetics of alcohol. Alcohol Clin Exp Res. 2001;25:502–507. [PubMed] [Google Scholar]
- 25.Everitt H, Hu M, Ajmo JM, Rogers CQ, Liang X, Zhang R, Yin H, Choi A, Bennett ES, You M. Ethanol administration exacerbates the abnormalities in hepatic lipid oxidation in genetically obese mice. Am J Physiol Gastrointest Liver Physiol. 2013;304:G38–G47. doi: 10.1152/ajpgi.00309.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raynard B, Balian A, Fallik D, Capron F, Bedossa P, Chaput JC, Naveau S. Risk factors of fibrosis in alcohol-induced liver disease. Hepatology. 2002;35:635–638. doi: 10.1053/jhep.2002.31782. [DOI] [PubMed] [Google Scholar]
- 27.Li TK. Quantifying the risk for alcohol-use and alcohol-attributable health disorders: present findings and future research needs. J Gastroenterol Hepatol. 2008;23 Suppl 1:S2–S8. doi: 10.1111/j.1440-1746.2007.05298.x. [DOI] [PubMed] [Google Scholar]
- 28.Jin M, Cai S, Guo J, Zhu Y, Li M, Yu Y, Zhang S, Chen K. Alcohol drinking and all cancer mortality: a meta-analysis. Ann Oncol. 2013;24:807–816. doi: 10.1093/annonc/mds508. [DOI] [PubMed] [Google Scholar]
- 29.Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Iron overload and cofactors with special reference to alcohol, hepatitis C virus infection and steatosis/insulin resistance. World J Gastroenterol. 2007;13:4699–4706. doi: 10.3748/wjg.v13.i35.4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganz T, Nemeth E. Iron imports. IV. Hepcidin and regulation of body iron metabolism. Am J Physiol Gastrointest Liver Physiol. 2006;290:G199–G203. doi: 10.1152/ajpgi.00412.2005. [DOI] [PubMed] [Google Scholar]
- 31.Reed T, Page WF, Viken RJ, Christian JC. Genetic predisposition to organ-specific endpoints of alcoholism. Alcohol Clin Exp Res. 1996;20:1528–1533. doi: 10.1111/j.1530-0277.1996.tb01695.x. [DOI] [PubMed] [Google Scholar]
- 32.Fisher NC, Hanson J, Phillips A, Rao JN, Swarbrick ET. Mortality from liver disease in the West Midlands, 1993-2000: observational study. BMJ. 2002;325:312–313. doi: 10.1136/bmj.325.7359.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 34.Trépo E, Gustot T, Degré D, Lemmers A, Verset L, Demetter P, Ouziel R, Quertinmont E, Vercruysse V, Amininejad L, et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J Hepatol. 2011;55:906–912. doi: 10.1016/j.jhep.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 35.Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, Rietschel M, Schafmayer C, Braun F, Hinrichsen H, et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology. 2011;53:86–95. doi: 10.1002/hep.24017. [DOI] [PubMed] [Google Scholar]
- 36.Stickel F, Hampe J. Genetic determinants of alcoholic liver disease. Gut. 2012;61:150–159. doi: 10.1136/gutjnl-2011-301239. [DOI] [PubMed] [Google Scholar]
- 37.Schuckit MA. Alcohol and alcoholism. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, et al., editors. Harrison’s Principles of internal medicine. New York: McGraw-Hill Medical Publishing Division; 2005. pp. 2562–2566. [Google Scholar]
- 38.Mello T, Ceni E, Surrenti C, Galli A. Alcohol induced hepatic fibrosis: role of acetaldehyde. Mol Aspects Med. 2008;29:17–21. doi: 10.1016/j.mam.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Kane AB, Kumar V. Environmental and nutritional pathology. In: Cotran RS, Kumar V, Collins T, et al., editors. Pathologic basis of disease. Philadephia: W.B. Saunders Company; 1999. pp. 40–457. [Google Scholar]
- 40.Israel Y, Kalant H, Khanna JM, Orrego H, Phillips MJ, Stewart DJ. Ethanol metabolism, oxygen availability and alcohol induced liver damage. Adv Exp Med Biol. 1977;85A:343–358. doi: 10.1007/978-1-4899-5181-6_22. [DOI] [PubMed] [Google Scholar]
- 41.Ugarte G, Iturriaga H, Pereda T. Possible relationship between the rate of ethanol metabolism and the severity of hepatic damage in chronic alcoholics. Am J Dig Dis. 1977;22:406–410. doi: 10.1007/BF01071886. [DOI] [PubMed] [Google Scholar]
- 42.Yamashita H, Kaneyuki T, Tagawa K. Production of acetate in the liver and its utilization in peripheral tissues. Biochim Biophys Acta. 2001;1532:79–87. doi: 10.1016/s1388-1981(01)00117-2. [DOI] [PubMed] [Google Scholar]
- 43.Sakakibara I, Fujino T, Ishii M, Tanaka T, Shimosawa T, Miura S, Zhang W, Tokutake Y, Yamamoto J, Awano M, et al. Fasting-induced hypothermia and reduced energy production in mice lacking acetyl-CoA synthetase 2. Cell Metab. 2009;9:191–202. doi: 10.1016/j.cmet.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 44.Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res. 2007;48:2736–2750. doi: 10.1194/jlr.M700378-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Moghe A, Joshi-Barve S, Ghare S, Gobejishvili L, Kirpich I, McClain CJ, Barve S. Histone modifications and alcohol-induced liver disease: are altered nutrients the missing link? World J Gastroenterol. 2011;17:2465–2472. doi: 10.3748/wjg.v17.i20.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kendrick SF, O’Boyle G, Mann J, Zeybel M, Palmer J, Jones DE, Day CP. Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology. 2010;51:1988–1997. doi: 10.1002/hep.23572. [DOI] [PubMed] [Google Scholar]
- 47.Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev. 1997;77:517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 48.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 49.Seth D, Haber PS, Syn WK, Diehl AM, Day CP. Pathogenesis of alcohol-induced liver disease: classical concepts and recent advances. J Gastroenterol Hepatol. 2011;26:1089–1105. doi: 10.1111/j.1440-1746.2011.06756.x. [DOI] [PubMed] [Google Scholar]
- 50.Soderberg BL, Salem RO, Best CA, Cluette-Brown JE, Laposata M. Fatty acid ethyl esters. Ethanol metabolites that reflect ethanol intake. Am J Clin Pathol. 2003;119 Suppl:S94–S99. doi: 10.1309/6F39-EAR2-L4GY-X5G6. [DOI] [PubMed] [Google Scholar]
- 51.Kaphalia BS, Cai P, Khan MF, Okorodudu AO, Ansari GA. Fatty acid ethyl esters: markers of alcohol abuse and alcoholism. Alcohol. 2004;34:151–158. doi: 10.1016/j.alcohol.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 52.Nagy LE. Molecular aspects of alcohol metabolism: transcription factors involved in early ethanol-induced liver injury. Annu Rev Nutr. 2004;24:55–78. doi: 10.1146/annurev.nutr.24.012003.132258. [DOI] [PubMed] [Google Scholar]
- 53.Salaspuro MP, Shaw S, Jayatilleke E, Ross WA, Lieber CS. Attenuation of the ethanol-induced hepatic redox change after chronic alcohol consumption in baboons: metabolic consequences in vivo and in vitro. Hepatology. 1981;1:33–38. doi: 10.1002/hep.1840010106. [DOI] [PubMed] [Google Scholar]
- 54.Day CP. Treatment of alcoholic liver disease. Liver Transpl. 2007;13:S69–S75. doi: 10.1002/lt.21336. [DOI] [PubMed] [Google Scholar]
- 55.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 56.Beutler B. TNF, immunity and inflammatory disease: lessons of the past decade. J Investig Med. 1995;43:227–235. [PubMed] [Google Scholar]
- 57.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 58.McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497–G502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- 59.Enomoto N, Ikejima K, Bradford BU, Rivera CA, Kono H, Goto M, Yamashina S, Schemmer P, Kitamura T, Oide H, et al. Role of Kupffer cells and gut-derived endotoxins in alcoholic liver injury. J Gastroenterol Hepatol. 2000;15 Suppl:D20–D25. doi: 10.1046/j.1440-1746.2000.02179.x. [DOI] [PubMed] [Google Scholar]
- 60.McClain C, Hill D, Schmidt J, Diehl AM. Cytokines and alcoholic liver disease. Semin Liver Dis. 1993;13:170–182. doi: 10.1055/s-2007-1007347. [DOI] [PubMed] [Google Scholar]
- 61.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 62.Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol. 2002;282:G937–G947. doi: 10.1152/ajpgi.00235.2001. [DOI] [PubMed] [Google Scholar]
- 63.Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gäbele E, Isayama F, Thurman RG. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol. 2002;168:2963–2969. doi: 10.4049/jimmunol.168.6.2963. [DOI] [PubMed] [Google Scholar]
- 64.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 65.Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 66.Feingold KR, Hardardóttir I, Grunfeld C. Beneficial effects of cytokine induced hyperlipidemia. Z Ernahrungswiss. 1998;37 Suppl 1:66–74. [PubMed] [Google Scholar]
- 67.Nachiappan V, Curtiss D, Corkey BE, Kilpatrick L. Cytokines inhibit fatty acid oxidation in isolated rat hepatocytes: synergy among TNF, IL-6, and IL-1. Shock. 1994;1:123–129. doi: 10.1097/00024382-199402000-00007. [DOI] [PubMed] [Google Scholar]
- 68.Stepniakowski KT, Sallee FR, Goodfriend TL, Zhang Z, Egan BM. Fatty acids enhance neurovascular reflex responses by effects on alpha 1-adrenoceptors. Am J Physiol. 1996;270:R1340–R1346. doi: 10.1152/ajpregu.1996.270.6.R1340. [DOI] [PubMed] [Google Scholar]
- 69.Grekin RJ, Dumont CJ, Vollmer AP, Watts SW, Webb RC. Mechanisms in the pressor effects of hepatic portal venous fatty acid infusion. Am J Physiol. 1997;273:R324–R330. doi: 10.1152/ajpregu.1997.273.1.R324. [DOI] [PubMed] [Google Scholar]
- 70.Liu J, Fujimiya T. Abrupt termination of an ethanol regimen provokes ventricular arrhythmia and enhances susceptibility to the arrhythmogenic effects of epinephrine in rats. Alcohol Clin Exp Res. 2010;34 Suppl 1:S45–S53. doi: 10.1111/j.1530-0277.2008.00851.x. [DOI] [PubMed] [Google Scholar]
- 71.Berg T, Walaas SI, Roberg BÅ, Huynh TT, Jensen J. Plasma Norepinephrine in Hypertensive Rats Reflects α(2)-Adrenoceptor Release Control Only When Re-Uptake is Inhibited. Front Neurol. 2012;3:160. doi: 10.3389/fneur.2012.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamasaki K, Nakashima M, Naito S, Akiyama Y, Ohtsuru A, Hamanaka Y, Hsu CT, Ito M, Sekine I. The sympathetic nervous system promotes carbon tetrachloride-induced liver cirrhosis in rats by suppressing apoptosis and enhancing the growth kinetics of regenerating hepatocytes. J Gastroenterol. 2001;36:111–120. doi: 10.1007/s005350170139. [DOI] [PubMed] [Google Scholar]
- 73.von Montfort C, Beier JI, Guo L, Kaiser JP, Arteel GE. Contribution of the sympathetic hormone epinephrine to the sensitizing effect of ethanol on LPS-induced liver damage in mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1227–G1234. doi: 10.1152/ajpgi.00050.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ajakaiye MA, Jacob A, Wu R, Zhou M, Ji Y, Dong W, Wang Z, Qiang X, Chaung WW, Nicastro J, et al. Upregulation of Kupffer cell α2A-Adrenoceptors and downregulation of MKP-1 mediate hepatic injury in chronic alcohol exposure. Biochem Biophys Res Commun. 2011;409:406–411. doi: 10.1016/j.bbrc.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Higashiyama R, Inagaki Y, Hong YY, Kushida M, Nakao S, Niioka M, Watanabe T, Okano H, Matsuzaki Y, Shiota G, et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology. 2007;45:213–222. doi: 10.1002/hep.21477. [DOI] [PubMed] [Google Scholar]
- 76.Kallis YN, Alison MR, Forbes SJ. Bone marrow stem cells and liver disease. Gut. 2007;56:716–724. doi: 10.1136/gut.2006.098442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parekkadan B, van Poll D, Megeed Z, Kobayashi N, Tilles AW, Berthiaume F, Yarmush ML. Immunomodulation of activated hepatic stellate cells by mesenchymal stem cells. Biochem Biophys Res Commun. 2007;363:247–252. doi: 10.1016/j.bbrc.2007.05.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Russo FP, Alison MR, Bigger BW, Amofah E, Florou A, Amin F, Bou-Gharios G, Jeffery R, Iredale JP, Forbes SJ. The bone marrow functionally contributes to liver fibrosis. Gastroenterology. 2006;130:1807–1821. doi: 10.1053/j.gastro.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 79.Fujimiya T, Liu J, Kojima H, Shirafuji S, Kimura H, Fujimiya M. Pathological roles of bone marrow-derived stellate cells in a mouse model of alcohol-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2009;297:G451–G460. doi: 10.1152/ajpgi.00055.2009. [DOI] [PubMed] [Google Scholar]
- 80.Lawler JF, Yin M, Diehl AM, Roberts E, Chatterjee S. Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J Biol Chem. 1998;273:5053–5059. doi: 10.1074/jbc.273.9.5053. [DOI] [PubMed] [Google Scholar]
- 81.Endo M, Masaki T, Seike M, Yoshimatsu H. TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c) Exp Biol Med (Maywood) 2007;232:614–621. [PubMed] [Google Scholar]
- 82.Donohue TM. Alcohol-induced steatosis in liver cells. World J Gastroenterol. 2007;13:4974–4978. doi: 10.3748/wjg.v13.i37.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tai ES, bin Ali A, Zhang Q, Loh LM, Tan CE, Retnam L, El Oakley RM, Lim SK. Hepatic expression of PPARalpha, a molecular target of fibrates, is regulated during inflammation in a gender-specific manner. FEBS Lett. 2003;546:237–240. doi: 10.1016/s0014-5793(03)00578-7. [DOI] [PubMed] [Google Scholar]
- 84.Pritchard MT, Nagy LE. Ethanol-induced liver injury: potential roles for egr-1. Alcohol Clin Exp Res. 2005;29:146S–150S. doi: 10.1097/01.alc.0000189286.81943.51. [DOI] [PubMed] [Google Scholar]
- 85.Thomes PG, Osna NA, Davis JS, Donohue TM. Cellular steatosis in ethanol oxidizing-HepG2 cells is partially controlled by the transcription factor, early growth response-1. Int J Biochem Cell Biol. 2013;45:454–463. doi: 10.1016/j.biocel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beier JI, Arteel GE. Alcoholic liver disease and the potential role of plasminogen activator inhibitor-1 and fibrin metabolism. Exp Biol Med (Maywood) 2012;237:1–9. doi: 10.1258/ebm.2011.011255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fearns C, Loskutoff DJ. Induction of plasminogen activator inhibitor 1 gene expression in murine liver by lipopolysaccharide. Cellular localization and role of endogenous tumor necrosis factor-alpha. Am J Pathol. 1997;150:579–590. [PMC free article] [PubMed] [Google Scholar]
- 88.Bergheim I, Guo L, Davis MA, Lambert JC, Beier JI, Duveau I, Luyendyk JP, Roth RA, Arteel GE. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterology. 2006;130:2099–2112. doi: 10.1053/j.gastro.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest. 1997;99:838–845. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sheng Z, Otani H, Brown MS, Goldstein JL. Independent regulation of sterol regulatory element-binding proteins 1 and 2 in hamster liver. Proc Natl Acad Sci USA. 1995;92:935–938. doi: 10.1073/pnas.92.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 92.Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101:2331–2339. doi: 10.1172/JCI2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010;16:1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lluis JM, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124:708–724. doi: 10.1053/gast.2003.50089. [DOI] [PubMed] [Google Scholar]
- 95.Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 97.García-Villafranca J, Guillén A, Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie. 2008;90:460–466. doi: 10.1016/j.biochi.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 98.You M, Crabb DW. Recent advances in alcoholic liver disease II. Minireview: molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1–G6. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]
- 99.Viollet B, Guigas B, Leclerc J, Hébrard S, Lantier L, Mounier R, Andreelli F, Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ruderman NB, Park H, Kaushik VK, Dean D, Constant S, Prentki M, Saha AK. AMPK as a metabolic switch in rat muscle, liver and adipose tissue after exercise. Acta Physiol Scand. 2003;178:435–442. doi: 10.1046/j.1365-201X.2003.01164.x. [DOI] [PubMed] [Google Scholar]
- 101.Shklyaev S, Aslanidi G, Tennant M, Prima V, Kohlbrenner E, Kroutov V, Campbell-Thompson M, Crawford J, Shek EW, Scarpace PJ, et al. Sustained peripheral expression of transgene adiponectin offsets the development of diet-induced obesity in rats. Proc Natl Acad Sci USA. 2003;100:14217–14222. doi: 10.1073/pnas.2333912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Supakul R, Liangpunsakul S. Alcoholic-induced hepatic steatosis--role of ceramide and protein phosphatase 2A. Transl Res. 2011;158:77–81. doi: 10.1016/j.trsl.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med. 2003;3:561–572. doi: 10.2174/1566524033479537. [DOI] [PubMed] [Google Scholar]
- 105.Munday MR, Hemingway CJ. The regulation of acetyl-CoA carboxylase--a potential target for the action of hypolipidemic agents. Adv Enzyme Regul. 1999;39:205–234. doi: 10.1016/s0065-2571(98)00016-8. [DOI] [PubMed] [Google Scholar]
- 106.Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD, Severson DL, Kelly DP, Lopaschuk GD. A role for peroxisome proliferator-activated receptor alpha (PPARalpha ) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098–4103. doi: 10.1074/jbc.M106054200. [DOI] [PubMed] [Google Scholar]
- 107.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53:1023–1034. doi: 10.1002/hep.24148. [DOI] [PubMed] [Google Scholar]
- 109.Galli A, Pinaire J, Fischer M, Dorris R, Crabb DW. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J Biol Chem. 2001;276:68–75. doi: 10.1074/jbc.M008791200. [DOI] [PubMed] [Google Scholar]
- 110.Lu Y, Zhuge J, Wang X, Bai J, Cederbaum AI. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology. 2008;47:1483–1494. doi: 10.1002/hep.22222. [DOI] [PubMed] [Google Scholar]
- 111.Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–28004. doi: 10.1074/jbc.M302140200. [DOI] [PubMed] [Google Scholar]
- 112.Lebrun V, Molendi-Coste O, Lanthier N, Sempoux C, Cani PD, van Rooijen N, Stärkel P, Horsmans Y, Leclercq IA. Impact of PPAR-α induction on glucose homoeostasis in alcohol-fed mice. Clin Sci (Lond) 2013;125:501–511. doi: 10.1042/CS20130064. [DOI] [PubMed] [Google Scholar]
- 113.Breitkopf K, Nagy LE, Beier JI, Mueller S, Weng H, Dooley S. Current experimental perspectives on the clinical progression of alcoholic liver disease. Alcohol Clin Exp Res. 2009;33:1647–1655. doi: 10.1111/j.1530-0277.2009.01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kruithof EK. Plasminogen activator inhibitors--a review. Enzyme. 1988;40:113–121. doi: 10.1159/000469153. [DOI] [PubMed] [Google Scholar]
- 115.Dimmitt SB, Rakic V, Puddey IB, Baker R, Oostryck R, Adams MJ, Chesterman CN, Burke V, Beilin LJ. The effects of alcohol on coagulation and fibrinolytic factors: a controlled trial. Blood Coagul Fibrinolysis. 1998;9:39–45. doi: 10.1097/00001721-199801000-00005. [DOI] [PubMed] [Google Scholar]
- 116.Tran-Thang C, Fasel-Felley J, Pralong G, Hofstetter JR, Bachmann F, Kruithof EK. Plasminogen activators and plasminogen activator inhibitors in liver deficiencies caused by chronic alcoholism or infectious hepatitis. Thromb Haemost. 1989;62:651–653. [PubMed] [Google Scholar]
- 117.Verrijken A, Francque S, Mertens I, Prawitt J, Caron S, Hubens G, Van Marck E, Staels B, Michielsen P, Van Gaal L. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2014;59:121–129. doi: 10.1002/hep.26510. [DOI] [PubMed] [Google Scholar]
- 118.Dimova EY, Kietzmann T. Metabolic, hormonal and environmental regulation of plasminogen activator inhibitor-1 (PAI-1) expression: lessons from the liver. Thromb Haemost. 2008;100:992–1006. [PubMed] [Google Scholar]
- 119.Ma LJ, Mao SL, Taylor KL, Kanjanabuch T, Guan Y, Zhang Y, Brown NJ, Swift LL, McGuinness OP, Wasserman DH, et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes. 2004;53:336–346. doi: 10.2337/diabetes.53.2.336. [DOI] [PubMed] [Google Scholar]
- 120.Sookoian S, Castaño GO, Burgueño AL, Rosselli MS, Gianotti TF, Mallardi P, Martino JS, Pirola CJ. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis. 2010;209:585–591. doi: 10.1016/j.atherosclerosis.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 121.Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 122.Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ, Stern DM. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- 123.Kishore R, Hill JR, McMullen MR, Frenkel J, Nagy LE. ERK1/2 and Egr-1 contribute to increased TNF-alpha production in rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol. 2002;282:G6–15. doi: 10.1152/ajpgi.00328.2001. [DOI] [PubMed] [Google Scholar]
- 124.Shi L, Kishore R, McMullen MR, Nagy LE. Chronic ethanol increases lipopolysaccharide-stimulated Egr-1 expression in RAW 264.7 macrophages: contribution to enhanced tumor necrosis factor alpha production. J Biol Chem. 2002;277:14777–14785. doi: 10.1074/jbc.M108967200. [DOI] [PubMed] [Google Scholar]
- 125.Bertolani C, Marra F. Role of adipocytokines in hepatic fibrosis. Curr Pharm Des. 2010;16:1929–1940. doi: 10.2174/138161210791208857. [DOI] [PubMed] [Google Scholar]
- 126.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 127.Suh SK, Hood BL, Kim BJ, Conrads TP, Veenstra TD, Song BJ. Identification of oxidized mitochondrial proteins in alcohol-exposed human hepatoma cells and mouse liver. Proteomics. 2004;4:3401–3412. doi: 10.1002/pmic.200400971. [DOI] [PubMed] [Google Scholar]
- 128.Sid B, Verrax J, Calderon PB. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem Pharmacol. 2013;86:200–209. doi: 10.1016/j.bcp.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 129.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 130.Gao B. Hepatoprotective and anti-inflammatory cytokines in alcoholic liver disease. J Gastroenterol Hepatol. 2012;27 Suppl 2:89–93. doi: 10.1111/j.1440-1746.2011.07003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.El-Assal O, Hong F, Kim WH, Radaeva S, Gao B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol. 2004;1:205–211. [PubMed] [Google Scholar]
- 132.Hong F, Radaeva S, Pan HN, Tian Z, Veech R, Gao B. Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology. 2004;40:933–941. doi: 10.1002/hep.20400. [DOI] [PubMed] [Google Scholar]
- 133.Miller AM, Wang H, Park O, Horiguchi N, Lafdil F, Mukhopadhyay P, Moh A, Fu XY, Kunos G, Pacher P, et al. Anti-inflammatory and anti-apoptotic roles of endothelial cell STAT3 in alcoholic liver injury. Alcohol Clin Exp Res. 2010;34:719–725. doi: 10.1111/j.1530-0277.2009.01141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun Z, Klein AS, Radaeva S, Hong F, El-Assal O, Pan HN, Jaruga B, Batkai S, Hoshino S, Tian Z, et al. In vitro interleukin-6 treatment prevents mortality associated with fatty liver transplants in rats. Gastroenterology. 2003;125:202–215. doi: 10.1016/s0016-5085(03)00696-6. [DOI] [PubMed] [Google Scholar]
- 135.Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, Osei-Hyiaman D, Moh A, Fu XY, Pacher P, et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology. 2008;134:1148–1158. doi: 10.1053/j.gastro.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE. The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology. 2010;51:1420–1429. doi: 10.1002/hep.23427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Miller AM, Wang H, Bertola A, Park O, Horiguchi N, Ki SH, Yin S, Lafdil F, Gao B. Inflammation-associated interleukin-6/signal transducer and activator of transcription 3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in interleukin-10-deficient mice. Hepatology. 2011;54:846–856. doi: 10.1002/hep.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.An L, Wang X, Cederbaum AI. Cytokines in alcoholic liver disease. Arch Toxicol. 2012;86:1337–1348. doi: 10.1007/s00204-012-0814-6. [DOI] [PubMed] [Google Scholar]
- 139.Kang L, Chen X, Sebastian BM, Pratt BT, Bederman IR, Alexander JC, Previs SF, Nagy LE. Chronic ethanol and triglyceride turnover in white adipose tissue in rats: inhibition of the anti-lipolytic action of insulin after chronic ethanol contributes to increased triglyceride degradation. J Biol Chem. 2007;282:28465–28473. doi: 10.1074/jbc.M705503200. [DOI] [PubMed] [Google Scholar]
- 140.Orman ES, Odena G, Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol. 2013;28 Suppl 1:77–84. doi: 10.1111/jgh.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dhanda AD, Lee RW, Collins PL, McCune CA. Molecular targets in the treatment of alcoholic hepatitis. World J Gastroenterol. 2012;18:5504–5513. doi: 10.3748/wjg.v18.i39.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lebrec D, Thabut D, Oberti F, Perarnau JM, Condat B, Barraud H, Saliba F, Carbonell N, Renard P, Ramond MJ, et al. Pentoxifylline does not decrease short-term mortality but does reduce complications in patients with advanced cirrhosis. Gastroenterology. 2010;138:1755–1762. doi: 10.1053/j.gastro.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 143.Hellerbrand C. Pathophysiological similarities and synergisms in alcoholic and non-alcoholic steatohepatitis. Dig Dis. 2010;28:783–791. doi: 10.1159/000324286. [DOI] [PubMed] [Google Scholar]
- 144.Miyake T, Kumagi T, Furukawa S, Tokumoto Y, Hirooka M, Abe M, Hiasa Y, Matsuura B, Onji M. Non-alcoholic fatty liver disease: factors associated with its presence and onset. J Gastroenterol Hepatol. 2013;28 Suppl 4:71–78. doi: 10.1111/jgh.12251. [DOI] [PubMed] [Google Scholar]
- 145.Cassano N, Vestita M, Apruzzi D, Vena GA. Alcohol, psoriasis, liver disease, and anti-psoriasis drugs. Int J Dermatol. 2011;50:1323–1331. doi: 10.1111/j.1365-4632.2011.05100.x. [DOI] [PubMed] [Google Scholar]
- 146.Iglesias E, Torrente-Segarra V, Bou R, Ricart S, González MI, Sánchez J, Calzada J, Antón J. Non-systemic juvenile idiopathic arthritis outcome after reaching clinical remission with anti-TNF-α therapy: a clinical practice observational study of patients who discontinued treatment. Rheumatol Int. 2014;34:1053–1057. doi: 10.1007/s00296-013-2884-z. [DOI] [PubMed] [Google Scholar]
- 147.Puthoor PR, de Zoeten EF. Pediatric Ulcerative Colitis: The Therapeutic Road to Infliximab. Biol Ther. 2013;3:1–14. doi: 10.1007/s13554-012-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]