

Abstract
Pancreatic cancer has a considerably poor prognosis with a 5-year survival probability of less than 5% when all stages are combined. Pancreatic cancer is characterized by its dense stroma, which is involved in the critical interplay with the tumor cells throughout tumor progression and furthermore, creates a barrier restricting efficient penetration of therapeutics. Alterations in a large number of genes are reflected by a limited number of signaling pathways, which are potential targets. Understanding more about the molecular basis of this devastating cancer type regarding tumor microenvironment, distinct subpopulations of cells, epithelial-to-mesenchymal transition and inflammation will lead to the development of various targeted therapies for controlling tumor growth and metastasis. In this complex scenario of pancreatic cancer, especially members of the “small integrin binding ligand N-linked glycoproteins” (SIBLINGs) and “secreted protein acidic and rich in cysteine” (SPARC) families have emerged due to their prominent roles in properties including proliferation, differentiation, apoptosis, adhesion, migration, angiogenesis, wound repair and regulation of extracellular matrix remodeling. SIBLINGs consist of five members, which include osteopontin (OPN), bone sialoprotein, dentin matrix protein 1, dentin sialophosphoprotein and matrix extracellular phosphoglycoprotein. The SPARC family of modular extracellular proteins is comprised of SPARC/osteonectin (ON) and SPARC-like 1 (hevin); secreted modular calcium binding proteins; testicans and follistatin-like protein. In this review, we especially focus on OPN and ON, elaborating on their special and growing importance in pancreatic cancer diagnosis and prognosis.
Keywords: Pancreatic cancer, Microenvironment, Signaling pathways, Osteopontin, Osteonectin, Hevin, Biomarker, Therapeutic targeting
Core tip: In this article we review the evidence that the protein families “small integrin binding ligand N-linked glycoproteins” (SIBLINGs) and “secreted protein acidic and rich in cysteine” (SPARC) modulate functions like proliferation, differentiation, apoptosis, adhesion, migration, angiogenesis, wound repair, and regulation of extracellular matrix remodeling. Moreover they play significant roles throughout each stage of pancreatic cancer formation and progression. We discuss, with special reference to osteopontin and osteonectin, how SIBLING and SPARC proteins have attracted growing importance as diagnostic and prognostic tools and discuss their fascinating potential as therapeutic targets.
INTRODUCTION
Pancreatic cancer, the fourth leading cause of cancer-related mortality, has a considerably poor prognosis, as reflected by a median survival of 5-8 mo and a 5-year survival probability of less than 5% when all stages are combined[1,2]. At the time of diagnosis, metastasis has already occurred in most of the patients. Early diagnosis may enable tumor resection; however relapse is still likely due to recurrence at the primary tumor site and distant metastasis. This is affirmed by the observation that the average survival time after neoadjuvant therapy and surgery in patients whose tumor was resectable before neoadjuvant therapy was similar to that of patients treated with chemotherapy and/or radiotherapy after surgery (23.3 and 20.5 mo, respectively)[2].
Recent advances in understanding the molecular basis of pancreatic cancer development and progression are expected to provide novel therapeutic opportunities. In this regard, the genomic diversity, tumor microenvironment, distinct populations of cancer stem cells (CSCs) resistant to chemo- and radiotherapies, and adaptation of cancer cells to the hypoxic conditions as well as to nutritional deficiency represent potential therapeutic challenges[3].
Pancreatic cancer is characterized by its dense and desmoplastic stroma which is critical for tumor progression and metastasis. Tumor stroma creates a barrier restricting efficient penetration of chemotherapeutics and targeted therapies[4,5]. Furthermore, the stroma and the tumor itself express various proteins, which have proven to be prognostic biomarkers and potential therapeutic targets, as well[6]. These cytokines secreted by pancreatic cancer cells, especially members of the small integrin binding ligand N-linked glycoprotein (SIBLING) and secreted protein acidic and rich in cysteine (SPARC) families are likely to draw a lot of interest for their prominent roles in pancreatic cancer growth.
SIBLINGs are a family of non-collagenous proteins consisting of five members, which include osteopontin (OPN), bone sialoprotein (BSP), dentin matrix protein (DMP1), dentin sialophosphoprotein (DSPP), and matrix extracellular phosphoglycoprotein (MEPE) (Table 1). The SIBLING family of proteins is principally located in bone and dentin and its members take part in extracellular matrix (ECM) formation and mineralization[7]. OPN is highly expressed in primary pancreatic cancer[8-10]. SIBLINGs are involved in tumor progression and metastasis by interacting with several integrins and with CD44 to mediate cellular signaling[11,12].
Table 1.
Small integrin binding ligand N-linked glycoproteins and secreted protein acidic and rich in cysteine gene families and their members[7,13-15]
| Gene family | Member (Aliases) |
| Small integrin binding ligand N-linked glycoproteins (SIBLING) | Osteopontin (OPN)/secreted phosphoprotein 1 (SPP1) |
| Bone sialoprotein II (BSP) | |
| Dentin matrix protein 1 (DMP1) | |
| Dentin sialophosphoprotein (DSPP) | |
| Matrix extracellular phosphoglycoprotein (MEPE) | |
| Secreted protein acidic and rich in cysteine (SPARC) | SPARC [osteonectin (ON)]/basement-membrane protein 40 (BM-40) |
| Hevin (SPARCL1; QR1) | |
| Secreted modular calcium binding proteins 1 and 2 (SMOC1 and SMOC2) | |
| Testican 1, 2 and 3/sparc/osteonectin, cwcv and kazal-like domains proteoglycan 1, 2 and 3 (SPOCK1, 2 and 3) | |
| Follistatin-like protein (FSTL-1)/TGF-beta-simulated clone-36/follistatin-like (TSC-36/Flik)/follistatin-related protein (FRP)/TGF-β inducible protein |
The SPARC family of modular extracellular proteins can phylogenetically be classified into four groups (Table 1), all of which contain the extracellular calcium-binding (EC) and follistatin-like (FS) domains (Table 1): (1) Osteonectin (ON) and SPARC-like 1 (hevin); (2) Secreted modular calcium binding proteins (SMOC) 1 and 2; (3) Testican 1, 2 and 3; and (4) Follistatin-like protein[13-15]. ON is overexpressed in pancreatic cancer stroma[10]. Low or absent stromal expression of ON was correlated with longer survival rates[6]. ON is involved in remodeling of ECM, morphogenesis, wound repair, and cell proliferation. The other SPARC family member hevin is down-regulated in pancreatic cancer, especially in the late stages and was suggested to function as a tumor suppressor and angiogenesis regulator[10,16-19].
In this review, we elaborate on the role of SIBLING and SPARC family members in pancreatic cancer progression and metastasis with specific emphasis on OPN and ON. We start by describing their signaling pathways, then elaborate on the critical interplay between tumor cells and their microenvironment, and outline the currently available targeted therapies in pancreatic cancer. In the following part, we discuss the impact of SIBLING and SPARC family proteins in pancreatic cancer, including their distribution, interference with signaling pathways, pro- and anti-tumorigenic effects, biomarker roles, and fascinating potential as therapeutic targets.
SIGNALING PATHWAYS IN PANCREATIC CANCER
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer. The development and progression of PDAC is mediated by the complex crosstalk between the tumor cells and the stromal components, involving various alterations in signaling pathways[20]. The first comprehensive genetic analysis of 24 pancreatic tumors revealed alterations of a large number of genes (63 per tumor). A more recent detailed analysis of 99 tumors identified 26 mutations per patient (range 1-116) as well as substantial heterogeneity with 2016 non-silent mutations and 1628 copy-number variations in 99 patients, affirming the previous findings and furthermore pointing out the potential involvement of axon guidance genes in pancreatic cancer progression[21]. However, dysregulation was limited to 12 core signaling pathways which therefore seem to be more preferable targets as compared to mutated genes[22]. These core pathways, which were genetically altered in most pancreatic cancers, include genes like KRAS and other monomeric GTPases, genes for apoptosis, DNA damage control, regulation of G1/S phase transition, Hedgehog, c-Jun N-terminal kinase, TGF-β, Wnt/Notch, genes for invasion, homophilic cell adhesion and integrin signaling[22].
PANCREATIC CANCER MICROENVIRONMENT
Critical interplay between the tumor cells and the microenvironment
The abundant stroma of pancreatic cancer, which has been termed desmoplasia, is composed of acellular (ECM, soluble proteins like cytokines and growth factors) and cellular [fibroblasts, myofibroblasts, pancreatic stellate cells (PSCs), vascular and immune cells] components[4]. The abundant infiltrating inflammatory cells are particularly polymorphonuclear neutrophils, macrophages, and lymphocytes. These immune cells are rich sources of various factors promoting tumor growth and EMT associated with enhanced migration capacity and metastasis[23,24].
The desmoplastic response is regulated throughout cancer initiation and progression stages by dynamic paracrine and autocrine signaling interactions between tumor and host stromal cells[25]. PSCs, the key players in desmoplastic reaction, are star shaped cells residing in periacinar, periductal and perivascular regions of the pancreas[26]. During malignant transformation, PSCs are transformed from a quiescent state into an activated (myofibroblast-like) phenotype which expresses α-smooth muscle actin and ECM proteins and acquires the capacity to proliferate, migrate, contract, phagocytose, and promote tissue repair[27]. Pancreatic cancer cells recruit PSCs to their immediate vicinity, while PSCs inhibit apoptosis and stimulate survival of cancer cells[26,28,29]. Human PSCs (hPSCs) have the capability to intravasate/extravasate, thus accompany cancer cells to distant metastatic sites[30]. The premalignant and malignant cells secrete many paracrine factors like transforming growth factor-beta (TGF-β), platelet derived growth factor (PDGF), vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), sonic hedgehog (SHH), epidermal growth factor (EGF), fibroblast growth factors (FGFs) and insulin-like growth factors (IGFs), which activate PSCs, which in turn secrete more ECM proteins, matrix metalloproteases (MMPs), PDGF, FGF, TGF-β, IGF1, small leucine-rich proteoglycans, periostin, collagen I, EGF and heparin sulfate proteoglycans (Figure 1)[25].
Figure 1.
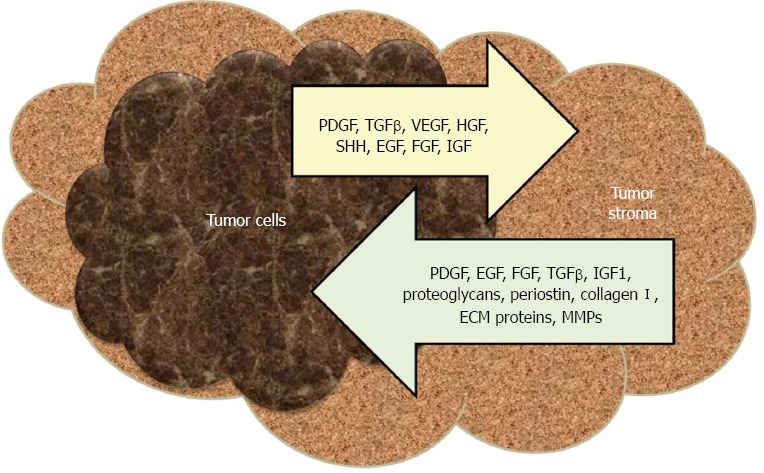
Critical interplay between the pancreatic cancer cells and the microenvironment. TGF-β: Transforming growth factor-beta; VEGF: Vascular endothelial growth factor; HGF: Hepatocyte growth factor; SHH: Sonic hedgehog; EGF: Epidermal growth factor; FGF: Fibroblast growth factor; IGF: Insulin-like growth factor; MMP: Matrix metalloprotease; PDGF: Platelet derived growth factor.
Targeted therapies in pancreatic cancer
Currently approved chemotherapeutic drugs for pancreatic cancer are pyrimidine analogs (fluorouracil, capecitabine, gemcitabine), platinum analogs (oxaliplatine), taxanes (paclitaxel and docetaxel), campthotecin analog irinotecan and mitomycine C[31]. The growing need for novel agents was met by understanding the molecular basis of pancreatic cancer which paved the way to modulate aberrant signaling pathways. EMT, a process whereby epithelial cells acquire mesenchymal characteristics which are associated with increased invasiveness, angiogenesis, resistance to chemotherapy and formation of CSCs, has also emerged as an immensely attractive target[32]. Suppression of tumor promoting inflammation presents another potential target. Inflammation is observed at the early stages of PDAC which progresses via an interplay between KRAS mutations and chemokines/cytokines. Upregulated oncogenic and inflammatory pathways intersect in the transcription factors STAT3 and NF-κB, designating them as excellent therapeutic targets[33]. In the light of these findings, recent research has focused on molecular targets like epidermal growth factor receptor (EGFR), VEGF, IGF-1R, mammalian target of rapamycin (mTOR), mitogen activated protein kinase (MEK), cyclooxygenase 2 (COX-2) or proteasome[34,35]. In addition, targeting c-MET or Alk-4/7 up-regulated in CSCs or pathways mediating EMT (Notch, Wnt, Hedhehog, Src and TGF-β) or transcription factors (Zeb1) emerge as viable strategies[36].
The role of EGFR, which is an overexpressed oncogene in 43%-69% of PDAC, is well established in pancreatic cancer progression[37]. EGFR belongs to the receptor tyrosine kinase (RTK) subfamily ErbB/EGFR and regulates downstream signaling pathways including the PI3K/AKT, RAS/MAPK, PLCγ/PKC and STATs pathways. A nuclear EGFR complex has also been reported in pancreatic cancer cell lines, Panc-1 and Colo-357 cells, but it’s not yet definitively identified as a true oncogene[38]. Several monoclonal antibodies (mAbs) namely cetuximab, matuzumab, panitumumab, and nimotuzumab which can bind to the extracellular domain of membrane-bound EGFR are under investigation. Smaller molecules like erlotinib and gefitinib can inhibit EGFR tyrosine kinase by competetive blockade of ATP binding. Today, erlotinib is the only targeted therapy which is approved as first line therapy in combination with gemcitabine for locally advanced or metastatic pancreatic cancer[35,39]. Human EGF receptor-2 (HER-2) is a commonly expressed oncogene in pancreatic cancer. Anti-HER-2 therapies include mAbs like transtuzumab and pertuzumab, and tyrosine kinase inhibitors (TKIs) like lapatinib[35].
VEGF is another overexpressed oncogene in 93% of PDAC[37]. Since overexpression of VEGF and its receptors are involved in angiogenic and mitogenic promotion of tumor growth, targeting this pathway with bevacizumab has been evaluated for the treatment of advanced pancreatic cancer combined with other chemotherapeutic regimens[39,40]. An alternative strategy for targeting the VEGF pathway has also been tested using anti-VEGF TKIs sorafenib, axitinib and vatalanib[35].
Other molecularly targeted therapies under investigation are farnesyltransferase inhibitors (tipifarnib), TGF-β signaling inhibitors (TGF-β2 inhibitor AP 12009, dual TGF-β type I/II receptor kinase selective inhibitor LY210976, TβR-I inhibitor LY364947 and selective kinase inhibitor SD-093), IGF-1R kinase inhibitors (NVP-AEW541 and BMS-754807), MMP inhibitors (marimastat and tanomastat), hedgehog signaling inhibitors (cyclopamine, saridegib and vismodegib), mTOR inhibitors (everolimus, temsirolimus, sirolimus), MEK1/2-ATP-uncompetitive inhibitors (selumetinib), COX-2 inhibitors (celecoxib), 26S proteasome inhibitors (bortezomib), NF-κB inhibitors (curcumin), integrin α5β1 inhibitors (volociximab), and a claudin-4 inhibitor (clostridium perfringens enterotoxin).
Pancreatic cancer development and progression is regulated by the interaction between various aforementioned pathways; hence targeting multiple pathways seems to be a novel therapeutic approach to interfere with this cross talk[34,35,41-49].
However, two very recent reviews on targeted therapies indicate a poor outcome in phase III trials in spite of numerous promising results from preclinical studies and phase I/II trials[34,35]. This insurmountable intrinsic and acquired resistance to the investigated therapeutics delineates the critical interplay between tumor cells and tumor microenvironment[5], anticipating the need to identify additional targets as well as novel agents and to specifically target the tumor stroma[34,50,51].
IMPACT OF SIBLING AND SPARC FAMILIES
Expression of SIBLING and SPARC family members has been associated with pancreatic cancer progression. These cytokines, secreted by pancreatic tumor stromal cells, interfere with various pathways and their expression is associated with survival rates[52-55].
Distribution
SIBLING: OPN is strongly expressed in tumor-associated macrophages especially at the invasive edge of the tumor[8,56,57], in the cytoplasm of tumor cells[53,57,58] and ECM of pancreatic cancer cell lines[59]. BSP is weakly to moderately detectable in islet and ductal cells of normal pancreatic tissues, and in the tubular complexes of PDAC and pancreatic cancer cell lines[60].
SPARC: ON is expressed at high levels by pancreatic acinar and islet cells of normal human tissues[61,62]. In chronic pancreatic inflammation, ON expression in acinar cells is transiently up-regulated but then lost at the final stages, which may favor acinar-to-ductal metaplasia[63]. The majority of pancreatic cancer cells and cell lines are ON negative[54,55,62,64,65]. Lack of ON expression in these cell lines was related to epigenetic silencing by aberrant methylation[62]. The aberrant ON methylation status was not different between sporadic and familial pancreatic cancers[66]. Low-to-absent ON expression levels in some pancreatic cancer cell lines was also associated with over-expression of runt-related transcription factor-2[67] and fibroblast growth factor receptor1-IIIc (FGFR1-IIIc)[68]. ON was overexpressed in stromal fibrocytes and endothelial cells of benign and malignant tissues, especially adjacent to the neoplastic epithelium but also in the distal stroma[55,61,62,65,69]. Hevin mRNA was expressed specifically within angioendothelium but not in adjacent tumor epithelium and stroma of invasive pancreatic cancer[70].
Interference with signaling pathways in cancer progression
SIBLING and SPARC proteins modulate many functions of healthy tissues, including cell proliferation, differentiation, apoptosis, adhesion, migration, angiogenesis, wound repair, and regulation of ECM remodeling. Mounting evidence suggested their significant functions in various cell-matrix interactions throughout each stage of cancer progression, which include, but are not limited to integrin linked kinase (ILK)/PI3K/Akt, Ras/Raf/MEK/ERK1/2/AP-1 and NF-κB as major signaling pathways[11,13,71].
OPN: OPN is a flexible protein in solution. This capability of OPN allows its binding, via Arg-Gly-Asp (RGD) motif-dependent and independent interactions, to different proteins like cell surface receptors, matrix metalloproteinases and ECM proteins[11]. OPN was shown to promote proliferation, invasion, angiogenesis, and metastasis in different types of malignant tumors[71-76]. OPN interacts mainly with various αv (αvβ1, αvβ3, αvβ5 and αvβ6) integrin receptors via the RGD sequence and with CD44v6 and v7-containing isoforms via the C-terminal fragment with a calcium binding site (Figure 2). Binding of OPN to integrin and CD44 initiates a downstream signaling cascade through the PI3K/Akt signaling pathway leading to NF-κB mediated cell proliferation and survival[71,73]. An OPN/integrin complex, through the Ras/Raf/MEK/ERK pathway, activates AP-1 dependent gene expression, hence plasmin and MMP-9 mediated ECM degradation and tumor invasion[71]. VEGF-induced OPN and integrin expression supports neovascularization processes by promoting endothelial cell migration and vascular lumen formation, activating monocytes to release pro-angiogenic cytokines and preventing endothelial cell apoptosis[73].
Figure 2.
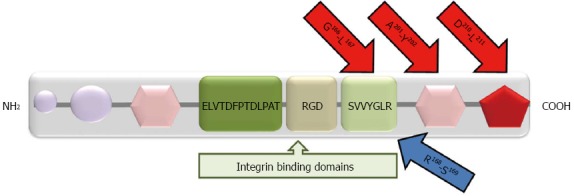
Structural domains of osteopontin. Purple circles: Matrix binding domains; pink hexagons: Calcium binding sites; Red pentagon: Heparin binding site. There are three integrin binding sequences: (1) Arginine-glycine-aspartic acid (RGD); (2) Serine-valine-valine-tyrosine-glutamate-leucine-arginine (SVVYGLR); and (3) ELVTDFPTDLPAT. MMP cleavage sites (G166-L167; A201-Y202; D210-L211) are shown by red arrows. The thrombin cleavage site (R168-S169) is shown by blue arrow.
ON: ON has three structural domains (Figure 3), each of which initiates differential processes in cancer progression. The N-terminal, highly acidic low affinity-calcium binding domain inhibits cell migration and chemotaxis, decreases fibronectin and thrombospondin-1 but increases plasminogen activator inhibitor-1 (PAI-1). The cysteine rich follistatin-like domain promotes de-adhesion, angiogenesis and proliferation and the high affinity-EC-binding domain inhibits migration, proliferation and adhesion, induces MMPs and regulates cell-matrix interactions[77,78]. Tumors overexpressing the N-terminal domain of ON were used as model to show that this domain has chemosensitizing properties. In fact, the N-terminal domain of ON caused a significantly greater reduction in cell viability than ON itself and this effect was related to enhancing the apoptotic cascade via activation of caspase 8[79].
Figure 3.
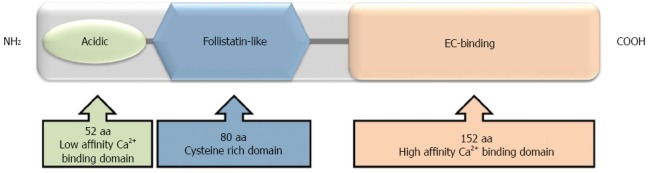
Structural domains of osteonectin. The N-terminal is a highly acidic, calcium binding domain (low affinity). The follistatin-like domain is rich in cysteine residues. The C-terminal is an extracellular calcium-binding domain (high affinity).
ON has a divergent effect in different cancer types. ON may be linked with a highly aggressive phenotype in some tumors, but it may function as a tumor suppressor in others[80]. This modulator effect, either positive or negative, on cell growth and migration was suggested to depend on the amount secreted by the tumor[81]. The microenvironment also determines whether ON will act as a de-adhesive or adhesive protein[82]. ON influences integrin signaling by reducing surface localization of integrin subunits and by directly interacting with ILK and it induces ILK/FAK/Akt activation to promote EMT, anti-apoptosis and cell migration[71,77]. Macrophage-derived ON is also involved in integrin-mediated metastasis[83]. ON can bind directly to collagens I-VIII and growth factors (VEGF-A, PDGF-AA and PDGF-BB) or modulate cell surface receptors of basic fibroblast growth factor and TGF-β[13,77,78,84,85]
Prominent roles in pancreatic cancer progression: experimental evidence
OPN: OPN has a pro-tumorigenic role and favors the metastatic growth of pancreatic cancer (Figure 4). OPN mRNA expression in human PDAC cell lines was significantly related to their growth in the liver of nude rats. Similarly, OPN knockdown was associated with reduced proliferation in rat pancreatic adenocarcinoma (AsML) cells[86]. OPN expression in AsML cells following explantation from the liver decreased gradually in time when grown in vitro for up to five weeks. However, co-culture of AsML cells and of human Suit2-007 PDAC cells with hepatocytes stimulated OPN expression. This two compartmental metastasis model clearly demonstrated the cross talk between PDAC cells and hepatocytes[86]. Comparison of OPN expression in two human pancreatic cell lines showed that OPN was eleven-fold up-regulated in cells of the highly liver metastatic cell line HPC-3H4, as compared to parental HPC-3 cells. In the same study, OPN RNAi and anti-OPN antibody treatment inhibited liver metastasis of pancreatic cancer cell line[87]. OPN was likely to play a role in the initial growth of PDAC cells in the liver while MMP-1 and EGF-1 were required for the maintenance of growth[88].
Figure 4.
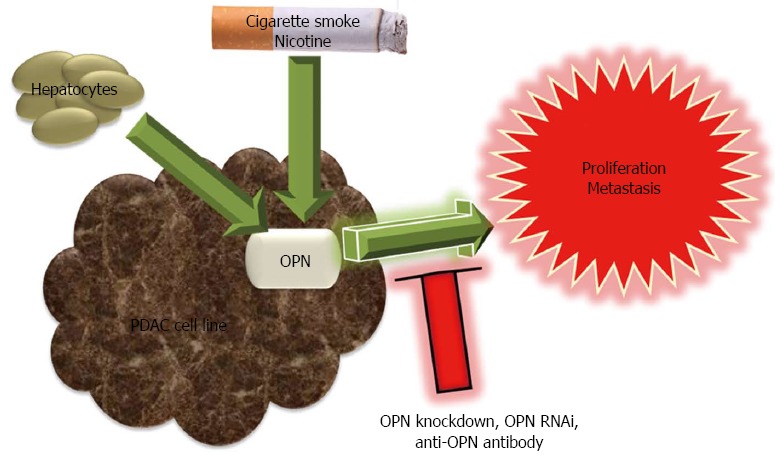
Protumorigenic role of osteopontin in pancreatic cancer development and progression. Osteopontin (OPN) expression in pancreatic cancer cell lines is associated with increased in vitro proliferation and enhanced growth and metastasis in vivo, which are reversed by OPN knockdown, OPN RNAi and anti-OPN antibody. Exposure to cigarette smoke (including nicotine) and hepatocytes induce OPN expression.
OPN was demonstrated to be a downstream mediator of nicotine in pancreatic cancer. Smoking and experimental exposure to cigarette smoke or nicotine stimulated OPN mRNA and protein expression in PDAC[89]. Nicotine exposure selectively induced splice variant OPNc and alpha7-nicotine acetylcholine receptor (α7-nAChR) expression[90]. Nicotine-induced OPN mRNA expression in pancreatic cancer cell lines was inhibited by a nicotinic acetylcholine receptor antagonist, a tyrosine kinase inhibitor or an ERK1/2 activation inhibitor, which implied that OPN was expressed by a nAChR-ERK1/2-dependent pathway[89]. OPN siRNA or antibody treatment inhibited nicotine enhanced expression of MMP-9 and VEGF[91].
ON: ON is a multifaceted protein with controversial functions of its structural domains. ON exhibited differential effects in pancreatic cancer models and displayed antitumorigenic as well as tumorigenic potential (Figure 5). The experimental models used providing differential environmental conditions as well as cancer cell line specific properties seem to contribute to its complex behavior.
Figure 5.
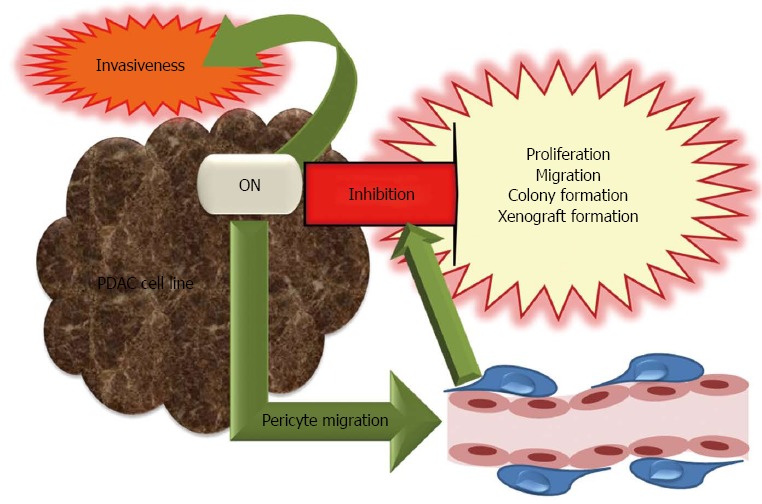
Pro- and anti-tumorigenic roles of osteonectin in pancreatic cancer development and progression. Osteonectin (ON) is a multifaceted protein with controversial functions of its structural domains. ON is anti-tumorigenic and inhibits proliferation, migration, colony and xenograft formation as well as invasiveness. Facilitation of pericyte migration by ON contributes to inhibition of tumor spread. However, ON is also pro-tumorigenic and induces invasiveness.
The antitumorigenic potential of ON in pancreatic cancer was demonstrated by in vivo and in vitro studies. An inverse relation exists between ON expression and growth of pancreatic cell lines in the liver of nude rats[86]. ON expressing human Suit2-007 and rat AsML cells were investigated in vitro to elucidate the antiproliferative role of ON. Knockdown of ON mRNA by an antisense oligonucleotide (ASO) in Suit2-007 cells was associated with increased proliferation as compared to a nonsense control. Accordingly, human recombinant ON decreased proliferation of AsML and Suit2-007 cells in a time and concentration dependent manner[86]. In other studies, exogenous ON also caused dose-dependent inhibition of proliferation in PDAC cell lines[65,68] and this effect was independent of endogenous ON expression[68]. ON-induced growth arrest was associated with increased p21 expression, which induces a G1 cell cycle block[65]. Furthermore, inhibition of endogenous ON by small hairpin RNA resulted in enhanced cell proliferation, migration, colony formation and xenograft formation[68]. The mechanism by which ON might promote apoptosis was investigated in the chemotherapy resistant pancreatic cell line MiaPaca/CPT, which showed that caspase 8-Bcl2 interaction was abolished following ON exposure and restored by treatment with ON antibodies[79]. The anti-tumorigenic effect of ON was confirmed by in vivo studies, e.g., ON expression reduced tumor invasiveness as shown in an orthotopic murine model of pancreatic adenocarcinoma. ON-null mice had larger and more invasive tumors with reduced MMP-9 expression, ECM deposition, and microvessel density as compared to the wild type[92]. However, reduced vessel density in ON-null mice was not accompanied with altered levels of VEGF and TGF-β1. Tumor spread in ON-null mice was explained by reduced ECM deposition (with less mature and/or collagen fibrils), decreased pericyte recruitment, disrupted vascular basement membrane and reduced apoptosis[93,94]. The interaction of ON with pericytes was investigated in an in vitro model utilizing primary pericytes isolated from ON positive or null mice pancreatic tissues or 10T1/2 cells which can differentiate into mesenchymal lineages. It was proposed that ON could facilitate pericyte migration by preventing interaction of endoglin, a TGF-β1 accessory receptor with αV integrins[95]. Accordingly, aberrant TGF-β1 activation in ON-null mice led to significant tumor progression[96].
However, ON also displayed tumorigenic potential. For example, ON treatment increased the invasive capacity of pancreatic adenocarcinoma cell lines in vitro. Specifically, the invasive capacity of the ON expressing human metastatic cell line Colo-357 was increased 14 fold in response to ON exposure. Down-regulation of ON expression by ASO reduced the in vitro invasiveness of Panc-1 cells[65]. Exogenous ON-induced invasiveness was observed in monoculture of Panc-1 cells as well as their co-culture with hPSCs[55]. Direct MMP-2 induction by exogenous ON was suggested to account for promotion of tumor invasion in PDAC[65].
Biomarker role for pancreatic cancer
A compendium of potential biomarkers for pancreatic cancer was developed, which includes 2516 genes. It was reported that 441 genes were overexpressed (defined as an at least two-fold increase or if shown by multiple methods) both at mRNA and protein levels. OPN was listed by more than four studies and therefore was among the best potential biomarker candidates for pancreatic cancer, deserving focused validation[10] in line with its proven value as a clinical tumor progression marker for several forms of cancer[97]. In the same compendium, among 266 genes overexpressed in cancer cells as well as in stroma, only 5 were expressed only in stroma. ON is among the small number of molecules which are overexpressed only in stroma[10].
OPN: The diagnostic and prognostic impact of OPN expression was highlighted by various studies. Using quantitative reverse transcription-polymerase chain reaction (qRT-PCR), OPN expression was shown to increase by 13.1 fold in the parenchyma adjacent to infiltrating cancer relative to normal pancreatic parenchyma, whereas this increase remained at 5.3 fold in the parenchyma adjacent to chronic pancreatitis. Therefore OPN was identified as a helpful predictor of pancreatic cancer lesions[98]. A recent study on surgical specimens from patients with PDAC demonstrated stronger immunostaining for OPN expression with advanced grades[99].
Serum OPN levels were also significantly elevated in pancreatic cancer patients as compared to healthy control subjects[8,100-103]. A pilot study evaluated 12 blood biomarker candidates for detection of pancreatic cancer and demonstrated that macrophage migration inhibition factor and OPN blood tests (with 100% and 95% sensitivity, respectively) were almost perfect to distinguish pancreatic cancer cases from healthy individuals[102]. Specifically, the OPN splice variants OPNb and OPNc were increased in pancreatic cancer when compared to non-cancer controls, as assessed by RT-PCR blood test[9]. OPNc, which supported anchorage independence, was suggested to be the most potent OPN isoform for the pro-metastatic behavior, hence a candidate marker for invasive PDAC[104].
Serum OPN levels in advanced stages III and IV were higher than in early stages I and II, indicating that OPN may be a useful diagnostic marker to distinguish resectable cases and to predict survival rates[101]. Cytoplasmic OPN expression was not correlated with average tumor size, tumor stage, and nodal status[53,58]. The improved survival when OPN was expressed in the cytoplasm (17.1 mo vs 11.6 mo) was linked to a relatively small size (< 2 cm) of OPN positive tumors. Therefore, it was suggested that OPN expression might be lost as tumors grow and turn into an aggressive phenotype[53].
ON: In experimental models based on pancreatic cell lines, both, an antitumorigenic and tumorigenic potential of ON was demonstrated, and studies in humans showed that stromal expression of ON is particularly important for the prognosis of these patients. The ON expression pattern was investigated by immunohistochemical analysis in 299 primary PDAC resection specimens from patients who underwent pancreatico-duodenectomy. ON expression by stromal fibroblasts was found to be a strong marker of poor prognosis. Median survival in ON (+) patients was decreased by 50% (15 mo) as compared to ON (-) patients (30 mo)[54]. Immunohistochemical analysis in 58 biopsy specimens from patients with locally advanced pancreatic cancer showed an inverse correlation between stromal ON expression and overall survival (OS). Therefore ON expression in non-resectable tumor stroma was strongly indicative of a poor prognosis[55]. In another trial, stromal ON expression on 5-year survival rate was evaluated. It was shown that the 5-year survival rate in patients with a low ON mRNA level was better (23%) as compared to those with high ON mRNA level. However, no significant correlation was found between stromal ON mRNA overexpression and depth of tumor invasion, lymph node metastasis, stage, histopathological tumor grade, lymphatic invasion, vascular invasion or surgical margin, (0%)[105]. A recent prospective randomized phase III study including 160 patients treated with curatively intended resection and receiving adjuvant treatment with gemcitabine, demonstrated that disease-free and overall survival decreased with strong ON expression. In contrast to previous reports, this finding was not only related to the peritumoral stroma (strong vs not strong DFS 9.0 mo vs 12.6 mo and OS 19.8 mo vs 26.6 mo) but also to the cytoplasmic ON expression of adenocarcinoma cells (positive vs negative DFS 7.4 mo vs 12.1 mo and OS 14.1 mo vs 25.6 mo)[106]. ON expression was shown to be a predictive marker independent of CA19-9 levels[107].
Altered methylation patterns of ON gene transcriptional regulation region were suggested for use as a tumorigenesis marker for early detection of pancreatic cancer. In a small scale study comprising 40 cases of pancreatic cancer and the adjacent normal tissues, 6 chronic pancreatitis tissues, and 6 acute pancreatic tissues, all were analyzed by bisulfite-specific PCR based sequencing. As a result, aberrant hyper-methylation of CpG region 1 and, especially, CpG region 2 might be an early step in pancreatic cancer development and progression and differentiate malignant tissues from healthy and chronic pancreatitis tissues[108].
Hevin: Hevin mRNA and protein levels were found to be high in bulk PDAC and pancreatic neoplasms[10,18,19]. However, its expression is related to the vascular content of a given lesion. Higher percentages of Hevin positive vessels were detected in chronic pancreatitis (32%) and benign and borderline pancreatic tumors (40%) as compared to PDAC (15%). Down-regulation of Hevin is observed in the late stages of pancreatic cancer progression[18].
Therapeutic targeting in pancreatic cancer
SIBLING and SPARC family members are matricellular proteins, which modulate many critical cellular processes like proliferation, migration, and angiogenesis. For this reason, they represent potential therapeutic targets for either stromal depletion or blockade of signaling pathways involved in pancreatic cancer progression.
Inhibition of metastasis: Since down-regulation of OPN reduces pancreatic cancer cell invasion, OPN has been suggested as a therapeutic target to inhibit metastasis[109].
Stromal depletion: ON and gp60 are functionally and immunologically related albumin binding proteins, which mediate trans-endothelial transportation of albumin via activation of caveolin-1 and formation of caveoli[110-112]. This function renders ON a promising target for stromal depletion of pancreatic tumors. Promising results were obtained by a new nanoparticle albumin-bound formulation of paclitaxel, namely nab-paclitaxel. Nab-paclitaxel is transported to malignant tissues by albumin, where it is sequestered by ON. Approximately 10-fold endothelial binding and 4-fold transport across the endothelial cell monolayer was achieved by nab-paclitaxel as compared to conventional paclitaxel[111]. The combination of gemcitabine plus nab-paclitaxel was evaluated in 36 patients with previously untreated advanced pancreatic cancer. Median OS increased significantly in the group with high ON expression when compared to the low-ON group (17.8 mo vs 8.1 mo). Improved survival was correlated with ON overexpression in the stroma but not in the tumor. In the same study, the intratumoral gemcitabine concentration was increased nearly 3-fold in mice harboring PANC265 xenografts, derived from 11 chemotherapy-naive patients, when nab-paclitaxel was added to gemcitabine treatment. Nab-paclitaxel alone and in combination with gemcitabine caused depletion of desmoplastic stroma with resultant vasodilation, which together helped to achieve an increased gemcitabine penetration into the tumor, hence a better response[107].
Improvement of oncolytic activity: Tumor-associated ON positive stromal cells were proposed as potential targets to improve the oncolytic efficacy of conditionally replicative adenoviruses (CRAd). ON positive transformed human microendothelial (HMEC-1) cells enhanced the oncolytic activity of CRAd, Ad(I)-F512-TK, on the ON-negative pancreatic cancer cell line MIA PaCa-2 in vivo. Similarly, the in vitro oncolytic activity of CRAd increased when MIA PaCa-2 cells were incubated in HMEC-1 and fibroblast (WI-38) conditioned media[113].
Modulation of pericyte migration: Modulation of pericyte recruitment may present a potential therapeutic strategy for increasing the effectiveness of an anti-angiogenic tumor therapy[95]. For example, PDGF-BB overexpression in subcutaneous or orthotopic pancreatic tumors in mice was accompanied with high pericyte content and decreased tumor growth. Therefore, increasing the pericyte content of the tumor microenvironment or targeting PDGFR signaling in tumor associated PDGFR+ pericytes with kinase inhibitors yielded promising results in experimental pancreatic cancer models[114,115]. Likewise, ON is involved in pericyte modulation. ON was shown to promote pericyte migration by inhibiting endoglin-dependent TGF-β1 activity in pancreatic cancer[95]. In parallel with this finding, losartan, which diminishes TGF-β1 activation, was able to slow pancreatic tumor progression[96]. Further studies will elaborate how pericyte modulation can be improved via ON targeting[95].
CONCLUSION
Experimental and clinical evidence denote the emerging role of the SIBLING and SPARC family of proteins in pancreatic cancer formation, progression, and metastasis. The differential expression of these proteins in healthy and tumor tissues and correlation of their serum or tumor expression levels with survival rates has shown that they can be useful diagnostic and prognostic biomarkers. ON seems to be a promising target for stromal depletion and anti-angiogenic therapy of pancreatic tumors. Future studies and development of novel agents targeting SIBLING and SPARC family of proteins may help to improve therapeutic response in pancreatic cancer.
Footnotes
P- Reviewer: Mori N, Sadik R, Wang YD S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Gillen S, Schuster T, Meyer Zum Büschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7:e1000267. doi: 10.1371/journal.pmed.1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hidalgo M, Von Hoff DD. Translational therapeutic opportunities in ductal adenocarcinoma of the pancreas. Clin Cancer Res. 2012;18:4249–4256. doi: 10.1158/1078-0432.CCR-12-1327. [DOI] [PubMed] [Google Scholar]
- 4.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamburrino A, Piro G, Carbone C, Tortora G, Melisi D. Mechanisms of resistance to chemotherapeutic and anti-angiogenic drugs as novel targets for pancreatic cancer therapy. Front Pharmacol. 2013;4:56. doi: 10.3389/fphar.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oikonomopoulos GM, Syrigos KN, Saif MW. Prognostic factors in pancreatic cancer. JOP. 2013;14:322–324. doi: 10.6092/1590-8577/1644. [DOI] [PubMed] [Google Scholar]
- 7.Staines KA, MacRae VE, Farquharson C. The importance of the SIBLING family of proteins on skeletal mineralisation and bone remodelling. J Endocrinol. 2012;214:241–255. doi: 10.1530/JOE-12-0143. [DOI] [PubMed] [Google Scholar]
- 8.Koopmann J, Fedarko NS, Jain A, Maitra A, Iacobuzio-Donahue C, Rahman A, Hruban RH, Yeo CJ, Goggins M. Evaluation of osteopontin as biomarker for pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2004;13:487–491. [PubMed] [Google Scholar]
- 9.Hartung F, Weber GF. RNA blood levels of osteopontin splice variants are cancer markers. Springerplus. 2013;2:110. doi: 10.1186/2193-1801-2-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harsha HC, Kandasamy K, Ranganathan P, Rani S, Ramabadran S, Gollapudi S, Balakrishnan L, Dwivedi SB, Telikicherla D, Selvan LD, et al. A compendium of potential biomarkers of pancreatic cancer. PLoS Med. 2009;6:e1000046. doi: 10.1371/journal.pmed.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellahcène A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. 2008;8:212–226. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudzki Z, Jothy S. CD44 and the adhesion of neoplastic cells. Mol Pathol. 1997;50:57–71. doi: 10.1136/mp.50.2.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradshaw AD. Diverse biological functions of the SPARC family of proteins. Int J Biochem Cell Biol. 2012;44:480–488. doi: 10.1016/j.biocel.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hambrock HO, Nitsche DP, Hansen U, Bruckner P, Paulsson M, Maurer P, Hartmann U. SC1/hevin. An extracellular calcium-modulated protein that binds collagen I. J Biol Chem. 2003;278:11351–11358. doi: 10.1074/jbc.M212291200. [DOI] [PubMed] [Google Scholar]
- 15.Vannahme C, Gösling S, Paulsson M, Maurer P, Hartmann U. Characterization of SMOC-2, a modular extracellular calcium-binding protein. Biochem J. 2003;373:805–814. doi: 10.1042/BJ20030532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan MM, Sage EH. Hevin/SC1, a matricellular glycoprotein and potential tumor-suppressor of the SPARC/BM-40/Osteonectin family. Int J Biochem Cell Biol. 2004;36:991–996. doi: 10.1016/j.biocel.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Claeskens A, Ongenae N, Neefs JM, Cheyns P, Kaijen P, Cools M, Kutoh E. Hevin is down-regulated in many cancers and is a negative regulator of cell growth and proliferation. Br J Cancer. 2000;82:1123–1130. doi: 10.1054/bjoc.1999.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esposito I, Kayed H, Keleg S, Giese T, Sage EH, Schirmacher P, Friess H, Kleeff J. Tumor-suppressor function of SPARC-like protein 1/Hevin in pancreatic cancer. Neoplasia. 2007;9:8–17. doi: 10.1593/neo.06646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu B, Jones J, Hollingsworth MA, Hruban RH, Kern SE. Invasion-specific genes in malignancy: serial analysis of gene expression comparisons of primary and passaged cancers. Cancer Res. 2001;61:1833–1838. [PubMed] [Google Scholar]
- 20.McCleary-Wheeler AL, McWilliams R, Fernandez-Zapico ME. Aberrant signaling pathways in pancreatic cancer: a two compartment view. Mol Carcinog. 2012;51:25–39. doi: 10.1002/mc.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Hänsch GM, Gaida MM. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. 2012;2012:720768. doi: 10.1155/2012/720768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardian K, Janczewska S, Durlik M. Microenvironment elements involved in the development of pancreatic cancer tumor. Gastroenterol Res Pract. 2012;2012:585674. doi: 10.1155/2012/585674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey JM, Leach SD. Signaling pathways mediating epithelial- mesenchymal crosstalk in pancreatic cancer: Hedgehog, Notch and TGFβ. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum (India): Transworld Research Network; 2012. [PubMed] [Google Scholar]
- 26.Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68:2085–2093. doi: 10.1158/0008-5472.CAN-07-2477. [DOI] [PubMed] [Google Scholar]
- 27.Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Podhajcer OL, Benedetti LG, Girotti MR, Prada F, Salvatierra E, Llera AS. The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 2008;27:691–705. doi: 10.1007/s10555-008-9146-7. [DOI] [PubMed] [Google Scholar]
- 30.Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, Biankin AV, Goldstein D, Pirola RC, Wilson JS, et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177:2585–2596. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herreros-Villanueva M, Hijona E, Cosme A, Bujanda L. Adjuvant and neoadjuvant treatment in pancreatic cancer. World J Gastroenterol. 2012;18:1565–1572. doi: 10.3748/wjg.v18.i14.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krantz SB, Shields MA, Dangi-Garimella S, Munshi HG, Bentrem DJ. Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J Surg Res. 2012;173:105–112. doi: 10.1016/j.jss.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steele CW, Jamieson NB, Evans TR, McKay CJ, Sansom OJ, Morton JP, Carter CR. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br J Cancer. 2013;108:997–1003. doi: 10.1038/bjc.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silvestris N, Gnoni A, Brunetti AE, Vincenti L, Santini D, Tonini G, Merchionne F, Maiello E, Lorusso V, Nardulli P, et al. Target therapies in pancreatic carcinoma. Curr Med Chem. 2014;21:948–965. doi: 10.2174/09298673113209990238. [DOI] [PubMed] [Google Scholar]
- 35.Zagouri F, Sergentanis TN, Chrysikos D, Zografos CG, Papadimitriou CA, Dimopoulos MA, Filipits M, Bartsch R. Molecularly targeted therapies in metastatic pancreatic cancer: a systematic review. Pancreas. 2013;42:760–773. doi: 10.1097/MPA.0b013e31827aedef. [DOI] [PubMed] [Google Scholar]
- 36.Castellanos JA, Merchant NB, Nagathihalli NS. Emerging targets in pancreatic cancer: epithelial-mesenchymal transition and cancer stem cells. Onco Targets Ther. 2013;6:1261–1267. doi: 10.2147/OTT.S34670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gnoni A, Licchetta A, Scarpa A, Azzariti A, Brunetti AE, Simone G, Nardulli P, Santini D, Aieta M, Delcuratolo S, et al. Carcinogenesis of pancreatic adenocarcinoma: precursor lesions. Int J Mol Sci. 2013;14:19731–19762. doi: 10.3390/ijms141019731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brand TM, Iida M, Li C, Wheeler DL. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med. 2011;12:419–432. [PMC free article] [PubMed] [Google Scholar]
- 39.Burris H, Rocha-Lima C. New therapeutic directions for advanced pancreatic cancer: targeting the epidermal growth factor and vascular endothelial growth factor pathways. Oncologist. 2008;13:289–298. doi: 10.1634/theoncologist.2007-0134. [DOI] [PubMed] [Google Scholar]
- 40.Büchler P, Reber HA, Büchler MW, Friess H, Hines OJ. VEGF-RII influences the prognosis of pancreatic cancer. Ann Surg. 2002;236:738–49; discussion 749. doi: 10.1097/00000658-200212000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlingensiepen KH, Fischer-Blass B, Schmaus S, Ludwig S. Antisense therapeutics for tumor treatment: the TGF-beta2 inhibitor AP 12009 in clinical development against malignant tumors. Recent Results Cancer Res. 2008;177:137–150. doi: 10.1007/978-3-540-71279-4_16. [DOI] [PubMed] [Google Scholar]
- 42.Subramanian G, Schwarz RE, Higgins L, McEnroe G, Chakravarty S, Dugar S, Reiss M. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004;64:5200–5211. doi: 10.1158/0008-5472.CAN-04-0018. [DOI] [PubMed] [Google Scholar]
- 43.Krantz SB, Shields MA, Dangi-Garimella S, Cheon EC, Barron MR, Hwang RF, Rao MS, Grippo PJ, Bentrem DJ, Munshi HG. MT1-MMP cooperates with Kras(G12D) to promote pancreatic fibrosis through increased TGF-β signaling. Mol Cancer Res. 2011;9:1294–1304. doi: 10.1158/1541-7786.MCR-11-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G, Abbruzzese JL, Chiao PJ. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7:829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michl P, Buchholz M, Rolke M, Kunsch S, Löhr M, McClane B, Tsukita S, Leder G, Adler G, Gress TM. Claudin-4: a new target for pancreatic cancer treatment using Clostridium perfringens enterotoxin. Gastroenterology. 2001;121:678–684. doi: 10.1053/gast.2001.27124. [DOI] [PubMed] [Google Scholar]
- 46.Deharvengt S, Marmarelis M, Korc M. Concomitant targeting of EGF receptor, TGF-beta and SRC points to a novel therapeutic approach in pancreatic cancer. PLoS One. 2012;7:e39684. doi: 10.1371/journal.pone.0039684. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Ouyang H, Gore J, Deitz S, Korc M. microRNA-10b enhances pancreatic cancer cell invasion by suppressing TIP30 expression and promoting EGF and TGF-β actions. Oncogene. 2014;33:4664–4674. doi: 10.1038/onc.2013.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M, Fujii T, Komuro A, Kiyono K, Kaminishi M, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci USA. 2007;104:3460–3465. doi: 10.1073/pnas.0611660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilbert JA, Adhikari LJ, Lloyd RV, Halfdanarson TR, Muders MH, Ames MM. Molecular markers for novel therapeutic strategies in pancreatic endocrine tumors. Pancreas. 2013;42:411–421. doi: 10.1097/MPA.0b013e31826cb243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spector I, Zilberstein Y, Lavy A, Nagler A, Genin O, Pines M. Involvement of host stroma cells and tissue fibrosis in pancreatic tumor development in transgenic mice. PLoS One. 2012;7:e41833. doi: 10.1371/journal.pone.0041833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S, Esposito I, Friess H, Gress TM, Habisch HJ, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61:172–178. doi: 10.1136/gutjnl-2011-301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bloomston M, Ellison EC, Muscarella P, Al-Saif O, Martin EW, Melvin WS, Frankel WL. Stromal osteonectin overexpression is associated with poor outcome in patients with ampullary cancer. Ann Surg Oncol. 2007;14:211–217. doi: 10.1245/s10434-006-9128-3. [DOI] [PubMed] [Google Scholar]
- 53.Collins AL, Rock J, Malhotra L, Frankel WL, Bloomston M. Osteopontin expression is associated with improved survival in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 2012;19:2673–2678. doi: 10.1245/s10434-012-2337-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C, Goggins M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–325. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 55.Mantoni TS, Schendel RR, Rödel F, Niedobitek G, Al-Assar O, Masamune A, Brunner TB. Stromal SPARC expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1806–1815. doi: 10.4161/cbt.7.11.6846. [DOI] [PubMed] [Google Scholar]
- 56.Brown LF, Papadopoulos-Sergiou A, Berse B, Manseau EJ, Tognazzi K, Perruzzi CA, Dvorak HF, Senger DR. Osteopontin expression and distribution in human carcinomas. Am J Pathol. 1994;145:610–623. [PMC free article] [PubMed] [Google Scholar]
- 57.Sedivy R, Peters K, Klöppel G. Osteopontin expression in ductal adenocarcinomas and undifferentiated carcinomas of the pancreas. Virchows Arch. 2005;446:41–45. doi: 10.1007/s00428-004-1142-x. [DOI] [PubMed] [Google Scholar]
- 58.Coppola D, Szabo M, Boulware D, Muraca P, Alsarraj M, Chambers AF, Yeatman TJ. Correlation of osteopontin protein expression and pathological stage across a wide variety of tumor histologies. Clin Cancer Res. 2004;10:184–190. doi: 10.1158/1078-0432.ccr-1405-2. [DOI] [PubMed] [Google Scholar]
- 59.Missiaglia E, Blaveri E, Terris B, Wang YH, Costello E, Neoptolemos JP, Crnogorac-Jurcevic T, Lemoine NR. Analysis of gene expression in cancer cell lines identifies candidate markers for pancreatic tumorigenesis and metastasis. Int J Cancer. 2004;112:100–112. doi: 10.1002/ijc.20376. [DOI] [PubMed] [Google Scholar]
- 60.Kayed H, Kleeff J, Keleg S, Felix K, Giese T, Berger MR, Büchler MW, Friess H. Effects of bone sialoprotein on pancreatic cancer cell growth, invasion and metastasis. Cancer Lett. 2007;245:171–183. doi: 10.1016/j.canlet.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 61.Porter PL, Sage EH, Lane TF, Funk SE, Gown AM. Distribution of SPARC in normal and neoplastic human tissue. J Histochem Cytochem. 1995;43:791–800. doi: 10.1177/43.8.7622842. [DOI] [PubMed] [Google Scholar]
- 62.Sato N, Fukushima N, Maehara N, Matsubayashi H, Koopmann J, Su GH, Hruban RH, Goggins M. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene. 2003;22:5021–5030. doi: 10.1038/sj.onc.1206807. [DOI] [PubMed] [Google Scholar]
- 63.Reding T, Wagner U, Silva AB, Sun LK, Bain M, Kim SY, Bimmler D, Graf R. Inflammation-dependent expression of SPARC during development of chronic pancreatitis in WBN/Kob rats and a microarray gene expression analysis. Physiol Genomics. 2009;38:196–204. doi: 10.1152/physiolgenomics.00028.2009. [DOI] [PubMed] [Google Scholar]
- 64.Tai IT, Dai M, Owen DA, Chen LB. Genome-wide expression analysis of therapy-resistant tumors reveals SPARC as a novel target for cancer therapy. J Clin Invest. 2005;115:1492–1502. doi: 10.1172/JCI23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guweidhi A, Kleeff J, Adwan H, Giese NA, Wente MN, Giese T, Büchler MW, Berger MR, Friess H. Osteonectin influences growth and invasion of pancreatic cancer cells. Ann Surg. 2005;242:224–234. doi: 10.1097/01.sla.0000171866.45848.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brune K, Hong SM, Li A, Yachida S, Abe T, Griffith M, Yang D, Omura N, Eshleman J, Canto M, et al. Genetic and epigenetic alterations of familial pancreatic cancers. Cancer Epidemiol Biomarkers Prev. 2008;17:3536–3542. doi: 10.1158/1055-9965.EPI-08-0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kayed H, Jiang X, Keleg S, Jesnowski R, Giese T, Berger MR, Esposito I, Löhr M, Friess H, Kleeff J. Regulation and functional role of the Runt-related transcription factor-2 in pancreatic cancer. Br J Cancer. 2007;97:1106–1115. doi: 10.1038/sj.bjc.6603984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen G, Tian X, Liu Z, Zhou S, Schmidt B, Henne-Bruns D, Bachem M, Kornmann M. Inhibition of endogenous SPARC enhances pancreatic cancer cell growth: modulation by FGFR1-III isoform expression. Br J Cancer. 2010;102:188–195. doi: 10.1038/sj.bjc.6605440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Binkley CE, Zhang L, Greenson JK, Giordano TJ, Kuick R, Misek D, Hanash S, Logsdon CD, Simeone DM. The molecular basis of pancreatic fibrosis: common stromal gene expression in chronic pancreatitis and pancreatic adenocarcinoma. Pancreas. 2004;29:254–263. doi: 10.1097/00006676-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 70.Iacobuzio-Donahue CA, Ryu B, Hruban RH, Kern SE. Exploring the host desmoplastic response to pancreatic carcinoma: gene expression of stromal and neoplastic cells at the site of primary invasion. Am J Pathol. 2002;160:91–99. doi: 10.1016/S0002-9440(10)64353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chong HC, Tan CK, Huang RL, Tan NS. Matricellular proteins: a sticky affair with cancers. J Oncol. 2012;2012:351089. doi: 10.1155/2012/351089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Denhardt DT, Guo X. Osteopontin: a protein with diverse functions. FASEB J. 1993;7:1475–1482. [PubMed] [Google Scholar]
- 73.Leali D, Naldini A. The role of osteopontin in angiogenesis. In: Rbatti D, editor. Recent advances in angiogenesis and antiangiogenesis. Sharjah: Bentham Science Publishers Ltd; 2009. pp. 10–19. [Google Scholar]
- 74.Adwan H, Bäuerle T, Najajreh Y, Elazer V, Golomb G, Berger MR. Decreased levels of osteopontin and bone sialoprotein II are correlated with reduced proliferation, colony formation, and migration of GFP-MDA-MB-231 cells. Int J Oncol. 2004;24:1235–1244. [PubMed] [Google Scholar]
- 75.Adwan H, Bäuerle TJ, Berger MR. Downregulation of osteopontin and bone sialoprotein II is related to reduced colony formation and metastasis formation of MDA-MB-231 human breast cancer cells. Cancer Gene Ther. 2004;11:109–120. doi: 10.1038/sj.cgt.7700659. [DOI] [PubMed] [Google Scholar]
- 76.Reufsteck C, Lifshitz-Shovali R, Zepp M, Bäuerle T, Kübler D, Golomb G, Berger MR. Silencing of skeletal metastasis-associated genes impairs migration of breast cancer cells and reduces osteolytic bone lesions. Clin Exp Metastasis. 2012;29:441–456. doi: 10.1007/s10585-012-9462-8. [DOI] [PubMed] [Google Scholar]
- 77.Nagaraju GP, Sharma D. Anti-cancer role of SPARC, an inhibitor of adipogenesis. Cancer Treat Rev. 2011;37:559–566. doi: 10.1016/j.ctrv.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yan Q, Sage EH. SPARC, a matricellular glycoprotein with important biological functions. J Histochem Cytochem. 1999;47:1495–1506. doi: 10.1177/002215549904701201. [DOI] [PubMed] [Google Scholar]
- 79.Rahman M, Chan AP, Tai IT. A peptide of SPARC interferes with the interaction between caspase8 and Bcl2 to resensitize chemoresistant tumors and enhance their regression in vivo. PLoS One. 2011;6:e26390. doi: 10.1371/journal.pone.0026390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tai IT, Tang MJ. SPARC in cancer biology: its role in cancer progression and potential for therapy. Drug Resist Updat. 2008;11:231–246. doi: 10.1016/j.drup.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 81.Rempel SA, Golembieski WA, Fisher JL, Maile M, Nakeff A. SPARC modulates cell growth, attachment and migration of U87 glioma cells on brain extracellular matrix proteins. J Neurooncol. 2001;53:149–160. doi: 10.1023/a:1012201300188. [DOI] [PubMed] [Google Scholar]
- 82.Cheng L, Sage EH, Yan Q. SPARC fusion protein induces cellular adhesive signaling. PLoS One. 2013;8:e53202. doi: 10.1371/journal.pone.0053202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, Cappetti B, Parenza M, Rumio C, Brekken RA, Chiodoni C, Colombo MP. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008;68:9050–9059. doi: 10.1158/0008-5472.CAN-08-1327. [DOI] [PubMed] [Google Scholar]
- 84.Kupprion C, Motamed K, Sage EH. SPARC (BM-40, osteonectin) inhibits the mitogenic effect of vascular endothelial growth factor on microvascular endothelial cells. J Biol Chem. 1998;273:29635–29640. doi: 10.1074/jbc.273.45.29635. [DOI] [PubMed] [Google Scholar]
- 85.Raines EW, Lane TF, Iruela-Arispe ML, Ross R, Sage EH. The extracellular glycoprotein SPARC interacts with platelet-derived growth factor (PDGF)-AB and -BB and inhibits the binding of PDGF to its receptors. Proc Natl Acad Sci USA. 1992;89:1281–1285. doi: 10.1073/pnas.89.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhivkova-Galunska M, Adwan H, Eyol E, Kleeff J, Kolb A, Bergmann F, Berger MR. Osteopontin but not osteonectin favors the metastatic growth of pancreatic cancer cell lines. Cancer Biol Ther. 2010;10:54–64. doi: 10.4161/cbt.10.1.12161. [DOI] [PubMed] [Google Scholar]
- 87.Ohno K, Nishimori H, Yasoshima T, Kamiguchi K, Hata F, Fukui R, Okuya K, Kimura Y, Denno R, Kon S, et al. Inhibition of osteopontin reduces liver metastasis of human pancreatic cancer xenografts injected into the spleen in a mouse model. Surg Today. 2010;40:347–356. doi: 10.1007/s00595-009-4082-x. [DOI] [PubMed] [Google Scholar]
- 88.Eyol E, Murtaga A, Zhivkova-Galunska M, Georges R, Zepp M, Djandji D, Kleeff J, Berger MR, Adwan H. Few genes are associated with the capability of pancreatic ductal adenocarcinoma cells to grow in the liver of nude rats. Oncol Rep. 2012;28:2177–2187. doi: 10.3892/or.2012.2049. [DOI] [PubMed] [Google Scholar]
- 89.Chipitsyna G, Gong Q, Anandanadesan R, Alnajar A, Batra SK, Wittel UA, Cullen DM, Akhter MP, Denhardt DT, Yeo CJ, et al. Induction of osteopontin expression by nicotine and cigarette smoke in the pancreas and pancreatic ductal adenocarcinoma cells. Int J Cancer. 2009;125:276–285. doi: 10.1002/ijc.24388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sullivan J, Blair L, Alnajar A, Aziz T, Chipitsyna G, Gong Q, Yeo CJ, Arafat HA. Expression and regulation of nicotine receptor and osteopontin isoforms in human pancreatic ductal adenocarcinoma. Histol Histopathol. 2011;26:893–904. doi: 10.14670/HH-26.893. [DOI] [PubMed] [Google Scholar]
- 91.Lazar M, Sullivan J, Chipitsyna G, Gong Q, Ng CY, Salem AF, Aziz T, Witkiewicz A, Denhardt DT, Yeo CJ, et al. Involvement of osteopontin in the matrix-degrading and proangiogenic changes mediated by nicotine in pancreatic cancer cells. J Gastrointest Surg. 2010;14:1566–1577. doi: 10.1007/s11605-010-1338-0. [DOI] [PubMed] [Google Scholar]
- 92.Arnold S, Mira E, Muneer S, Korpanty G, Beck AW, Holloway SE, Mañes S, Brekken RA. Forced expression of MMP9 rescues the loss of angiogenesis and abrogates metastasis of pancreatic tumors triggered by the absence of host SPARC. Exp Biol Med (Maywood) 2008;233:860–873. doi: 10.3181/0801-RM-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arnold SA, Rivera LB, Miller AF, Carbon JG, Dineen SP, Xie Y, Castrillon DH, Sage EH, Puolakkainen P, Bradshaw AD, et al. Lack of host SPARC enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis Model Mech. 2010;3:57–72. doi: 10.1242/dmm.003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Puolakkainen PA, Brekken RA, Muneer S, Sage EH. Enhanced growth of pancreatic tumors in SPARC-null mice is associated with decreased deposition of extracellular matrix and reduced tumor cell apoptosis. Mol Cancer Res. 2004;2:215–224. [PubMed] [Google Scholar]
- 95.Rivera LB, Brekken RA. SPARC promotes pericyte recruitment via inhibition of endoglin-dependent TGF-β1 activity. J Cell Biol. 2011;193:1305–1319. doi: 10.1083/jcb.201011143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arnold SA, Rivera LB, Carbon JG, Toombs JE, Chang CL, Bradshaw AD, Brekken RA. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFβ activation. PLoS One. 2012;7:e31384. doi: 10.1371/journal.pone.0031384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weber GF, Lett GS, Haubein NC. Osteopontin is a marker for cancer aggressiveness and patient survival. Br J Cancer. 2010;103:861–869. doi: 10.1038/sj.bjc.6605834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fukushima N, Koopmann J, Sato N, Prasad N, Carvalho R, Leach SD, Hruban RH, Goggins M. Gene expression alterations in the non-neoplastic parenchyma adjacent to infiltrating pancreatic ductal adenocarcinoma. Mod Pathol. 2005;18:779–787. doi: 10.1038/modpathol.3800337. [DOI] [PubMed] [Google Scholar]
- 99.Tsai WC, Lin CK, Yang YS, Chan DC, Gao HW, Chang FN, Jin JS. The correlations of LMX1A and osteopontin expression to the clinicopathologic stages in pancreatic adenocarcinoma. Appl Immunohistochem Mol Morphol. 2013;21:395–400. doi: 10.1097/PAI.0b013e318277d9de. [DOI] [PubMed] [Google Scholar]
- 100.Koopmann J, Rosenzweig CN, Zhang Z, Canto MI, Brown DA, Hunter M, Yeo C, Chan DW, Breit SN, Goggins M. Serum markers in patients with resectable pancreatic adenocarcinoma: macrophage inhibitory cytokine 1 versus CA19-9. Clin Cancer Res. 2006;12:442–446. doi: 10.1158/1078-0432.CCR-05-0564. [DOI] [PubMed] [Google Scholar]
- 101.Poruk KE, Firpo MA, Scaife CL, Adler DG, Emerson LL, Boucher KM, Mulvihill SJ. Serum osteopontin and tissue inhibitor of metalloproteinase 1 as diagnostic and prognostic biomarkers for pancreatic adenocarcinoma. Pancreas. 2013;42:193–197. doi: 10.1097/MPA.0b013e31825e354d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen R, Crispin DA, Pan S, Hawley S, McIntosh MW, May D, Anton-Culver H, Ziogas A, Bronner MP, Brentnall TA. Pilot study of blood biomarker candidates for detection of pancreatic cancer. Pancreas. 2010;39:981–988. doi: 10.1097/MPA.0b013e3181dac920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bünger S, Laubert T, Roblick UJ, Habermann JK. Serum biomarkers for improved diagnostic of pancreatic cancer: a current overview. J Cancer Res Clin Oncol. 2011;137:375–389. doi: 10.1007/s00432-010-0965-x. [DOI] [PubMed] [Google Scholar]
- 104.Sullivan J, Blair L, Alnajar A, Aziz T, Ng CY, Chipitsyna G, Gong Q, Witkiewicz A, Weber GF, Denhardt DT, et al. Expression of a prometastatic splice variant of osteopontin, OPNC, in human pancreatic ductal adenocarcinoma. Surgery. 2009;146:232–240. doi: 10.1016/j.surg.2009.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miyoshi K, Sato N, Ohuchida K, Mizumoto K, Tanaka M. SPARC mRNA expression as a prognostic marker for pancreatic adenocarcinoma patients. Anticancer Res. 2010;30:867–871. [PubMed] [Google Scholar]
- 106.Sinn M, Sinn BV, Striefler JK, Stieler J, Pelzer U, Prinzler J, Neuhaus P, Dietel M, Dörken B, Oettle H, et al. SPARC in pancreatic cancer: Results from the CONKO-001 study. J Clin Oncol. 2013;31 suppl:Abstr 4016. [Google Scholar]
- 107.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–4554. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gao J, Song J, Huang H, Li Z, Du Y, Cao J, Li M, Lv S, Lin H, Gong Y. Methylation of the SPARC gene promoter and its clinical implication in pancreatic cancer. J Exp Clin Cancer Res. 2010;29:28. doi: 10.1186/1756-9966-29-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kolb A, Kleeff J, Guweidhi A, Esposito I, Giese NA, Adwan H, Giese T, Büchler MW, Berger MR, Friess H. Osteopontin influences the invasiveness of pancreatic cancer cells and is increased in neoplastic and inflammatory conditions. Cancer Biol Ther. 2005;4:740–746. doi: 10.4161/cbt.4.7.1821. [DOI] [PubMed] [Google Scholar]
- 110.Schnitzer JE, Oh P. Antibodies to SPARC inhibit albumin binding to SPARC, gp60, and microvascular endothelium. Am J Physiol. 1992;263:H1872–H1879. doi: 10.1152/ajpheart.1992.263.6.H1872. [DOI] [PubMed] [Google Scholar]
- 111.Stinchcombe TE. Nanoparticle albumin-bound paclitaxel: a novel Cremphor-EL-free formulation of paclitaxel. Nanomedicine (Lond) 2007;2:415–423. doi: 10.2217/17435889.2.4.415. [DOI] [PubMed] [Google Scholar]
- 112.Talekar M, Kendall J, Denny W, Garg S. Targeting of nanoparticles in cancer: drug delivery and diagnostics. Anticancer Drugs. 2011;22:949–962. doi: 10.1097/CAD.0b013e32834a4554. [DOI] [PubMed] [Google Scholar]
- 113.Lopez MV, Viale DL, Cafferata EG, Bravo AI, Carbone C, Gould D, Chernajovsky Y, Podhajcer OL. Tumor associated stromal cells play a critical role on the outcome of the oncolytic efficacy of conditionally replicative adenoviruses. PLoS One. 2009;4:e5119. doi: 10.1371/journal.pone.0005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, Bucana C, Ellis LM. Overexpression of PDGF-BB decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J Clin Invest. 2007;117:2114–2122. doi: 10.1172/JCI31334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]