

Abstract
Activity at the vertebrate nerve—muscle synapse creates large macroendosomes (MEs) via bulk membrane infolding. Visualized with the endocytic probe FM1-43, most (94%) of the ~25 MEs/terminal created by brief (30-Hz, 18-second) stimulation dissipate rapidly (~1 minute) into vesicles. Others, however, remain for hours. Here we study these “ late” MEs by using 4D live imaging over a period of ~1 hour after stimulation. We find that some (51/398 or 13%) disappear spontaneously via exocytosis, releasing their contents into the extracellular milieu. Others (at least 15/1,960 or 1%) fuse or closely associate with a second class of endosomes that take up acidophilic dyes (acidic endosomes [AEs]). AEs are plentiful (~47/terminal) and exist independent of stimulation. Unlike MEs, which exhibit Brownian motion, AEs exhibit directed motion (average, 83 nm/sec) on microtubules within and among terminal boutons. AEs populate the axon as well, where movement is predominantly retrograde. They share biochemical and immunohistochemical markers (e.g., lysosomal-associated membrane protein [LAMP-1]) with lysosomes. Fusion/association of MEs with AEs suggests a sorting/degradation pathway in nerve terminals wherein the role of AEs is similar to that of lysosomes. Based on our data, we propose that MEs serve as sorting endosomes. Thus their contents, which include plasma membrane proteins, vesicle proteins, and extracellular levels of Ca2+, can be targeted either toward the reformation and budding of synaptic vesicles, toward secretion via exocytosis, or toward a degradation process that utilizes AEs either for lysis within the terminal or for transport toward the cell body.
INDEXING TERMS: endocytosis, vesicle processing, synapse
Processing of spent vesicles in motor nerve terminals often involves creation of large endosomal intermediates, which we call macroendosomes (MEs). In early processing models, similar structures (vacuoles or cisternae) were thought to result from fusion of 50-nm vesicles internalized by clathrin-mediated endocytosis (CME; Heuser and Reese, 1973). Although vesicle fusion with permanent endosomes remains a possibility (e.g., Hoopmann et al., 2010; Uytterhoeven et al., 2011), MEs as we define them originate directly from the plasma membrane via infolding and subsequent clathrin-independent fission (Kadota et al., 1994; Koenig and Ikeda, 1996). This bulk endocytosis has been reported in association with high-intensity stimulation in a variety of nerve terminals, precisely the conditions under which CME from the plasma membrane alone might not keep pace with exocytosis (Miller and Heuser, 1984; Takei et al., 1996; Teng et al., 1999; Richards et al., 2000; Gaffield et al., 2011; reviewed by Clayton and Cousin, 2009). However, we have found that MEs appear after even very brief stimulation in a bath containing the endocytic probes sulforhodamine (Teng et al., 1999) or FM1-43 (Teng et al., 2007). Thus bulk endocytosis occurs along with CME under all conditions of use, including response to low-intensity stimulation, under which CME alone would seem adequate for vesicle reformation. Moreover, although some MEs undergo clathrin-mediated budding into vesicles, others, which we call “ late” MEs, remain intact well after compensatory endocytosis is complete (Teng et al., 2007).
If not used exclusively for compensatory endocytosis and vesicle reformation, bulk endocytosis must serve some additional function. One possibility is that MEs participate in a constitutive process not directly related to recycling. Sorting and degradative functions are two possibilities. Both are associated with receptor-mediated endocytosis in the soma of neurons and other cell types. Analogues of these processes within motor terminals have already been described (Weible et al., 2001; Qualmann and Kelly, 2000; Hoopmann et al., 2010; Uytterhoeven et al., 2011). Sorting in particular might be required for long-term homeostasis within motor terminals, which are isolated from the cell body due to transit times required for axonal transport. One function, for which the existence of sorting endosomes has long been inferred, is correction of errors made in reformation of vesicles by CME (reviewed by Shupliakov and Brodin, 2010).
Regardless of its function, bulk endocytosis challenges the nerve terminal with a set of unique homeostatic problems. Vesicle membrane proteins are known to reside within permanent pools at the presynaptic membrane of the central nervous system (CNS) (Fernandez-Alfonso and Ryan, 2004) and motor terminals (Tabares et al., 2007), even after completion of compensatory endocytosis following activity. Endosomes internalized from the cleft contain not only these proteins but plasma membrane proteins and high (extracellular) levels of Ca2+ as well. After vesicle budding from MEs is complete, what ultimately happens to these various “leftover” internalized components of the extracellular milieu is unclear.
To investigate the function of MEs and the disposition of their contents, we have imaged living late MEs over time. We find evidence for two processing and degradation pathways—fusion of MEs with the plasma membrane, and fusion (or extremely close association) of MEs with a second class of endosomes within the terminal. Both occur after brief periods of low-level synaptic activity. Part of this work has appeared in abstract form (Stewart and Wilkinson, 2010).
MATERIALS AND METHODS
The single-fiber-thick transversus abdominis muscle of the garter snake was used in most experiments. This simple segmental muscle (100 fibers) provides unequaled visual and electrophysiological access to living nerve terminals, particularly at the level of individual terminal boutons (reviewed by Wilkinson and Teng, 2003).
Animals
All procedures followed the Washington University Guidelines for Animal Studies. Garter snakes (Thamnophis sirtalis) were housed as previously described (Teng et al., 2007). Snakes were anesthetized by xylazine injection (1.1 mg/kg i.p.). Snakes were rapidly sacrificed by decapitation, and several pieces of body-wall muscle were washed in normal reptilian Ringer’s solution (millimolar concentrations: NaCl, 145; KCl, 2.5; CaCl2, 3.6; MgSO4, 1.8; KH2PO4, 1.0; HEPES, 5.0; pH 7.2). Tissues were stored at 4°C until use (24–48 hours maximum). The dissection procedure has been described previously (Wilkinson and Lichtman, 1985). The dissected muscle–nerve preparations were pinned in Sylgard dishes for staining, and imaged in a glass-bottomed (25-mm circular #1 coverslip) metal dish, held in place by magnetic pins. The preparations were mounted upside down to image the neuromuscular junctions, and the material was stretched to lay as flat as possible.
Mouse tissues were provided by the laboratory of R. Mecham (Washington University). Mouse diaphragms and hindlimb muscles were pinned as above, and incubated at 37°C in mammalian Ringer’s solution (millimolar concentrations: NaCl, 155; KCl, 3.0; CaCl2, 2.0; MgSO4, 1.0; KH2PO4, 3.0, HEPES, 5.0; pH 7.4).
Supravital dye staining
Dyes (Molecular Probes/Invitrogen, Carlsbad, CA unless indicated) were dissolved in dimethyl sulfoxide (DMSO) or normal Ringer’solution, diluted in normal Ringer’s solution, applied at room temperature (22°C) for 30 minutes, and then washed in normal Ringer’s solution before imaging to remove unbound dyes. In some cases LysoTracker and FM1-43FX were applied at 4°C to prechilled preparations. FM 1-43FX was applied in conjunction with high-potassium shock by using Ringer’s solution containing 60 mM KCl (NaCl reduced to 86 mM) for 2 minutes, and then washed in normal Ringer’s solution for varying times (typically 10–15 minutes). Dye concentrations were as follows: FM1-43FX, 1:500 (7 μM); LysoTracker Red, 1:5,000 (0.2 μM); Oregon Green–488–taxol, 1:1,000 (1 μM); Alexa 488–bungarotoxin, 1:2,000 (0.25 μg/ml).
Electrical stimulation
Preparations not depolarized with KCl were pinned into a dish containing reptilian Ringer’s solution. A nerve was placed within a suction electrode connected to an isolated stimulator. The two adjacent muscles served as unstimulated controls. A single stimulus was delivered to confirm that the muscle twitched; the preparation was then rested for 5 minutes. Stimulus trains (200-μs negative-going rectangular pulses) were delivered at 30 Hz for 18 seconds.
Imaging
Living preparations were imaged by using a Zeiss Axiovert 200 microscope equipped with a 63×/0.9 NA water-immersion objective. Images were collected on a Cascade 512B CCD camera (Photometrics, Tucson, AZ), and analyzed by using Slidebook 5.0 software (Intelligent Imaging Innovations, Boulder, CO). In some experiments, snake preparations were cooled to reduce background from labeled 50-nm vesicles (Teng et al., 1999). For cold applications, all materials were prechilled in the refrigerator, and images were collected within 10 minutes of removal. For mouse diaphragm and for cell lines used as controls, dishes and solutions were prewarmed to 37°C by using an incubator, and warm media was reintroduced as necessary. Deconvolution (no neighbors algorithm) and other data transformations (such as, aximum-z projection of z-stack data sets) were performed with Slide-book software. Some 3D and 4D data sets were analyzed by using Imaris Bitplane software (Bitplane Scientific Software, St. Paul, MN). Contrast and brightness settings were adjusted for optimal viewing. All data sets, including control images, were adjusted identically.
Images were exported as TIFF files, red to magenta conversion was performed by using Adobe Photoshop, and text annotation was performed by using Microsoft PowerPoint. Unless otherwise stated, all still images and video frames of living preparations presented were deconvolved. When necessary, videos were corrected for drift due to slight muscle activity, and for fading due to repeated imaging. Some fixed and immunostained tissues were imaged with a Zeiss LSM 510 confocal microscope, by using a 63×/1.4 NA oil immersion objective and LSM software (Zeiss, Thornwood, NY).
Although fading (photobleaching) was slight (see Supplementary Video S1), we found evidence for phototoxicity in preliminary experiments. Specifically, preparations imaged repeatedly over time contained more MEs per terminal than those imaged after remaining in the dark for the same time. The disparity suggested that either dissipation into vesicles or exocytosis of MEs (or both) was inhibited by light. The effect was greatest with conventional confocal imaging, slightly less with two-photon confocal imaging (long-wavelength incident light), still less with our photomicroscope (described above) attached to a SensiCam camera (Cooke, Romulus, MI), and least with the Cascade 512B camera used in the present work. With significant light exposure, we found that up to two-thirds of MEs expected to exocytose failed to do so. In the present study, light exposure was limited and phototoxicity was not detectable. The average number of MEs per terminal remaining after 1 hour was ~20 whether terminals were imaged in 4D with our protocol or kept dark (see Discussion).
Statistics
Acidic endosomes (positive for LysoTracker or lysosomal-associated membrane protein [LAMP]) were counted manually from multiple terminals. Only clearly visible structures were included in the analysis. Vesicle diameters were determined with the calibration function of the LSM software. The velocities of acidic endosomes were manually calculated from 2D time-lapse images by using the ruler function in Slidebook 5.0; again, only clearly distinguished vesicles were included. Statistical analyses (mean ± SD, Mann–Whitney paired t-tests, and nonlinear regressions) were performed and graphed by using GraphPad (La Jolla, CA). Nonlinear regression curves generated for Figure 4A were created by using a one-phase decay model. Direct comparison of one-phase and two-phase decay models showed that the former was preferred. Criteria for constraints and verification of the curve fits (residual plots and P values for runs tests) were applied with guidance from the GraphPad manual (www.graphpad.com).
Figure 4.
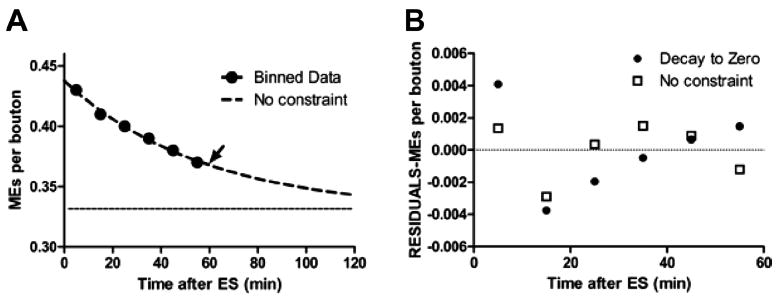
The macroendosome (ME) exocytosis rate decays exponentially to a plateau. A: The total number of MEs remaining as function of time after electrical stimulation (ES) was compiled from all 4D data sets. Data were binned into 10-minute intervals (black circles). The frequency of exocytic events is best fit by a single exponential (dashed line; arrow indicates the time constant at 58 minutes) that decays to a plateau (dotted line) of 0.33 MEs/bouton, or 20 MEs per average-sized terminal. B: Residual analyses of Gaussian least-squares fits to unconstrained exponential in A versus fit to exponential constrained to decay to zero MEs/bouton. Note systematic error in constrained fit (linear trend in filled circles).
Immunohistochemistry
Snake nerve–muscle preparations were fixed in 4% paraformaldehyde/0.1 M sodium phosphate, pH 7.2, for 20 minutes, and then washed in reptilian Ringer’s solution for 30 minutes. Snake and mouse whole tissues were fixed in the same paraformaldehyde solution for 4 hours, and then washed in phosphate-buffered saline (PBS; pH 7.2) overnight. Sections (100 μm) were cut on a Vibratome and stored in PBS. Tissues were permeabilized in either 100 μg/ml digitonin or 0.2 % Triton-X-100 in Ringer’s solution for 15 minutes, followed by a brief wash in Ringer’s solution before addition of primary antibody. Blocking and antibody incubations were performed for at least 1 hour at room temperature in blocking buffer (normal Ringer’s solution plus 2% horse serum). Alexa Fluor–coupled secondary antibodies (Invitrogen; 1:2,000) were applied for 60 minutes in blocking buffer. Bis-benzamide (1 μg/ml) was added for 10 minutes in the last wash to stain nuclei. Tissues were further dissected as needed to allow for flat mounting in Fluoroshield (Sigma, St. Louis, MO), and stored flat at 4°C until use.
Antibody caracterization
The following antibodies were used in this study, as listed in Table 1.
TABLE 1.
Primary Antibodies Used in This Study
| Antigen (reference) | Immunogen | Manufacture, species, cat. no. | Dilution |
|---|---|---|---|
| SV2 | Synaptic vesicles purified from Discogype omnata | Developmental Studies Hybridoma Bank (Iowa City, IA), # SV2 Mouse monoclonal IgG1 | 1:1,000 |
| Cathepsin D | Human Cathepsin D, purified | Gift of Stuart Kornfeld, Washington University Rabbit polyclonal, affinity isolated | 1:500 |
| LAMP-1 | NIH/3T3 mouse embryo, fibroblast tissue culture cell membranes | Developmental Studies Hybridoma Bank, #1D4B Rat monoclonal IgG2a | 1:100 |
| LAMP-2 | BALB/c 3T3 mouse embryo fibroblast tissue culture cell glycoproteins | Developmental Studies Hybridoma Bank, #ABL-93 Rat monoclonal IgG2a | 1:100 |
| LAMP-2 | Mouse liver homogenate | Developmental Studies Hybridoma Bank, #GL2A7 Rat monoclonal IgG2a | 1:100 |
| LAMP-1 | Amino acids 405–416 of human LAMP-1 | Sigma (St. Louis, MO), # L1418 Rabbit polyclonal, affinity Isolated | 1:1,000 |
Monoclonal antibody SV2 was raised against Discopyge omnata (electric eel) synaptic vesicles, and specifically recognizes a glycoprotein of ~100 kDa (Buckley et al., 1983; Buckley and Kelly, 1985). Subsequent analyses have demonstrated that it is a synaptic vesicle marker in multiple phyla, including amphibians and reptiles (Mandell et al., 1990; Wibowo et al., 2009). In the peripheral nervous system, SV2 reactivity is highly concentrated at the presynaptic nerve terminal (Macleod et al., 1999; Misgeld et al., 2002; Sylvain et al., 2011). We have replicated these results by using the garter snake motor nerve terminal (see Fig. 7) and garter snake brain (data not shown). We have also verified these results by using two other synaptic resident proteins, synapsin-1 and syntaxin-2 (data not shown).
Figure 7.
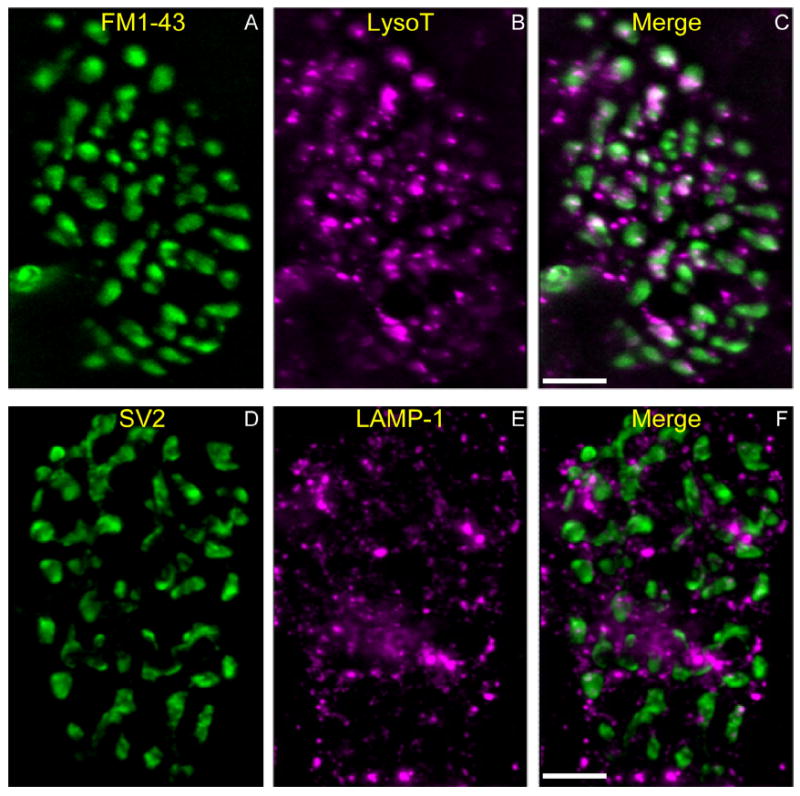
LAMP-1–positive structures in fixed snake terminals resemble living AEs. A–C: Living snake terminal labeled with FM1-43 (A; green) by KCl depolarization (60 mM, 3 minutes) to reveal location of bouton vesicular compartment. AEs are labeled with LysoTracker (B; magenta). Merged image (C) indicates that most AEs are within boutons but not within the vesicular compartments. Note AEs in axon (entering at the lower left). The axon (green) is labeled by FM1-43 because of the dye’s affinity to myelin membrane. D–F: Another snake terminal fixed and immunostained with the synaptic vesicle marker SV2 (D; green) to reveal vesicular compartments. The terminal is also stained with anti-LAMP-1 (Sigma), a marker for lysosomes (E; magenta; F is merged). Note the similar size and number of LysoTracker and LAMP-1–positive vesicles (compare B and E), and the similar distributions of LAMP-1 and LysoTracker mainly outside the vesicular compartment (compare C and F). Scale bar = 10 μm in C (applies to A–C) and F (applies to D–F).
The antibodies described below, Cathepsin D, LAMP-1 (1D4B), LAMP-2 (ABL-93), and LAMP-2 (GL2A7), all produced similar specific staining patterns in mouse brain, mouse muscle and mouse N2a cells. The staining pattern is punctate or (for larger structures) vesicular and concentrated near the nucleus, in N2a cells (see Fig. 12). This staining pattern is consistent with many previous reports (Zhou et al., 2001; Agulhon et al., 2003; Ohmi et al., 2003; Pimpinelli et al., 2005). We have used two different LAMP-1 and LAMP-2 antibodies, and they both produced highly similar immunostaining patterns in mouse brain, mouse muscle, and N2a cells. An additional level of confidence in our results is provided by the use of Cathepsin D; this protein is known to reside in the lumen of the lysosome, rather than in the membrane. These antibodies did not react with snake tissue.
Figure 12.
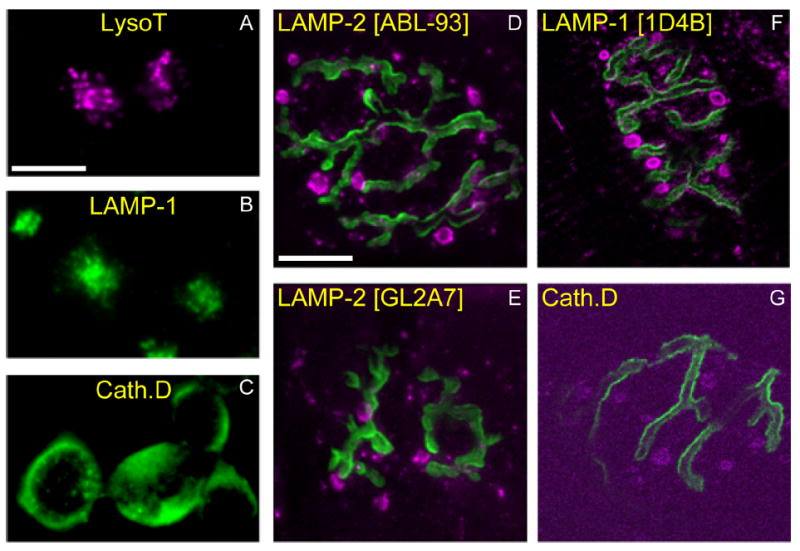
AEs contain lysosomal marker proteins. A–C: Mouse neuroblastoma (N2a) cells were incubated vitally with LysoTracker (A; magenta), or fixed and immunostained with an antibody against LAMP-1 (1D4B) (B; green) or Cathepsin D (C; green). As expected, small (300–500-nm) vesicles (lysosomes) are seen concentrated near the nuclear compartment. D–G: Mouse muscles (gastrocnemius) were incubated with 488–Bungarotoxin (green) to identify AChRs apposing motor terminals and fixed. Immunostaining with antibodies against various lysosomal proteins (magenta; specific antibodies indicated at top of each panel) revealed a pattern similar to LysoTracker staining of AEs (see Fig. 11F), including large structures outside the terminal. Scale bar = 10 μm in A (applies to A–C) and D (applies to D–G).
Rabbit polyclonal antiserum Cathepsin D (raised against human Cathepsin D) recognizes a 45-kDa precursor species, and 39- and 27-kDa processed species, by immunoblot (Boonen et al., 2011) and by immunoprecipitation (Faust et al., 1987). Preimmune control sera do not recognize any of these species (Faust et al., 1987). Cathepsin D antisera have been used to identify lysosomes by immunohistochemistry and electron microscopy (Roberg and Ollinger, 1998; Koike et al., 2000).
Rat monoclonal antibody 1D4B reacts specifically with mouse LAMP-1 (but not LAMP-2) by immunoblot (Andrejewski et al., 1999; Moreno, 2003), immunohistochemistry, and electron microscopy (Chen et al., 1985). LAMP-1 is a glycosylated protein that migrates at ~120 kDa by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Moreno, 2003).
Rat monoclonal antibody ABL-93 reacts specifically with the mouse lysosomal protein LAMP-2 (but not LAMP-1) by immunoblot (Andrejewski et al., 1999; Moreno, 2003) and by immunohistochemistry (Chen et al., 1985). LAMP-2 is a glycosylated protein that migrates at ~100 kDa by SDS-PAGE (Moreno, 2003).
Rat monoclonal antibody GL2A7 specifically reacts with the mouse lysosomal protein LAMP-2 by immunoblot (Saravanan et al., 2004) and by immunohistochemistry (Gondre-Lewis et al., 2004; Saravanan et al., 2007).
The rabbit polyclonal antiserum anti-LAMP-1 (Sigma L-1418) is specific to the C-terminal “tail” sequence of LAMP-1, which is essential for targeting the protein to lysosomes (Conesa et al., 2003) and is highly conserved across species (Granger et al., 1990). It is likely that this property allows this antiserum to specifically react with snake tissues. Immunostaining of both snake and mouse brain tissue produces nearly identical staining patterns as described above (data not shown). This antiserum specifically recognizes a protein of ~120 kDa by immunoblot and by immunohistochemistry (Yogalingam et al., 2008; Yogalingam and Pendergast, 2008; Mosser et al., 2008; Studer et al., 2010).
Drugs
All chemicals were from Sigma unless noted. Working concentrations of drugs used were as follows: bafilomycin A, 20 nM; Leu-Gly-B-naphthylamide, 0.5 mM; NH4Cl, 50 mM; nocodazole, 20 μM; colchicine, 40 μM; latrunculin A, 10 μM.
Cell culture
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen) plus 10% calf serum and antibiotics. N2a cells were maintained in Opti-MEM (Invitrogen) plus 10% calf serum plus antibiotics. Cells were grown on glass coverslips for live imaging as described above. LysoTracker staining was performed in regular media at 1:5,000 at 37°C. For immunohistochemistry, cells were fixed and stained as described above for snake muscle.
RESULTS
We previously studied the time course of ME formation and dispersion into vesicles via clathrin-mediated budding (Teng et al., 2007). We used chemical fixation to arrest activity at precise time points after brief stimulation. FM1-43 or other endocytic probes were then rinsed from the fixed tissue. Approximately four MEs were present 20 seconds after stimulation (30 Hz, 3 seconds) in an average bouton (each terminal contains ~58 boutons; Wilkinson and Lichtman, 1985). With continued budding, this number decreased rapidly to 1.2 at 30 seconds, and then more slowly, to 1.0 at 60 seconds. Here we studied these late MEs that remain after the budding process is largely complete. On average, 25 ± 11 MEs/terminal (mean ± SD; n = 16 terminals live-imaged in 4D), or ~0.43/bouton, were present throughout the observation period (5–60 minutes) which began 2–5 minutes after electrical stimulation (see Materials and Methods). Observed in time-lapse, late MEs either remain stable for the duration of imaging or appear to undergo one of two disparate fates: exocytosis, or apparent fusion with a separate population of endosomes that was previously unknown to us. We refer to the latter as acidic endosomes (AEs). Figure 1 shows MEs and AEs as they typically appear in a living motor terminal after brief stimulation. A time-lapse video of the same terminal (Supplementary Video S1) illustrates the slight movement of some MEs, the disappearance by putative exocytosis of two MEs (arrows in Fig. 1; see also Fig. 2), and the typical directed movement of some AEs. These and other features of late MEs and of AEs are described below.
Figure 1.
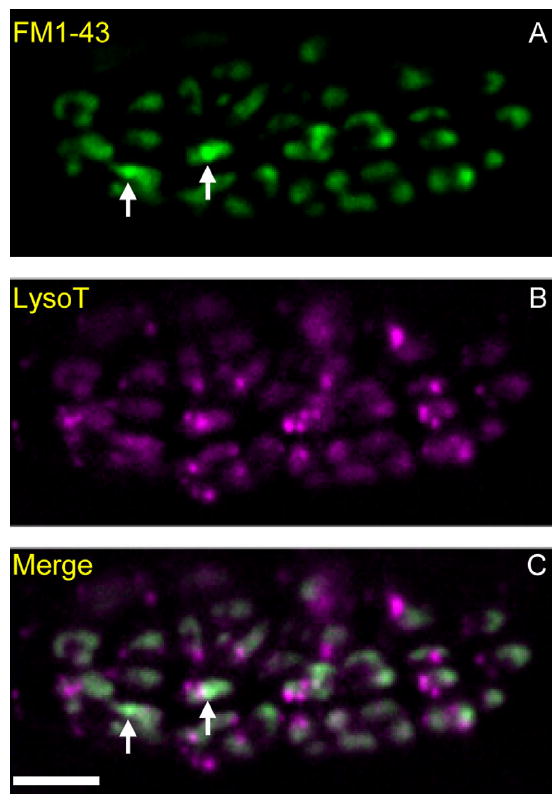
Macroendosomes (MEs) and acidic endosomes (AEs) in a snake motor nerve terminal. A–C: Preparations were incubated with LysoTracker (B; magenta), rinsed, and then electrically stimulated with bath-applied FM1-43 (A; green; C is merged). Punctate acidic endosomes (AEs, magenta in B and C) are visible throughout the terminal. Two macroendosomes (MEs, green) are marked with arrows in A and C. Background FM1-43 “haze” (green) obscures MEs and is due to recently endocytosed 50-nm vesicles that also contain FM1-43. Scale bar = 10 μm in C (applies to A–C).
Figure 2.
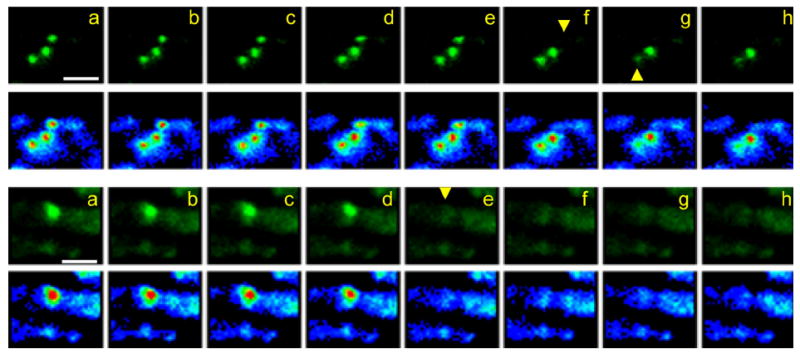
MEs vanish between sequential time-lapse frames. Shown are two examples of living terminals stimulated with bath-applied FM1-43 as in Figure 1. a–h, Top: Time-lapse views of consecutive frames from a 4D sequence (interval, 40 seconds), are presented from left to right. Images are compressed from z-stacks (six image planes, 1.5-μm spacing) and magnified such that only one of ~60 terminal boutons is in view. Each sequence is shown normally (green; upper images in each pair) and in pseudocolor (red, yellow, green, blue, purple in order of decreasing brightness). Yellow arrowheads indicate disappearance of two MEs whereas a third, between them, remains. a–h, Bottom: A single ME disappears from a bouton. Recording parameters are as described above except frame interval is 1 minute. Scale bar = 5 μm in a (applies to a–h).
Evidence for exocytosis of MEs
We observed late MEs over various times (10–60 minutes, beginning 2–5 minutes after electrical stimulation (30 Hz, 18 seconds) or KCl depolarization (60 mM, 2 minutes). Data presented below are from 16 nerve terminals (16 muscles from 10 snakes). All were electrically stimulated in an FM1-43–containing bath (30 Hz, 18 seconds). For each terminal, 4D time-lapse sequences were collected for 50–60 minutes. A total of 398 MEs were visualized via FM1-43 fluorescence at the beginning of data collection. We recorded the disappearance of 51 MEs or 13% of MEs initially present. Thus on average, late MEs disappear from a terminal at a rate of ~4/h. Disappearances in KCl-stimulated preparations and/or in single-focal-plane time lapse sequences were similar (data not shown).
An ME that was fully visible in one time-lapse frame (and previous ones) was completely absent from the subsequent frame (Fig. 2; see also Fig. 3, Supplementary Videos S1 and S2). Thus the time required for an ME to disappear is likely to be small compared with our frame intervals (10–120 seconds). We saw no evidence for ME bursting, which would increase intraterminal Ca2+ as stored extracellular fluid was released. There was no movement suggesting muscle fiber contraction due to release of transmitter, nor change in terminal appearance. Because optical data at each time point was recorded as a 3D image stack, the entire thickness of the terminal was in view. It is therefore unlikely that an ME vanished because it left the plane of focus. In contrast, because z-axis resolution is limited even after deconvolution, MEs appeared to occupy most or all of the thickness of the bouton that contained them. Thus we could not determine whether MEs moved toward the bouton’s membrane in the z-direction before they disappeared, nor could we confirm that an ME about to disappear was definitely not near the membrane. Occasionally, an ME was seen to move predominantly in the x–y plane of focus and then disappear at the edge of a bouton, consistent with exocytosis (Fig. 3; Supplementary Video S2).
Figure 3.
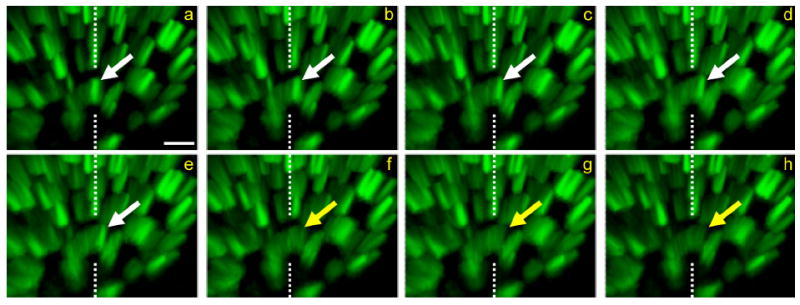
Exocytosis of MEs continues long after stimulation. Time-lapse sequence of disappearing ME. The preparation was briefly stimulated with bath-applied FM1-43. a–h: Eight sequential frames from a 4D data set (six image planes, 1.5-μm spacing, 1-minute interval). The arrow follows the lateral movement and disappearance (indicated by change of arrow color from white to yellow) of one ME between frames e and f, approximately 55 minutes after stimulation. The dashed lines reference the position of the ME in frame a. At light-level resolution, the ME disappears as it contacts the membrane, consistent with exocytosis. The terminal is viewed obliquely (Imaris software) so that both the x-y plane and the z-axis depth can be appreciated. Scale bar = 1 μm in a (applies to a–h).
In our previous study using fixed tissue, we observed that some MEs persisted for 30 minutes after stimulation, with decline in ME number appearing to reach a plateau of ~0.3/bouton (Teng et al., 2007). Live imaging results confirm this and extend the observation period to 1 hour. The time course of ME disappearance pooled from all 4D experiments with electrical stimulation is shown in Figure 4A. The data were fit best (R2 = 0.99; runs test P = 0.80) by a model in which the number of remaining MEs decayed exponentially (time constant, 58 minutes) to a plateau of 0.33 MEs/bouton. Data were fit less well by a single exponential model in which ME numbers were assumed to reach 0/bouton (R2 = 0.98; runs test P = 0.30; time constant, 377 minutes; Fig. 4B). We also tested and rejected linear and double-exponential fits to the time course of ME disappearance.
Acidic endosomes
Synaptic vesicles of various types are reported to be about pH 5.7 (Miesenböck et al., 1998). Presumably, MEs contain some vesicular proton pumps and are acidic as well. To test whether late MEs in nerve terminals might acidify further, as do similar structures in cell bodies (late endosomes; pH < 5.5), we stained stimulated preparations with the pH-sensitive supravital dyes LysoTracker, Neutral Red, and Acridine Orange. Without exception, MEs were not labeled by these dyes, nor were synaptic vesicles. We assumed that either their pH was not sufficiently acidic to stain with the probes we used, or some other feature of these structures precluded staining. Lysosomes, for example, which are the intended target of LysoTracker, have a pH about one unit lower than synaptic vesicles (pH 4.5–4.8; Ohkuma and Poole, 1978). However, we were surprised to observe with all three dyes a large population of previously unknown, brightly labeled acidic structures, which, as noted above, we call acidic endosomes (AEs; Figure 5). Each terminal contained 48.3 ± 11.7 (mean ± SD; n = 21) AEs, a number that did not change significantly after 2 minutes of depolarization with 60 mM KCl (40.7 ± 14.4; n = 18; P = 0.19; see Fig. 8A). The structures are labeled due to their acidity and not due to some artifact. Neutralization of pH by bath application of ammonium chloride destained LysoTracker-labeled AEs (Fig. 6B), as did application of bafilomycin, a specific inhibitor of proton ATPases (Bowman et al., 1988; Fig. 6C). Neither agent affected the FM labeling of MEs or vesicles (arrowheads in Fig. 6A,B).
Figure 5.
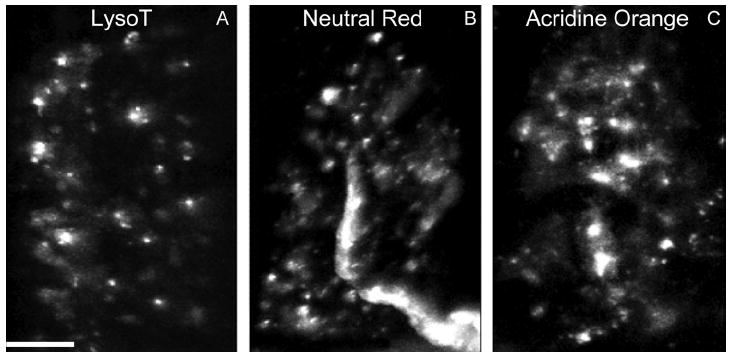
AEs are recognized by conventional supravital acidophilic probes. A–C: Living snake nerve–muscle preparations were incubated with Neutral Red (B) or Acridine Orange (C; both 1 μg/ml, 30 minutes), followed by washing with reptilian saline and imaging as in Figure 4. Characteristics of the small puncta (white) stained by the dyes are similar to LysoTracker-positive structures (AEs) (compare with A; see also Fig. 1B). Scale bar = 10 μm in A (applies to A–C).
Figure 8.
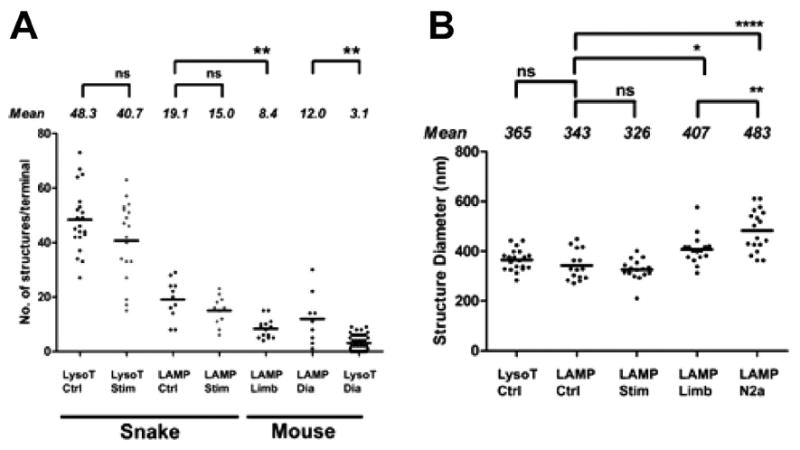
Properties of AEs in snake and mouse terminals. A: The number of living AEs in both snake and mouse (see Fig. 11) terminals decreased slightly, but not significantly, after depolarization of the terminal with KCl (Stim) (60 mM, 2 minutes). AEs in living snake terminals were more numerous than detectable LAMP-1–positive structures viewed after fixation. In contrast, detectable living AEs in mouse terminals (limb and diaphragm) were less numerous than LAMP-1–positive structures (see text). Comparisons marked ** were significant (P = 0.001). B: Size of snake AEs viewed at light level did not differ significantly between living (LysoT) and fixed (LAMP) preparations. Size did not change significantly after KCl depolarization (Stim; 60 mM, 2 minutes). Both snake and mouse (LAMP Limb) AEs were significantly smaller than anti-LAMP-1–positive lysosomes in N2a (mouse neuroblastoma) cells (****, P < 0.0001; **, P = 0.008; *, P = 0.01; ns, not significant). Means for each condition are indicated by solid lines.
Figure 6.
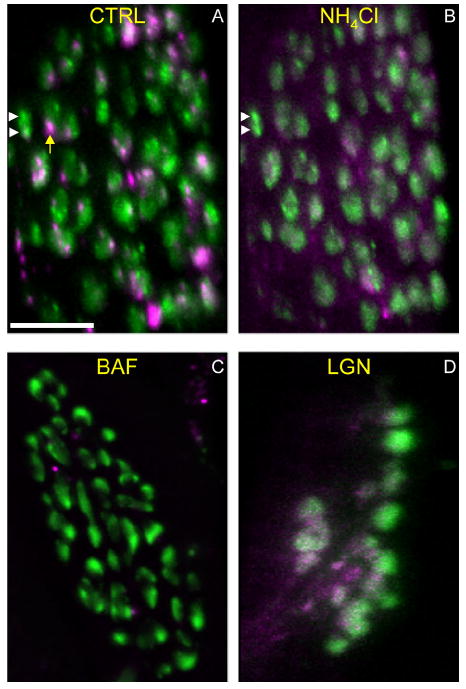
AEs are deacidified by agents that disrupt lysosomal function. Preparations were incubated with LysoTracker and then depolarized with potassium chloride (KCl; 60 mM, 3 minutes) in the presence of FM1-43. A: Control (CTRL) showing MEs (green; white arrowheads) and AEs (magenta; yellow arrow) in a typical terminal. Green background is 50-nm vesicles endocytosed during the stimulation. B: The same preparation in A was treated with ammonium chloride (NH4Cl; 50 mM, 15 minutes) and reimaged. The LysoTracker signal is largely abolished, indicating pH increase or other disruption within AEs. MEs (white arrowheads) and vesicle haze were unaffected by the treatment. C,D: Additional preparations were incubated with LysoTracker and FM1-43 as above, and were then incubated with 20 nM Bafilomycin (BAF; C) or 0.5 mM Leu-Gly-β-naphthylamide (LGN; D) for 30 minutes and reimaged in the presence of the drug. Most of the LysoTracker-positive structures (AEs) were no longer visible. Each image is compressed from a z-stack (maximum-z projection). Scale bar = 10 μm in A (applies to A–D).
The AE compartment
Synaptic vesicles and mitochondria are segregated into separate regions, or compartments, of living snake boutons (Lichtman et al., 1989). The compartment occupied by most AEs is distinct from both. It includes (mainly) the axon, axon connectives, and regions of boutons that seem to be near connectives. In living terminals, the separation was apparent by comparing the vesicular compartment (labeled with FM1-43) and the location of most AEs (labeled with LysoTracker; Fig. 7B). We performed similar comparisons of the mitochondrial compartment (labeled with 4-Di-2-ASP; Lichtman et al., 1989) with the AE compartment to confirm their separation and to confirm that LysoTracker does not stain mitochondria (data not shown). After testing LysoTracker as well as several antibodies (see below) we found that one antibody (an anti-LAMP-1 lysosomal marker) cross-reacted with snake and stains AEs with relatively low background in fixed preparations. Figure 7D-F shows a comparison of the AE compartment with the vesicular compartment (immunostained for the vesicular protein SV2) after fixation. The distribution of LAMP-1-positive structures resembled that of supravitally stained AEs, as did both the number (Fig. 8A) and size (Fig. 8B) of the structures.
Some AEs move on microtubules
Approximately 15% of AEs moved perceptively under our time-lapse conditions. The average speed of an AE in motion within snake terminals was 81 ± 51 nm/s (n = 38; Fig. 9A). The fastest AE speed recorded was 201 nm/s. The motion of AEs was often back-and-forth, along straight or curved lines as if on tracks (Supplementary Video S3). This is in contrast to the more random (Brownian) motion of MEs (see Supplementary Videos S1, S2). AE movement in straight lines or gentle curves was about equal in both directions, whether within boutons or within connectives that joined boutons. Within the axon, however, movement favored the retrograde direction (75% retrograde, 25% anterograde; n = 55 AEs in five axons). As shown in Figure 9A, velocity of anterograde axonal movement was similar to that of directed movement within the terminal whereas retrograde axonal movement was significantly faster. Treatment of preparations with colchicine (40 μM, 2 hours) or nocodazole (20 μM, 2 hours; Fig. 9B), agents that disrupt microtubules in mammals, reversibly arrested movement of some, but clearly not all AEs. The effect of these treatments was more easily seen among AEs in axons than in terminals. The remaining movement was largely random, but a substantial amount of track-like movement persisted. We found that AE movement also persisted after cooling (4°C, 12 hours). Thus, although mammalian microtubules are disrupted by cold (Banks et al., 1975), reptilian microtubules are evidently less sensitive (Pannese et al., 1982). This structural difference might also convey some immunity to disruption by drugs that target mammalian microtubules. In control experiments with N2a cells, directed movement of LysoTracker-positive vesicles (presumed lysosomes) was largely abolished by nocodazole and colchicine treatment (data not shown).
Figure 9.
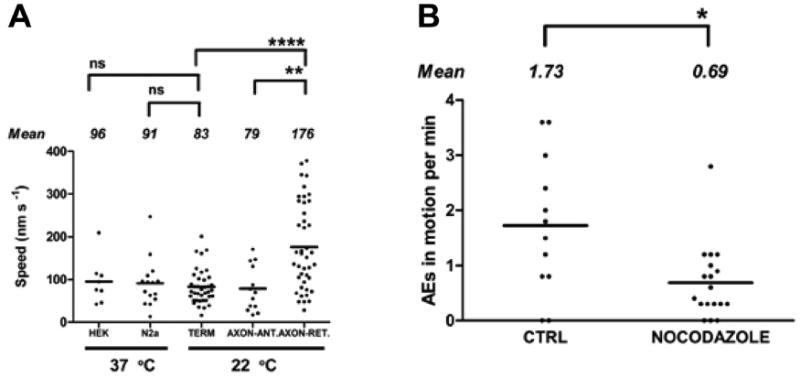
AE velocities in axons and terminals. A: Time-lapse sequences were analyzed to determine distance traveled per unit time. Directed movement of AEs within snake terminals (TERM), and within the motor axon moving toward the terminal (AXON-ANT), did not differ significantly from directed movement of lysosomes within HEK (human embryonic kidney) and N2a cells. Movement of AEs within the axon and directed toward the cell body (AXON-RET) was significantly faster (****, P < 0.0001; **, P = 0.002; ns, not significant). B: Treatment with nocodazole (20 μM, 2 hours) partially inhibits movement of AEs. The number of moving AEs (shown normalized per minute of observation) observed over identical 20X 10-seconds time-lapse records in treated versus control preparations decreased significantly (*, P = 0.02). Means for each condition are indicated by solid lines.
Application of fluorescently labeled taxol to the bath (1 μM) revealed a pattern of putative microtubules that occupied the same region of terminals that was occupied by AEs. Some of the AEs colocalized with taxol-stained microtubules in our experiments (Fig. 10C) and appeared to move along them (Supplementary Video S4). Bath application of latrunculin A (15 μM) had no discernable effect on AE movement. This agent disrupts actin filaments in snake motor terminals and elsewhere (Cole et al., 2000).
Figure 10.
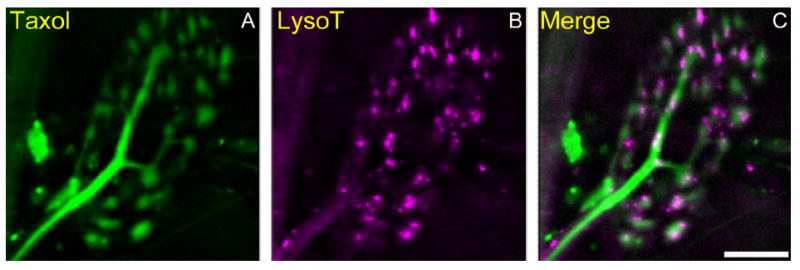
Some but not all AEs are associated with microtubules. A–C: Living snake terminals were incubated with Oregon green 488–taxol (A; green) and LysoTracker (B; magenta) and imaged as in Figure 6 (maximum z projection). Taxol-labeled tubulin was most visible within the axon and within thin connectives between terminal boutons. Note the close association of several AEs with the tubulin arbor (C; merged). Scale bar = 10 μm in C (applies to A–C).
AEs resemble lysosomes
To help characterize snake AEs, we used the mouse diaphragm as a positive control for our observations in snake, and mouse N2a cells to compare AEs with known lysosomes. LysoTracker stained acidic structures (AEs) in living mouse motor terminals that resemble those in snake, although the imaging was much poorer (Fig. 11B). We also noted larger acidic structures that were associated with neuromuscular junctions (NMJs) but located in other cells (presumably Schwann cells and muscle; arrows in Fig. 11B,E). These structures were not seen in snake. We screened antibodies known or thought to react with mouse lysosomes by using mouse N2a cultured neuronal cell lines. We then tested antibody reactivity of both snake and mouse motor terminals. Four antibodies against lysosomal proteins (one anti-LAMP-1, two anti-LAMP-2, and anti-Cathepsin D) labeled LysoTracker-positive structures in the mouse cell lines (Fig. 12B,C). Although only one antibody cross-reacted with snake (an anti-LAMP-1; Fig. 7E), all of these antibodies labeled AEs in mouse motor terminals as well (Figs. 11E, 12D-G), indicating that mouse AEs contained lysosomal proteins. Compared with snake, there were fewer LysoTracker-positive AEs at the living mouse NMJ that were definitely located within the motor terminal (3.1 ± 2.9; n = 45 terminals; Fig. 8A). The number of LAMP-positive structures in fixed mouse limb terminals viewed at confocal resolution was similarly smaller than the number in fixed snake terminals, although the difference was only weakly significant (P < 0.01; Fig. 8A). We consider it likely that poor visualization of living mouse terminals contributed to this discrepancy.
Figure 11.
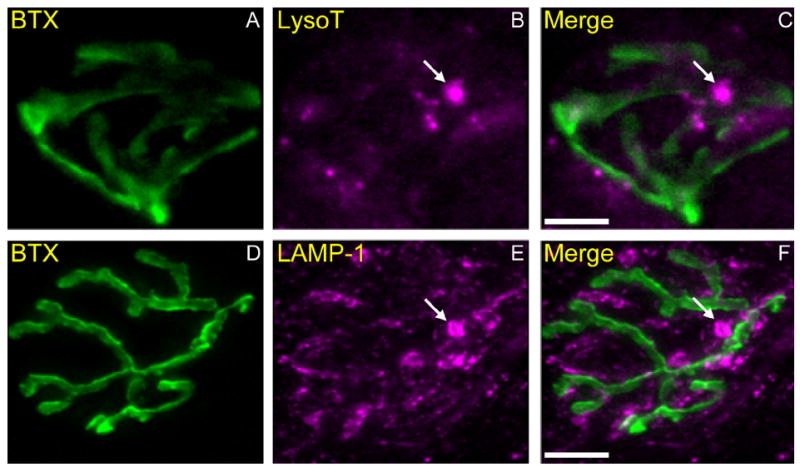
AEs are found within mouse motor terminals. A–C: Neuromuscular junctions (NMJs) in living mouse diaphragm incubated with 488-Bungarotoxin (BTX; 0.5 μg/ml, 30 minutes, 37°C) to label postsynaptic acetylcholine receptors (nAChRs) that appose the vesicular compartment of the terminal (A; green). The preparation was simultaneously incubated with LysoTracker (B; magenta), and then imaged as in Figure 4 (maximum z projection). Several small LysoTracker-positive AEs are visibly aligned with AChRs and presumably within the terminal (C; merged). In addition, there are occasional much larger acidic structures within or adjacent to the terminal that are absent from snake NMJs (arrow in B and C; compare with Fig. 7B). D–F: NMJs in fixed mouse muscle (gastrocnemius) immunostained with 488–bungarotoxin (D; green) and LAMP-1 (Sigma) antibody (E; magenta) as in Figure 7D-F. Note the presence of very large (>1-μm diameter) LAMP-1–positive structures (arrows in E and F) near the NMJs, in addition to more regularly sized (300–500 nm) LAMP-1–positive structures. Size and number distributions of mouse AEs are shown in Figure 8. Scale bar = 10 μm in C (applies to A–C) and F (applies to D–F).
Size distributions of AEs and lysosomes are compared in Figure 8B. Measured sizes were near the diffraction limit, ranging from 200 nm to 600 nm. Snake AEs were of virtually identical size on average whether imaged supravitally with LysoTracker or in fixed tissue with immunofluorescence (365 ± 43 nm, mean ± SD; n = 20; Fig. 8B). Size of AEs were similar in snake (363 ± 99 nm; n = 15) and mouse limb terminals (407 ± 60 nm; n = 16), but both were significantly smaller than lysosomes in mouse N2a cells (483 ± 83 nm; n = 18; Fig. 8B). Movement of lysosomes in N2a and HEK cells was also similar to that of snake AEs (Fig. 9A), excepting AEs within axons as described above. Directed velocities of LysoTracker-stained structures did not differ significantly among snake AEs (81 ± 51 nm/s, n = 18), mouse N2a lysosomes (91 ± 56 nm/s, n = 15), and HEK lysosomes (96 ± 50 nm/s, n = 18) (Fig. 9A).
We also tested the specific inhibitor of lysosome function LGN (Leu-Gly-B-naphthylamide; Jadot et al., 1990). This drug diminished or eradicated acidophilic staining of lysosomes in mouse N2a cells as well as AEs in both mouse (data not shown) and snake terminals (Fig. 6D). Thus there is at least partial similarity of hydrolytic function between lysosomes and AEs. LGN had no effect on FM-labeled MEs.
Fusion or association of late MEs with AEs
As noted above, most MEs and AEs occupied separate compartments within terminals (Fig. 7). However, we saw several examples of close proximity between the two structures, often with AEs moving near or even circling MEs. We also observed double-labeled structures that indicated either fusion events between AEs and MEs or very close association at distances below the diffraction limit. An example is in Figure 13. Only a small part (4 of ~60 boutons) of one nerve terminal is shown. One of two AEs present in the field had colocalized with an ME (white arrow in Fig. 13C). By utilizing software to view the 3D image stack from any position, we confirmed that the same structure contained both labels (see Fig. 13 legend). Further description of our method, plus another example of a putatively fused ME–AE, is given in Figure 14. All 3D images of double-labeled structures such as those in Figures 13 and 14 showed precise overlap at light resolution, suggesting fusion. However, because the sizes of MEs and AEs are near the optical diffraction limit, it remains possible that the structures were intertwined in some way so that they appeared fused. We consider this unlikely, because MEs reconstructed from serial EM sections exhibit fairly regular shapes (Teng et al., 2007), as do conventional lysosomes.
Figure 13.
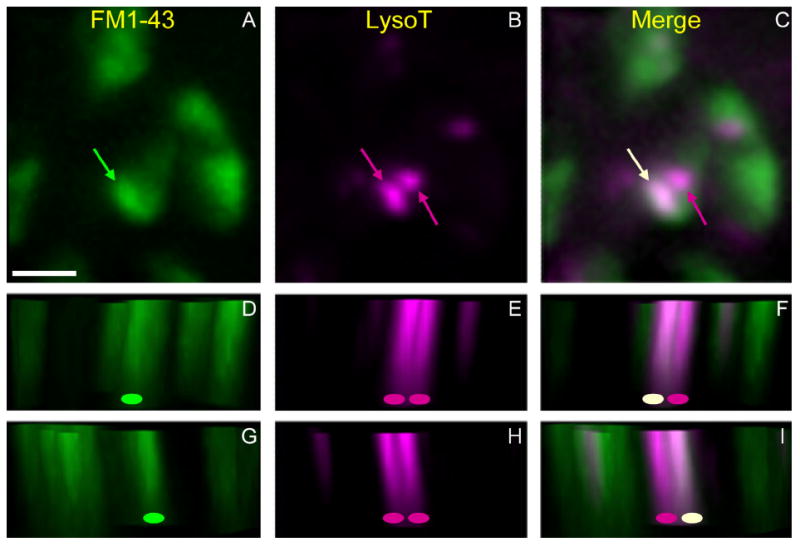
Some MEs internalized by stimulation appear to fuse with preexisting AEs. Preparations were incubated with LysoTracker to label AEs (magenta), rinsed, and then stimulated with FM1-43 to create and label MEs (green). Shown is a small region of one terminal (four boutons) imaged in three dimensions (eight image planes, 0.55 μm per plane). A–C: Normal x–y view. The compressed image stacks show vesicle haze, one FM1-43-labeled structure (A, green arrow), and two LysoTracker-labeled structures (B, magenta arrows). Merged image (C) reveals that ME and leftmost AE are fused (white arrow; magenta arrow is the other AE). D–I: lateral views of image stack with z-axis vertical (green oval, ME; magenta oval, AE; off-white oval, fused; see Materials and Methods). The view is of parallel slices along a diagonal line, from the lower left to the upper right in x–y panels and either just below (D–F) or just above (G–I) the fused endosome. Note colocalization of green and magenta voxels (white or near-white) for the fused endosome when viewed from either direction. Scale bar = 5 μm in A (applies to A–I).
Figure 14.
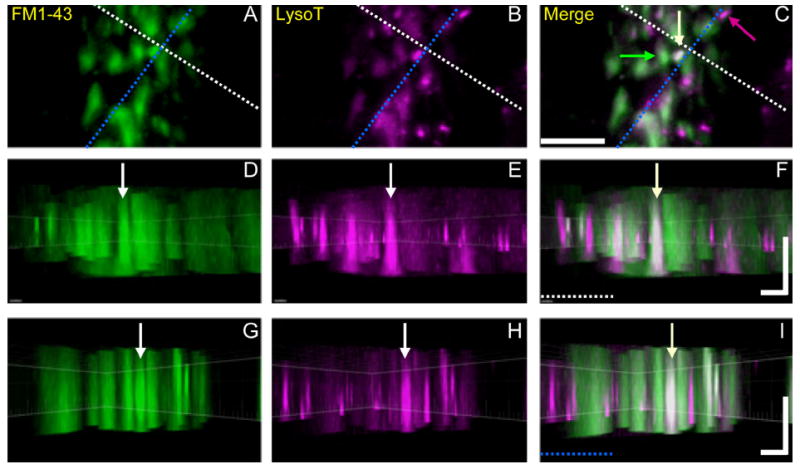
Orthogonal views of voxels representing putative ME-AE fusion events. Snake nerve–muscle preparations were incubated in LysoTracker and stimulated with FM1-43 and imaged as in Figure 13 (13 image planes, 0.55 μm per plane). A–C: Conventional views show part of a nerve terminal whose boutons contain MEs, AEs, and at least one putative fusion event (white arrow in C). The orthogonal dashed lines just above (white) and to the right (blue) of the yellow endosome indicate the orientation of z-slices shown in the side-view panels below (note dashed colored lines in F and I), and also indicate the location of the fused endosome as it appears in the left (FM1-43, green) and center (LysoTracker, magenta) panels. D–F: Slice view in a plane whose horizontal dimension is along the white dashed line in A–C, and whose vertical dimension is along the z-axis. G–I: Slice view along the blue line in A–C. Thus each vertical column depicts the fused endosome (vertical arrows) in all three orthogonal planes, indicating that magenta and green voxels are colocalized (white or near-white). Image was processed by using Imaris Bitplane software (see Materials and Methods). Scale bar = 5 μm in C (applies to A–C); 2 μm (horizontal) and 3 μm (vertical) in F (applies to D–F) and I (applies to G–I).
We have not yet observed endosomes confirmed to be AEs at EM level. We confirmed in time-lapse recordings that double-labeled structures persisted in all frames of the movie and did not separate. Double-labeled structures were relatively rare compared with the overall population of MEs (25/terminal; see above). We observed a total of only 15 in 98 terminals (1.5/terminal) stimulated and held at 22°C, and 11 in 71 terminals (1.5/terminal) that were stimulated and held at 4°C to decrease background haze from vesicles (see Materials and Methods).
Double labeling indicated ME–AE fusion or association in our protocol because neither dye had access to endosomes of both types. We labeled acidic structures first, followed by rinsing. Replacement of the bath with pH 5.0 reptilian saline after rinsing, to promote LysoTracker fluorescence, revealed no visible LysoTracker staining of the plasma membrane (data not shown). Thus visible quantities of LysoTracker did not adhere to the membrane after rinsing, and therefore could not have been endocytosed along with FM1-43 when boutons were subsequently depolarized to create MEs. Conversely, although some MEs might have been present before stimulation (see Discussion), they did not have access to bath-applied FM1-43 (Teng et al., 2007; Gaffield et al., 2011). As expected, each apparently fused structure we observed was stained with FM1-43 and LysoTracker, respectively, with the same brightness as typical unfused MEs and AEs. Although it is possible that fusion or association occurred between AEs and FM-labeled 50 nm vesicles, these structures would contain far less FM1-43 and probably not be detected. With some assumptions, we estimate that fusion of 100–1,000 vesicles with a single AE would be required to introduce the same quantity of FM1-43 as a single ME. Therefore, we conclude that each double-labeled living structure was a recently formed ME containing FM1-43 that, after internalization, fused or associated with a preexisting AE containing LysoTracker. We failed to observe fusion/association of MEs with AEs as events occurring between time-lapse frames, despite hours of observation time. Those that we observed had evidently occurred before recording began, during the period of vesicle budding. The earliest observations possible, limited by rinsing time to remove FM1-43, were 2–5 minutes after completion of stimulation or KCl depolarization.
DISCUSSION
We have used 4D deconvolution imaging of motor nerve terminals to characterize the behavior and interaction of two classes of endosomes: macroendosomes (MEs), internalized by bulk endocytosis, and acidic endosomes (AEs), a class of permanent organelles that we describe for the first time. We conclude that bulk endocytosis serves as the entry point for at least three processes. One is to assist membrane CME via additional vesicle fission from MEs (e.g., Richards et al., 2000; Teng and Wilkinson, 2000). The second, not previously observed in quiescent terminals, is fusion of MEs with the plasma membrane. These exocytic events continue to occur for up to at least an hour after activity and vesicle budding have ceased. The third process occurs mostly or exclusively <2–3 minutes after stimulation: some MEs (and perhaps some vesicles as well) fuse or associate with AEs, a class of organelles that resemble lysosomes. Based on these observations, we suggest that some (late) MEs do not dissipate into vesicles but remain to serve as sorting endosomes.
Exocytosis of MEs
In our previous study of MEs in fixed preparations, the number of MEs per bouton increased and stabilized during stimulation, and then decreased rapidly over time for ~1 minute after stimulation was terminated. In the present study, only those MEs that remained ~3 minutes or more after stimulation were visualized in living terminals because of the time required to rinse FM1-43 from the bath. Exocytic events were inferred from the disappearance of MEs between time-lapse frames.
Exocytosis of MEs has been reported in goldfish retinal bipolar terminals by using evanescent field microscopy (Coggins et al., 2007). The MEs in that study were created (and loaded with FM1-43) by bulk endocytosis during intense and prolonged stimulation, and their subsequent exocytosis was observed during an additional round of stimulation. In snake, MEs are also continuously formed during long-duration stimulation whereas others dissipate into vesicles and vanish, presumably via exocytosis (Teng et al., 2007). However, Coggins et al. (2007) provided evidence that ME exocytosis in their preparation was stimulus-induced, suggesting a mechanism similar to the exocytosis of vesicles. In contrast, the ME exocytosis reported here was not directly triggered by stimulation, and was observed at times well after intracellular Ca2+ levels had returned to baseline (Teng and Wilkinson, 2003).
Putative exocytosis of MEs has also been reported in Drosophila. In motor terminals, loss-of-function mutations in proteins associated with CME promote a shift toward increased bulk endocytosis (Zhang et al., 1998; Koh et al., 2004). Large vesicles (endosomes) of various sizes are seen in EM. The vesicles can be exocytosed and contain transmitter, as evidenced by large spontaneous endplate potentials. These observations support our evidence for ME exocytosis, provided that the mutated proteins that inhibit CME (the AP180 homolog LAP [Zhang et al., 1998] and the intersectin homolog dap160 [Koh et al., 2004]) also inhibit analogous clathrin-mediated budding from MEs. Without budding, vesicle membrane proteins responsible for acidification and transport of transmitter would remain in the ME, allowing it to accumulate transmitter and secrete it when exocytosed. Combining our data with data from Drosophila mutants suggests that MEs undergo exocytosis regardless of whether or not budding is permitted to occur.
Acidic endosomes
We found no substantial difference between snake and mouse AEs. The number, size, and size distributions of AEs in snake and mouse terminals are similar. Both contain the lysosomal proteins LAMP-1, and both are disrupted by LGN, a specific lytic agent for lysosomes (Jadot et al., 1990). Mouse AEs contain LAMP-2 and Cathepsin D, a lumenal lysosomal protease; these proteins are either absent in snake AEs or, more likely, our antibodies did not cross-react.
AEs de- and reacidify after reagents are bath-applied in a manner identical to that of lysosomes in cultured cells. AEs and lysosomes in cells move at similar velocities, but a larger fraction of AEs, compared with lysosomes, is stationary at a given time. Consistent with this, tubulin is present in the AE compartment of terminals, but only a fraction of AEs is associated with it. Moreover, the microtubule-disrupting agents nocodazole and colchicine have more limited effects on AEs than on lysosomes in cultured cells. Within terminals, AEs move approximately equally in both directions within and between boutons, similar to the bidirectional movement of lysosomes. Within the axon, however, retrograde movement is about three times as prevalent and twice as fast as anterograde movement. This finding suggests that the complement of motors carried by AEs favors the retrograde direction, a property not comparable to lysosomes within cell bodies.
Lysosome-like vesicles in motor nerve terminals are likely to be specialized. Some products of lysosomal hydrolysis might be recycled, but others, such as amino acids, could presumably serve only as an energy source for mitochondria unless they are transported to the cell body. Homeostatic functions, such as preparation of molecules for axonal transport, have been associated with lysosomes in nerve terminals (e.g., delivery of trophic factors via endolysosomes; Zhang et al., 2009). The speed of retrograde AE movement in snake at room temperature is comparable to or somewhat slower than that associated with fast axonal transport in mammals at 37°C. We did not study AEs in the axon once they moved away from terminals.
MEs and vesicle reformation
As discussed above, two observations suggest that the role of MEs extends beyond that of providing “overload protection” at high levels of transmitter release. First, MEs are formed even with low levels of release. Second, some (“ late”) MEs remain long after vesicle reformation is complete. One possibility is that MEs are analogues of sorting endosomes found in cell bodies. Permanent endosomal intermediates, or cisternae, have been proposed to serve this function (Heuser and Reese, 1973). However, most cisternae are in fact MEs created by bulk endocytosis (Miller and Heuser, 1984; Teng and Wilkinson, 2000; reviewed by Clayton and Cousin, 2009). Moreover, the speed with which spent vesicles are returned to the releasable pool indicates that at least most vesicles are competent after reformation by CME alone, without need for endosomal intermediates (Takei et al., 1996). There is recent evidence of how this is done, despite the complexity of the vesicle proteome (Takamori et al., 2006). In particular, CME adaptors select vesicle proteins over membrane proteins not just by virtue of affinity but because vesicle proteins are both concentrated and segregated near active zones where both exo- and endocytosis occurs (Teng et al., 1999; Tabares et al., 2007; Hoopmann et al., 2010).
Nevertheless, it seems unlikely that CME can provide repeated error-free recycling, and a parallel endosomal pathway has therefore been re-proposed (reviewed by Shupliakov and Brodin, 2010). Evidence for conventional sorting endosomes already exists for invertebrate (Drosophila) motor terminals (Uytterhoeven et al., 2011). Sorting endosomes are also present in rat hippocampal neurons and synaptosome fractions. Synaptic vesicles can apparently fuse with them (Rizzoli et al., 2006; Hoopmann et al., 2010), suggesting that a similar scheme exists within intact nerve terminals. Notably, in both of the above preparations, the endosomes described are inferred to be a separate population from MEs. In Drosophila, putative sorting endosomes contain Rab5, a marker of vesicle–endosome fusion that is lacking in Drosophila MEs. In rat hippocampus, some synaptic vesicles contain SNARE proteins that are appropriate for fusion with endosomes, as opposed to fusion with the membrane as required for exocytosis (see also below).
Although there is no evidence for permanent sorting endosomes in vertebrate motor terminals, our data suggest that late MEs may play a similar role. The time course of ME exocytosis after stimulation reaches a non-zero plateau (~20 MEs/terminal) after ~1 hour. We also fit data from previous fixed tissue experiments (Fig. 8D in Teng et al., 2007) and found a similar exponential decay plateau of ~20 MEs/terminal, not zero. We propose that these persistent late MEs are semipermanent sorting endosomes that retain the capacity to dissipate and exocytose when the terminal becomes active, perhaps hours later. In this context, exocytosis of MEs is one sorting function, whereas fusion/association events between MEs and AEs potentially mediate two others—degradation and axonal transport. Analogous fusion occurs between conventional late endosomes and lysosomes (e.g., Mullock et al., 1998), whereas a recent model derived from observations in Drosophila Sky mutants suggests interaction between sorting endosomes and apparent lysosomes in motor terminals (Uytterhoeven et al., 2011). We propose that a third, putative function is re-formation of vesicles in parallel with CME using late MEs, not dedicated sorting endosomes, as intermediates.
Physiological and molecular biological evidence from a variety of synaptic preparations supports the above view. At some synapses, vesicles budded from MEs preferentially enter a reserve pool with low release probability, whereas those budded directly from the membrane enter the releasable pool (Richards et al., 2000; reviewed by Wilkinson and Cole, 2001). If MEs serve only as an additional pathway for budding vesicles, these release probabilities would not be expected to differ. Indeed, the association of ME formation with high activity is itself paradoxical, given that more, not fewer, viable vesicles are needed for immediate reuse. An alternative interpretation is that high activity and consequent rapid recycling increases CME error rate and thus increases need for correction via a parallel sorting pathway. That pathway is enhanced by formation of additional MEs to augment those already present (late MEs). Correction could begin by tagging vesicles budded from MEs with SNAREs or other proteins appropriate for endosome fusion with the same or another ME. Consistent with such tagging, low release-probability vesicles budded from MEs in frog motor terminals are not anatomically segregated from releasable ones, and therefore must differ biochemically (Rizzoli and Betz, 2004). Indeed, some synaptic vesicles contain proteins associated with trafficking (Takamori et al., 2006), and certain clathrin adapters are specialized to participate in budding from endosomes but not from the plasma membrane (Blumstein et al., 2001; Voglmaier et al., 2006; Glyvuk et al., 2010).
Diversity among v-SNARES also suggests endosomal sorting, although not specifically by MEs. Endosome-specific v-SNAREs have been detected in synaptosomes (Hoopman et al., 2010). Similar SNARE composition differences exist between releasable and reserve pool vesicles in cultured rat hippocampal neurons (Hua et al., 2011). Presumably, v-SNARES associated with the releasable pool promote exocytosis, whereas reserve pool v-SNARES could mediate vesicle fusion with late MEs for error correction before reentry into the releasable pool. Further work is needed to determine whether these and other required proteins are associated with MEs in vertebrate motor terminals.
Supplementary Material
Acknowledgments
We thank J. Huettner, L. Xia, and J. Lange for providing cells, R. Roth and J. Heuser for consultation regarding electron microscopy, and G. Grajales (Confocal Imaging Facility, University of Puerto Rico) for 4D image rendering and analysis.
Grant sponsor: National Institutes of Health; Grant number: NS-024752 (to R.S.W.).
Footnotes
Additional Supporting Information may be found in the online version of this article.
Study concept and design: R.S., H.T., and R.W. Collection, analysis and interpretation of data: R.S., H.T., and R.W. Drafting of the manuscript: R.S. and R.W. The experiments were performed in the laboratory of R.W. at Washington University (St. Louis, MO). All authors approved the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors have no competing interests to declare.
LITERATURE CITED
- Agulhon C, Rostaing P, Ravassard P, Sagne C, Triller A, Giros B. Lysosomal amino acid transporter LYAAT-1 in the rat central nervous system: an in situ hybridization and immunohistochemical study. J Comp Neurol. 2003;462:71–89. doi: 10.1002/cne.10712. [DOI] [PubMed] [Google Scholar]
- Andrejewski N, Punnonen E-L, Guhde G, Tanaka Y, Lullmann-Rauchl R, Hartmann D, von Figura K, Saftig P. Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem. 1999;274:12692–12701. doi: 10.1074/jbc.274.18.12692. [DOI] [PubMed] [Google Scholar]
- Banks P, Mayor D, Owen T. Effects of low temperatures on microtubules in the non-myelinated axons of post-ganglionic sympathetic nerves. Brain Res. 1975;83:277–292. doi: 10.1016/0006-8993(75)90936-1. [DOI] [PubMed] [Google Scholar]
- Blumstein J, Faundez V, Nakatsu F, Saito T, Ohno H, Kelly RB. The neuronal form of adaptor protein-3 is required for synaptic vesicle formation from endosomes. J Neurosci. 2001;21:8034–8042. doi: 10.1523/JNEUROSCI.21-20-08034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonen M, van Meel E, Oorschot V, Klumperman J, Kornfeld S. Vacuolization of mucolipidosis type II mouse exocrine gland cells represents accumulation of autolysosomes. Mol Biol Cell. 2011;22:1135–1147. doi: 10.1091/mbc.E10-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85:7912–7916. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley KM, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley KM, Schweitzer ES, Miljanich GP, Clift-O’Grady L, Drosten Kushner P, Reichardt LF, Kelly RB. A synaptic vesicle antigen is restricted to the junctional region of the presynaptic plasma membrane. Proc Nat Acad Sci U S A. 1983;80:7342–7346. doi: 10.1073/pnas.80.23.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Murphy TL, Willingham MC, Pastan I, August JT. Identification of two lysosomal membrane glycoproteins. J Cell Biol. 1985;101:85–95. doi: 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EM, Cousin EL. The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. J Neurochem. 2009;111:901–914. doi: 10.1111/j.1471-4159.2009.06384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins MR, Grabner CP, Almers W, Zenisek D. Stimulated exocytosis of endosomes in goldfish retinal bipolar neurons. J Physiol. 2007;584:853–865. doi: 10.1113/jphysiol.2007.140848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JC, Villa BRS, WIlkinson RS. Disruption of actin impedes transmitter release in snake motor terminals. J Physiol. 2000;525:579–586. doi: 10.1111/j.1469-7793.2000.t01-2-00579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa M, Prat A, Mort JS, Marvaldi J, Lissitsky JC, Seidah NG. Down-regulation of alpha-v/beta-3 integrin via misrouting to lysosomes by overexpression of a beta-3/Lamp1 fusion protein. Biochem J. 2003;370:703–711. doi: 10.1042/BJ20021301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust PL, Wall DA, Perara E, Lingappa VR, Kornfeld S. Expression of human cathepsin D in Xenopus oocytes: phosphorylation and intracellular targeting. J Cell Biol. 1987;105:1937–1945. doi: 10.1083/jcb.105.5.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA. The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron. 2004;41:943–953. doi: 10.1016/s0896-6273(04)00113-8. [DOI] [PubMed] [Google Scholar]
- Gaffield MA, Romberg CF, Betz WJ. Live imaging of bulk endocytosis in frog motor terminals using FM dyes. J Neurophysiol. 2011;106:599–607. doi: 10.1152/jn.00123.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glyvuk NY, Tsytsyura Y, Geumann C, D’Hooge R, Huve J, Kratzke M, Baltes J, Boning D, Klingauf J, Schu P. AP-1/sigma1B-adaptin mediates endosomal synaptic vesicle recycling, learning and memory. EMBO J. 2010;29:1318–1330. doi: 10.1038/emboj.2010.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondre-Lewis TA, Moquin AE, Drake JR. Prolonged antigen persistence within nonterminal late endocytic compartments of antigen-specific B lymphocytes. J Immunol. 2001;166:6657–6664. doi: 10.4049/jimmunol.166.11.6657. [DOI] [PubMed] [Google Scholar]
- Granger BL, Green SA, Gabel CA, Howe CL, Mellman I, Helenius A. Characterization and cloning of lgpll0, a lysosomal membrane glycoprotein from mouse and rat cells. J Biol Chem. 1990;265:12036–12043. [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann P, Punge A, Barysch SV, Westphal V, Bückers J, Opazo F, Bethani I, Lauterbach MA, Hell SW, Rizzoli SO. Endosomal sorting of readily releasable synaptic vesicles. Proc Natl Acad Sci U S A. 2010;107:19055–19060. doi: 10.1073/pnas.1007037107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, Edwards RH. v-SNARE composition distinguishes synaptic vesicle pools. Neuron. 2011;71:474–481. doi: 10.1016/j.neuron.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadot M, Colmant C, Wattiaux-De Coninck S, Wattiaux R. Intralysosomal hydrolysis of glycyl-L-phenylalanine 2-naphthylamide. Biochem J. 1984;219:965–970. doi: 10.1042/bj2190965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota T, Mizote M, Kadota K. Dynamics of presynaptic endosomes produced during transmitter release. J Electron Microsc. 1994;43:62–71. [PubMed] [Google Scholar]
- Koenig JH, Ikeda K. Synaptic vesicles have two distinct recycling pathways. J Cell Biol. 1996;135:797–808. doi: 10.1083/jcb.135.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TW, Verstreken P, Bellen HJ. Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron. 2004;43:193–205. doi: 10.1016/j.neuron.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami E, Peters C, von Figura K, Uchiyama Y. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Sunderland WJ, Wilkinson RS. High-resolution imaging of synaptic structure with a simple confocal microscope. New Biologist. 1989;1:75–82. [PubMed] [Google Scholar]
- Macleod GT, Gan J-B, Bennett MR. Vesicle-associated proteins and quantal release at single active zones of amphibian (Bufo marinus) motor-nerve terminals. J Neurophysiol. 1999;82:1133–1146. doi: 10.1152/jn.1999.82.3.1133. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Townes-Anderson E, Czernik AJ, Cameron R, Greengard P, De Camilli P. Synapsins in the vertebrate retina: absence from ribbon synapses and heterogeneous distribution among conventional synapses. Neuron. 1990;5:19–33. doi: 10.1016/0896-6273(90)90030-j. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Miller TM, Heuser JE. Endocytosis of synaptic vesicle membrane at the frog neuromuscular junction. J Cell Biol. 1984;98:685–698. doi: 10.1083/jcb.98.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–648. doi: 10.1016/s0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- Moreno RD. Differential expression of lysosomal associated membrane protein (LAMP-1) during mammalian spermiogenesis. Mol Reprod Dev. 2003;66:202–209. doi: 10.1002/mrd.10342. [DOI] [PubMed] [Google Scholar]
- Mosser VA, Jones KT, Hoffman KM, McCarty NA, Jackson DA. Differential role of beta-arrestin ubiquitination in agonist-promoted down-regulation of M1 vs. M2 muscarinic acetylcholine receptors. J Mol Signaling. 2008;3:20–33. doi: 10.1186/1750-2187-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullock BM, Bright NA, Fearon CW, Gray SR, Luzio JP. Fusion of lysosomes with late endosomes produces a hybrid organelle of intermediate density and is NSF dependent. J Cell Biol. 1998;140:591–601. doi: 10.1083/jcb.140.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannese E, Arcidiacono G, Rigamonti L, Procacci P, Ledda M. Stability at low temperatures of neuronal microtubules in spinal ganglia and dorsal roots of the lizard (Lacerta muralis) J Ultrastruct Res. 1982;79:18–30. doi: 10.1016/s0022-5320(82)90049-1. [DOI] [PubMed] [Google Scholar]
- Pimpinelli F, Lehmann S, Maridonneau-Parini I. The scrapie prion protein is present in flotillin-1-positive vesicles in central- but not peripheral-derived neuronal cell lines. Eur J Neurosci. 2005;21:2063–2072. doi: 10.1111/j.1460-9568.2005.04049.x. [DOI] [PubMed] [Google Scholar]
- Qualmann B, Kelly RB. Syndapin isoforms participate in receptor-mediated endocytosis and actin organization. J Cell Biol. 2000;148:1047–1061. doi: 10.1083/jcb.148.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Guatimosim C, Betz WJ. Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron. 2000;27:551–559. doi: 10.1016/s0896-6273(00)00065-9. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. The structural organization of the readily releasable pool of synaptic vesicles. Science. 2004;303:2037–2039. doi: 10.1126/science.1094682. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Bethani I, Zwilling D, Wenzel D, Siddiqui TJ, Brandhorst D, Jahn R. Evidence for early endosome-like fusion of recently endocytosed synaptic vesicles. Traffic. 2006;7:1163–1176. doi: 10.1111/j.1600-0854.2006.00466.x. [DOI] [PubMed] [Google Scholar]
- Roberg K, Ollinger K. A pre-embedding technique for immunocytochemical visualization of cathepsin D in cultured cells subjected to oxidative stress. J Histochem Cytochem. 1998;46:411–418. doi: 10.1177/002215549804600316. [DOI] [PubMed] [Google Scholar]
- Saravanan K, Schaeren-Wiemers N, Klein D, Sandhoff R, Schwarz A, Yaghootfam A, Gieselmann V, Franken S. Specific downregulation and mistargeting of the lipid raft-associated protein MAL in a glycolipid storage disorder. Neurobiol Dis. 2004;16:396–406. doi: 10.1016/j.nbd.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Saravanan K, Bussow H, Weiler N, Gieselmann V, Franken S. A spontaneously immortalized Schwann cell line to study the molecular aspects of metachromatic leukodystrophy. J Neurosci Methods. 2007;161:223–233. doi: 10.1016/j.jneumeth.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Brodin L. Recent insights into the building and cycling of synaptic vesicles. Exp Cell Res. 2010;316:1344–1350. doi: 10.1016/j.yexcr.2010.02.035. [DOI] [PubMed] [Google Scholar]
- Stewart RS, Wilkinson RS. Acidic structures in snake motor boutons. Society for Neuroscience Annual Meeting; San Diego, CA. 2010. [Google Scholar]
- Studer D, Palankar R, Bédard M, Winterhalter M, Springer S. Retrieval of a metabolite from cells with polyelectrolyte microcapsules. Small. 2010;6:2412–2419. doi: 10.1002/smll.200901997. [DOI] [PubMed] [Google Scholar]
- Sylvain NJ, Brewster DL, Ali DW. Embryonic ethanol exposure alters synaptic properties at zebrafish neuromuscular junctions. Neurotoxicol Teratol. 2011;33:313–321. doi: 10.1016/j.ntt.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Tabares L, Ruiz R, Linares-Clemente P, Gaffield MA, Alvarez de Toledo G, Fernandez-Chacon R, Betz WJ. Monitoring synaptic function at the neuromuscular junction of a mouse expressing synaptopHluorin. J Neurosci. 2007;27:5422–5430. doi: 10.1523/JNEUROSCI.0670-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, De Camilli P. The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. J Cell Biol. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Wilkinson RS. Clathrin-mediated endocytosis near active ones in snake motor boutons. J Neurosci. 2000;20:7986–7993. doi: 10.1523/JNEUROSCI.20-21-07986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Wilkinson RS. ‘Delayed’ endocytosis is regulated by extracellular Ca2+ in snake motor boutons. J Physiol. 2003;551(1):103–114. doi: 10.1113/jphysiol.2003.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Cole JC, Roberts RL, Wilkinson RS. Endocytic active zones: hot spots for endocytosis in vertebrate neuromuscular terminals. J Neurosci. 1999;19:4855–4866. doi: 10.1523/JNEUROSCI.19-12-04855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Lin MY, Wilkinson RS. Macroendocytosis and endosome processing in snake motor boutons. J Physiol. 2007;582:243–262. doi: 10.1113/jphysiol.2007.130989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell. 2011;145:117–132. doi: 10.1016/j.cell.2011.02.039. [DOI] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Weible MA, II, Bartlett SE, Reynolds AJ, Hendry IA. Prolonged recycling of internalized neurotrophins in the nerve terminal. Cytometry. 2001;43:182–188. doi: 10.1002/1097-0320(20010301)43:3<182::aid-cyto1047>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Wibowo E, Brockhausen J, Koppl C. Efferent innervation to the auditory basilar papilla of scincid lizards. J Comp Neurol. 2009;516:74–85. doi: 10.1002/cne.22101. [DOI] [PubMed] [Google Scholar]
- Wilkinson RS, Cole JC. Resolving the Heuser–Ceccarelli debate. Trends Neurosci. 2001;24:195–197. doi: 10.1016/s0166-2236(00)01769-0. [DOI] [PubMed] [Google Scholar]
- Wilkinson RS, Lichtman JW. Regular alternation of fiber types in the transversus abdominis muscle of the garter snake. J Neurosci. 1985;5:2979–2998. doi: 10.1523/JNEUROSCI.05-11-02979.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson RS, Teng H. The nerve-muscle synapse of the garter snake. J Neurocytol. 2003;32:523–528. doi: 10.1023/B:NEUR.0000020608.82235.3e. [DOI] [PubMed] [Google Scholar]
- Yogalingam G, Pendergast AM. Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. J Biol Chem. 2008;283:35941–35953. doi: 10.1074/jbc.M804543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogalingam G, Bonten EJ, van de Vlekkert D, Hu H, Moshiach S, Connell SA, d’Azzo A. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev Cell. 2008;15:74–86. doi: 10.1016/j.devcel.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetzky B, Bellen HJ. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron. 1998;21:1465–1475. doi: 10.1016/s0896-6273(00)80664-9. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wang F, Cao J, Shen Y, Huang Q, Bao L, Zhu X. Nudel promotes axonal lysosome clearance and endo-lysosome formation via dynein-mediated transport. Traffic. 2009;10:1337–1349. doi: 10.1111/j.1600-0854.2009.00945.x. [DOI] [PubMed] [Google Scholar]
- Zhou CJ, Zhao LX, Inagaki N, Guan JL, Nakajo S, Hirabayashi T, Kikuyama S, Shioda S. ATP-binding cassette transporter ABC2/ABCA2 in the rat brain: a novel mammalian lysosome-associated membrane protein and a specific marker for oligodendrocytes but not for myelin sheaths. J Neurosci. 2001;21:849–857. doi: 10.1523/JNEUROSCI.21-03-00849.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.