

Abstract
The blood-brain barrier (BBB) protects brain tissue from potentially harmful plasma components. Small vessel disease ([SVD], arteriolosclerosis) is common in the brains of older people and is associated with lacunar infarcts, leukoaraiosis and vascular dementia. To determine whether plasma extravasation is associated with SVD, we immunolabeled the plasma proteins fibrinogen and IgG, which are assumed to reflect BBB dysfunction, in deep grey matter (anterior caudate-putamen, [DGM]) and deep subcortical white matter (DWM) in the brains of a well-characterized patient cohort with minimal Alzheimer disease pathology (Braak stage 0-II) (n = 84; age ≥65 years). Morphometric measures of fibrinogen labeling were compared between people with neuropathologically defined SVD and aged control subjects. Parenchymal cellular labeling with fibrinogen and IgG was detectable in DGM and DWM in many subjects (>70%). Quantitative measures of fibrinogen were not associated with SVD in DGM or DWM; SVD severity was correlated between DGM and DWM (p < 0.0001). Fibrinogen in DGM showed a modest association with a history of hypertension; DWM fibrinogen was associated with dementia and cerebral amyloid angiopathy (all p < 0.05). In DWM, SVD was associated with leukoaraiosis identified in life (p < 0.05), but fibrinogen was not. Our data suggest that in aged brains plasma extravasation and hence local BBB dysfunction is common but do not support an association with SVD.
Keywords: Arteriolosclerosis; Blood-brain barrier; Dementia, Fibrinogen; Leukoaraiosis; Small vessel disease
INTRODUCTION
The blood-brain barrier (BBB) is a specialized physical and functional barrier comprised of cerebral endothelial cells with intercellular tight junctions, cell-cell signaling with astrocyte end-feet, and efflux pumps in their apical membranes (1, 2). Studies in experimental animals show that in healthy brain tissue with a fully functional BBB, trans-endothelial permeability is exceedingly low and that there is minimal passive extravasation of plasma proteins, inorganic solutes or even water molecules (1, 3, 4). The BBB is disrupted in various animal models of CNS disorders, including acute ischemia (5), multiple sclerosis (6, 7), chronic white matter ischemia (8), and acute cold injury (9).
Several lines of evidence suggest imperfect BBB function in human brain tissue that may be limited in duration and location. Quantitative magnetic resonance imaging (MRI) studies suggest penetration of circulating contrast agent into brain tissue and cerebrospinal fluid (10-12). Immunohistochemical labeling of plasma proteins (e.g. fibrinogen, IgG, albumin, and prothrombin) has been used to assess plasma leakage (13-20). Numerous studies have demonstrated evidence of plasma extravasation in brain disease states, including multiple sclerosis (21-24), HIV encephalitis (25), cerebral malaria (26), epilepsy (27), Alzheimer disease (AD) (14, 19, 28-30), and cerebral ischemic lesions (13-18). While some groups have reported the absence of extravascular plasma markers in healthy brain tissue (15, 23, 25), others have found evidence of paradoxical plasma leakage in “normal” control brain samples (14, 16, 21, 22, 26-28, 31).
Small vessel disease ([SVD]; also termed arteriolosclerosis) is a common brain vasculopathy in older people, associated with lacunar infarcts, vascular cognitive impairment and diffuse white matter lesions that are identified in CT and MRI scans as “leukoaraiosis” (32-35).
While age and hypertension are prominent risk factors, the pathogenesis of SVD remains obscure (33, 34, 36). Some investigators have hypothesized that BBB dysfunction is associated with SVD (37, 38).
We examined fibrinogen and IgG immunohistochemical labeling in a well-characterized cohort of donated brains of older people who had minimal AD (39). We compared those with neuropathologically defined SVD with an age-matched control group (i.e. older people without AD, SVD or other documented brain disease). All cases had in-life clinical and cognitive assessment (40, 41), and detailed neuropathological examination postmortem, including assessment of SVD (39). We tested whether fibrinogen labeling was associated with age, hypertension, dementia or SVD severity.
MATERIALS AND METHODS
Human Tissue
Paraffin-embedded tissue samples of formalin-fixed deep grey matter ([DGM]; anterior caudate-putamen) and deep frontal cortical white matter (DWM) from individuals aged ≥65 years (n = 84) (Supplementary Table S1) were examined. This cohort was composed of all cases neuropathologically graded as Braak stage II or less for neurofibrillary tangle pathology within the Thomas Willis Oxford Brain Collection, John Radcliffe Hospital, Oxford, UK. None had more than 1 ApoE4 or ApoE2 allele. Most cases (76%) were part of the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort (www.medsci.ox.ac.uk/optima) (41). This study had approval of the UK National Research Ethics Service. All tissue was donated following written informed consent by donors or next of kin.
Clinical Data
Medical history, including a documented history of hypertension, the use of antihypertensive medication (never/former/current) were collected from subjects and checked with family doctors’ computerized records. History of hypertension was defined as systolic blood pressure >140 mm Hg, or diastolic blood pressure >90 mm Hg, or use of anti-hypertensive medication (41). Cognitive assessments were performed with the CAMCOG scale (part of the Cambridge Examination for Mental Disorders of the Elderly, CAMDEX). For CAMCOG (range 0 – 107) a score below 80 is rated as dementia. Subjects with dementia were assessed cognitively every 6 months and others were assessed annually. In no case was the interval between cognitive assessment and death greater than 2 years.
Rating of Leukoaraiosis Severity
Severity of leukoaraiosis from in-life CT scans was independently rated by 2 radiologists who were blinded to clinical data, as previously described (40, 41). Briefly, leukoaraiosis was graded in the anterior frontal, posterior frontal, parietal, and occipital cortex, for severity (ranging from 0 [none] to 3 [severe]) and extent (0 indicates none; 1, periventricular leukoaraiosis; 2, periventricular and deep white matter leukoaraiosis; and 3, all white matter involved). Inter-rater agreement for these ratings was substantial (κ = 0.63–0.79 over the different regions) (40). For each area, severity × extent was summed to obtain a total leukoaraiosis score (range 0–36). Leukoaraiosis scores were included if they were assigned within 5 years prior to death (n = 47).
Antibodies
Primary antibodies against human fibrinogen (rabbit polyclonal A-0080) and human IgG (rabbit polyclonal A-0423) were from DakoCytomation, Ely, Cambridgeshire, UK. Human IgG monoclonal antibody (mouse IgG1, Clone RWP49) was from Novocastra-Leica Microsystems, Newcastle-upon-Tyne, UK; the immunogen was a recombinant protein corresponding to 327 residues of human IgG. Anti-fibrinogen polyclonal (1:50,000), anti-human IgG polyclonal (1:120,000) and anti-human IgG monoclonal (1:2,000) antibodies were diluted on the day of use in phosphate buffered saline containing 0.1% v/v Triton-X100 and 3% (w/v) bovine serum albumin (PBT-BSA).
Immunohistochemical Methods
Six-μm-thick sections were de-waxed and processed for standard immunohistochemical labeling (36, 42). Endogenous peroxidase activity was blocked by exposure to H2O2 (3% v/v, aqueous solution) for 8 minutes. After high-pressure heat-induced antigen retrieval (30 seconds, 125°C, in pH 7.8 Tris-citrate buffer), non-specific binding was blocked with PBT-BSA for 60 minutes at room temperature and sections were exposed to primary antibodies at 4°C overnight. Antibody labeling was visualized using a peroxidase-conjugated secondary reagent (Envision® kit, K4065, Dako, Carpinteria, CA) and diaminobenzidine chromogen, then counterstained with Mayer’s hematoxylin. As a negative control, neighboring sections were treated with irrelevant primary antibody (rabbit anti-sheep IgG; BD-Pharmingen, Oxford, UK). Sections were examined on a Zeiss Axioplan-2 microscope driven by Axiovision v4.7 software.
Neuropathological Assessment of SVD
Assignment to “SVD” or “aged control” groups was based on microscopic examination of hematoxylin and eosin (H&E)-stained sections by a registered neuropathologist (M.M.E. or C.J.). Characteristics of the SVD and aged control groups are shown in Table 1. SVD was defined by vasculopathy-oriented criteria, as in previous studies (36, 43). These included hyaline thickening of arteriolar walls; widened perivascular spaces; and parenchymal changes considered to result from SVD (perivascular pallor of myelin staining, loosening with attenuation of nerve fibers with gliosis in white matter or loss of nerve cells and gliosis in deep grey matter) in 1 or more sections (36, 43) (Fig. 1A-D).
Table 1. Characteristics of Aged Control and Small Vessel Disease Subjects.
| Aged control (n = 33) | Small vessel disease (n = 51) | |
|---|---|---|
| Age at death (y) Mean ± SD |
81 ± 8a | 83 ± 8a |
| % Female | 45 % | 39 % |
| History of hypertension | 25 % | 46 % |
| History of dementia | 12 % | 29 % |
| Postmortem interval (h) Mean ± SD |
48 ± 31b | 50 ± 30b |
p = 0.21
p = 0.79
Figure 1.
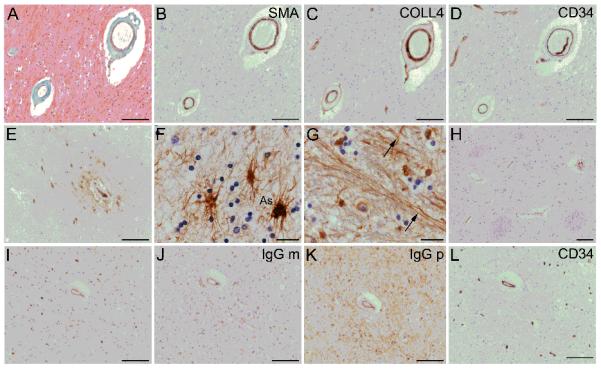
Histopathological evidence of small vessel disease (SVD) and plasma extravasation in human brain tissue of older people. (A-D) Vascular changes representative of SVD (arteriolosclerosis) in 2 small arteries within subcortical white matter. Masson trichrome stain shows concentric, fibrohyaline thickening due to collagen and other connective tissue (green) in the medial layer, with loss of nuclei (nuclei stained black) (A). The smooth muscle marker α-actin (SMA) confirms partial loss of myocytes from the medial layer. Labeling for collagen-4 (COLL4) shows concentric, “doughnut” labeling, characteristic of small vessel disease (C). CD34 immunolabeling confirms intact endothelium (D). (E) Extravascular fibrinogen is seen around blood vessels in parenchymal cells and as perivascular “collars”. (F, G) Cellular fibrinogen is seen in cells with astrocytic morphology (As) (F), and within neural fibers (arrows, G). (H) Some sections lack extravascular fibrinogen. (I-K) Neighboring sections are immunolabeled for the plasma markers, i.e. fibrinogen (I) or immunoglobulin G monoclonal antibody (IgG m, J) or immunoglobulin G polyclonal antibody (IgG p, K). A neighboring section labeled with anti-CD34 demonstrates endothelial labeling and supports specificity of the labeling with plasma markers. In B-L, the chromogen is 3,3′ diaminobenzidine (brown); the nuclear chromatin counterstain hematoxylin (blue). Panels A-D, F-G, I-L are from subcortical white matter; E and H are from anterior putaminal grey matter. Scale bars: F, G, 10 μm; all other panels, 100 μm.
Sections were also independently graded using a more recent SVD severity scale that is oriented to parenchymal pathology (39). Deep grey matter structures were evaluated in sections of basal ganglia and thalamus stained with H&E. White matter structures were evaluated in sections of frontal and occipital white matter stained with H&E and Luxol fast blue/Cresyl violet. Semiquantitative scores for subcortical SVD (0–3) were assigned for each region as follows: 0, normal appearing white or grey matter; 1, slight pallor of myelin staining in white matter, and/or slight loosening of parenchymal tissue on H&E stain, and/or some mild dilatation of perivascular spaces; 2, more marked loss of myelin and/or loosening of parenchymal tissue, sometimes with a bubbly appearance to white matter and/or more markedly widened perivascular spaces; 3, regions of almost complete myelin loss in white matter, severe loosening of parenchymal tissue extending in places to cavitation and severely dilated perivascular spaces. White matter SVD score (0–6) and DGM SVD score (0–6) were obtained as the summed scores of 2 sections for each region.
Neuropathological Assessment of Cerebral Amyloid Angiopathy
Cerebral amyloid angiopathy (CAA) severity was graded from 0 to 4 in cortical tissue as 0 = vessels devoid of amyloid to 4 = severe deposition accompanied by projection of amyloid into the adjacent parenchyma (44). Leptomeningeal and cortical CAA were graded in 3 regions (frontal, temporal and parietal lobes); mean leptomeningeal, cortical and composite CAA scores were then calculated (44). Composite CAA score was used for analysis.
Quantification of Fibrinogen Labeling
An unbiased protocol was used to sample images from each fibrinogen-labeled section, giving a final sampled area ~21 mm2 for each section (Supplementary Fig. S1). Three high-resolution TIFF images (1.73 × 106 pixels/mm2) were sampled at pre-determined locations, under a 2x objective lens. Examples are shown in Supplementary Figure S2. In DGM these were non-overlapping fields within the grey matter strip bounded by the ependymal lining of the lateral ventricle and the internal capsular white matter. Fields were sampled at least 100 μm distant from these boundaries and approximately equally spaced in a lateral to medial orientation. In DWM, non-overlapping fields were equally spaced along the longest axis of the section within deep subcortical white matter. Mean illumination intensity was adjusted to a constant value of 225 (arbitrary units) and white balance was normalized by the imaging software. Images were sampled on a constant 1-ms exposure and stored as 3840 × 3072 8-bit TIFF files with spatial resolution of 0.76 μm/pixel.
Fibrinogen labeling was quantified by 2 independent methods. First, categorical scores for parenchymal cellular labeling (“CELL” score) were assigned on visual inspection of the 3 TIFF files by a registered neuropathologist (M.M.E.) who was blinded to all clinical and experimental data. Each image file was scored “1” if it contained a minimum of 20 clearly labeled cells within parenchymal tissue, and “0” otherwise, giving a range of possible CELL scores of 0, 1, 2 or 3 for each section. All cells were considered, that is, no attempt was made to discriminate neurons, astrocytes, oligodendrocytes or microglia, for quantitation purposes. When grading was repeated for all cases after an interval of 40 days, intra-rater repeatability was high (κ > 0.80). Agreement with another independent blinded rater (A.H.H.) was also high (κ = 0.81). As a second, independent measure of fibrinogen labeling, the fibrinogen-positive area fraction (AF) in each TIFF image was calculated using a densitometry algorithm (NIH ImageJ free software, http://imagej.nih.gov/ij). Briefly, labeled pixels were detected using a fixed threshold detection method and the AF expressed as 100* (number of positive pixels/total pixels). This approach has no observer input (i.e. it is unbiased), but does not exclude intravascular fibrinogen. Absence of intravascular fibrinogen labeling was assumed to reflect loss of antigenicity, and was used as an exclusion criterion (7 cases were excluded for this reason and are not included).
Statistical Analysis
Statistical analysis was carried out in R (v2.14; http://www.R-project.org/). Kendall’s tau-b rank correlation coefficient (τ) was used to test for association, given the presence of collapsed ordinal variables.
RESULTS
Neuropathological Assessment of Serum Markers
All cases reported were positive for intra-vascular fibrinogen, suggesting that antigenicity was intact (n = 84). Some degree of extravascular fibrinogen labeling was a frequent finding in DGM and DWM, either as perivascular “collars” or parenchymal cellular or axonal labeling (Fig. 1E-G; Supplementary Fig. S2). In some instances cellular labeling was restricted to a perivascular distribution. In perivascular collars the labeling intensity for fibrinogen was well-fitted by a decaying exponential function of distance from the outer aspect of the vessel wall (Supplementary Fig. S3), consistent with a pattern of diffusion away from the vessel. Cellular labeling was seen in glial cells and axons (Fig. 1F, G), and rarely in neuronal somata (not shown).
The patterns of human IgG labeling in neighboring sections, using either a polyclonal or monoclonal IgG antibody, were similar to that of fibrinogen (Fig. 1I-K). Neighboring sections treated with no primary antibody, or with an irrelevant primary antibody (anti-CD34 monoclonal antibody, Fig. 1L) or polyclonal rabbit anti-sheep IgG (not shown) did not exhibit this extravascular labeling pattern.
As a positive control for fibrinogen and IgG, 2 cases with neuromyelitis optica (NMO) were also examined. In NMO tissue, extravascular labeling with fibrinogen and IgG were a feature. These were particularly evident within NMO lesional areas, defined by depletion of AQP4 and of glial fibrillary acidic protein-positive astroglia (Supplementary Fig. S4).
Postmortem processes are a potential confound in exploring serum markers within brain tissue. We therefore examined tissue from a collection of aged brains with very short postmortem interval (PMI) derived from another tissue bank (UCI-MIND; PMI <6 hours; n = 10). Fibrinogen labeling of parenchymal cells, perivascular collars and axonal fibers were confirmed as a frequent finding in these cases with short PMI (Supplementary Fig. S5).
Quantitative Assessment of Fibrinogen Labeling
For the Oxford-based cohort of aged cases (n = 84) (39), we performed a semiquantitative analysis of fibrinogen labeling using 2 independent measures (CELL score and fibrinogen-positive AF). Fibrinogen was used for all quantitation due to high potency, low background labeling and robust antigen survival.
Categorical CELL scores were strongly associated with machine-derived fibrinogen AF, suggesting that the 2 assessment methods were robust (Table 2). Fibrinogen-positive AF for DGM was not associated with that for DWM (τ = 0.04, p = 0.74). Fibrinogen-derived AF and CELL score did not differ between SVD and aged control cases (Fig. 2A, B; Table 2). A recently validated regional SVD severity scale based on SVD-associated parenchymal tissue changes (39) showed no association with fibrinogen AF or CELL score (Table 2). Neuropathological SVD severity (39) in DGM was strongly correlated with that in DWM (τ = 0.59, p < 0.0001). In a subset of cases, endothelial cell labeling with the tight junction marker claudin-5, a tight junction protein specific for brain endothelia, did not differ between SVD and aged control cases (n = 11, 11; Supplementary Fig. S6).
Table 2. Test of Association with Fibrinogen-Positive Area Fraction and Cellular Labelling Score for Deep Grey Matter and Deep Subcortical White Matter.
| AF, τ | AF, p | CELL, τ | CELL, p | |
|---|---|---|---|---|
| Deep grey matter | ||||
| Age at death | −0.037 | 0.65 | −0.037 | 0.69 |
| History of hypertension | 0.19 | 0.047* | 0.27 | 0.013 * |
| Subgroup (SVD or AC) | −0.11 | 0.25 | 0.045 | 0.68 |
| SVD severity score1 | −0.038 | 0.66 | 0.088 | 0.36 |
| CAA severity2 | −0.031 | 0.73 | 0.003 | 0.98 |
| History of dementia | −0.07 | 0.48 | 0.005 | 0.90 |
| Postmortem interval | 0.121 | 0.13 | 0.022 | 0.81 |
| AF | -- | -- | 0.52 | <0.001** |
| Deep white matter | ||||
| Age at death | 0.001 | 0.92 | −0.04 | 0.70 |
| History of hypertension | −0.03 | 0.78 | −0.062 | 0.61 |
| Subgroup (SVD or AC) | −0.072 | 0.53 | −0.043 | 0.72 |
| SVD severity score1 | 0.002 | 0.99 | 0.026 | 0.82 |
| Leukoaraiosis severity3 | −0.02 | 0.89 | −0.002 | 0.99 |
| CAA severity2 | 0.16 | 0.14 | 0.24 | 0.036 * |
| History of dementia | 0.25 | 0.043* | 0.055 | 0.69 |
| Postmortem interval | −0.015 | 0.88 | 0.09 | 0.39 |
| AF | -- | -- | 0.50 | <0.001** |
The table lists Kendall’s tau-b rank correlation coefficient (τ) as a measure of association, and the level of significance (p).
p < 0.05;
p < 0.001.
AF, fibrinogen-positive area fraction; CAA, cerebral amyloid angiopathyi; CELL, cellular labelling score for deep grey matter (caudate-putamen) and for deep subcortical white matter; SVD, small vessel disease.
See (39).
See (44).
See (40).
Figure 2.
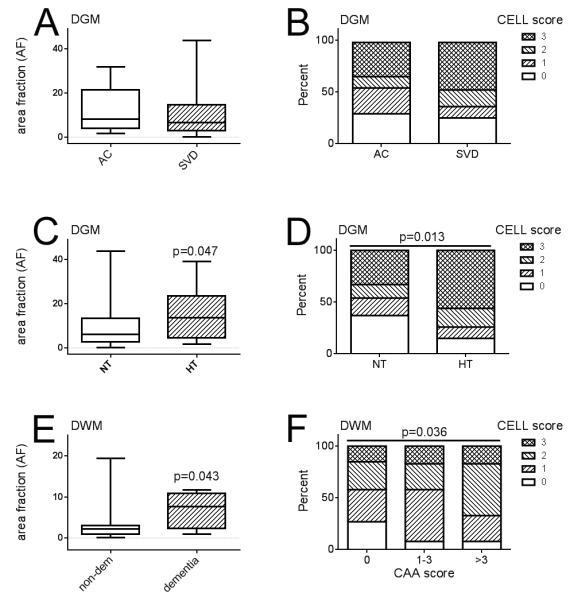
Quantitative assessment of fibrinogen immunolabeling. (A, B) Fibrinogen-positive area fraction ([AF]; range 0-100%) and distribution of fibrinogen labeled cell scores (CELL, range 0, 1, 2 or 3) did not differ between small vessel disease (SVD) subjects and aged control subjects (AC). Data are shown for deep grey matter (DGM). (C, D) Within DGM, fibrinogen-positive AF (C) and distribution of fibrinogen labeled CELL scores (D) were significantly higher in subjects with documented histories of hypertension (HT) vs. those who were documented as being normotensive (NT). (E) Within deep white matter (DWM), fibrinogen-positive AF was greater in those subjects with a documented history of dementia than in subjects without dementia (non-dem). (F) Within DWM, the distribution of fibrinogen labeled CELL scores increased with severity of neuropathological cerebral amyloid angiopathy (CAA) score (range 0–24). Box-whisker plots show median, interquartile range and full range.
DWM fibrinogen measures were unrelated to the leukoaraiosis severity score derived from in-life CT scans (Table 2). The SVD severity score for DWM was correlated with the leukoaraiosis severity score (τ = 0.27, p = 0.032).
In-life history of hypertension was associated with AF (Fig. 2C) and CELL score (Fig. 2D) in DGM but not in DWM (Table 2). PMI and age at death did not differ between aged controls and SVD groups (Table 1), and both showed no association with fibrinogen (Table 2). Specifically, extravascular fibrinogen was absent in some “oldest-old” cases (age >80 years; example shown in Fig. 1H). History of dementia showed modest association with AF in DWM (Fig. 2E), but not in DGM (Table 2). Severity of CAA based on neuropathological assessment (44) showed a modest association with CELL score in DWM (Fig, 2F) but not in DGM (Table 2).
DISCUSSION
We assume that large plasma proteins (fibrinogen and IgG) are histological markers for BBB dysfunction and that parenchymal cell labeling reflects cellular uptake of plasma components, as demonstrated in animal studies (45-47). Some degree of extra-vascular fibrinogen and IgG labeling was a frequent finding in this study. In both DGM and DWM, no measure of fibrinogen labeling differed between aged control subjects and those with SVD, or was associated with a neuropathological score of SVD severity (39), or with in-life leukoaraiosis severity.
Fibrinogen As a Marker of BBB Dysfunction
Fibrinogen is a large plasma glycoprotein (340 kDa) that is synthesized in the liver (48). It was assumed to be a faithful marker of BBB dysfunction for several reasons. First, many sections exhibited a clearly perivascular pattern of labeling, either intracellular or diffuse, suggesting vascular leakage. Second, in tissue from NMO cases. Fibrinogen labeling overlapped with astrocytic lesions. NMO is a rare condition with known molecular pathology, specifically auto-antibodies to the water transporter AQP4. Because AQP4 is expressed in the end-feet of perivascular astrocytes, NMO is characterized by focal damage to astrocytic end-feet, with local BBB failure and plasma extravasation. Thus NMO appears to be an ideal positive control for assessing local BBB dysfunction. Third, fibrinogen labeling was consonant with the unrelated serum marker IgG in aged brain samples as well as in NMO cases. Previous reports from multiple laboratories support fibrinogen as a marker of plasma leakage in diverse CNS pathological conditions (13-15, 18, 21, 23, 24, 27).We found that independent measures of fibrinogen labeling (i.e. observer-derived CELL score and machine-derived AF) were strongly associated.
It is unlikely that our findings reflect postmortem artefact. The intracellular labeling that we and others have observed suggests active uptake, which is unlikely to have occurred postmortem. No morphometric measures of fibrinogen labeling were associated with PMI and cellular labeling was abundant in tissues with very low PMI. In view of the similar pattern observed with IgG, in situ synthesis of fibrinogen within brain tissue also appears very unlikely.
Extravascular Fibrinogen was a Common Finding
Our data suggest that BBB abnormalities are a common feature in DGM and DWM of older people but are possibly limited in location and duration. While this finding conflicts with the BBB concept, numerous previous reports support some tight junction abnormality and BBB dysfunction in “healthy” human brains (14, 16, 21, 22, 26-28, 31). In a blinded, quantitative study in which quantitation of cell labeling was performed, substantial IgG-positive parenchymal cell numbers (approximately 400 cells/mm2) were noted in normal, non-lesional DWM (16). In parenchymal vessels (>20 μm diameter) of frontal cortex, the fraction of vessels that were positive for intramural fibrinogen was high (approximately 30%) in aged, non-demented control subjects, further supporting the notion that plasma extravasation is common in aged brain tissue (28). These reports (and ours) on plasma leakage agree with quantitative studies of tight junction integrity. On average, 14% of small vessels with structurally abnormal tight junctions (labeled for the tight junction protein ZO-1) were detected in healthy white matter from control subjects (22).
Penetration of the BBB by circulating IgG is assumed in the concept of immunotherapy for brain disease (49-52). Brain uptake of IgG and other macromolecules may be trans-cellular or via “leaky” tight junctions (50, 51). Although brain endothelial cells express the IgG receptor FcR, brain penetration by IgG in mice deficient in FcR is similar to that in wild type animals (53). This suggests that trans-junctional entry via tight junctions that are leaky (possibly temporarily) is a more likely route for brain access by circulating antibodies.
Fibrinogen and its breakdown products are cleared from brain tissue by a local tissue plasminogen activator/plasminogen system (24, 54). The parenchymal cell labeling seen in our study and by others (13-15, 18) may be a cytotoxic process, as suggested in animal models (47, 55). Alternatively, it could reflect a protective mechanism used by long-lived brain cells to sequester potentially-harmful plasma proteins (1, 22).
BBB Dysfunction and Small Vessel Disease
Neuropathological SVD severity was strongly correlated between DGM and DWM, suggesting that SVD proceeds in parallel in these 2 tissue regions. We found no association of fibrinogen with neuropathological measures of SVD, either in DGM or in DWM, or with in-life leukoaraiosis severity in DWM. Similarly, coverage with the tight junction protein claudin-5 did not differ between SVD and aged control groups. Thus, our data do not support the idea that ongoing BBB dysfunction is a feature of SVD (12, 37, 56). Our findings conflict with some prior reports (14, 15) and agree with others (16, 20). The lack of association seen here does not exclude a possible role for BBB abnormality earlier in life, possibly as an SVD-initiating “trigger” event.
Comparison with Neuroimaging Data
Several radiological studies have reported partition of intravascular contrast agent into brain tissue, interpreted as BBB dysfunction, in patient groups with SVD or vascular cognitive impairment (11, 12, 38, 56, 57). Some of these did not support a significant association of marker extravasation with disease (38, 56, 57). It appears most likely that brain endothelia handle MRI contrast agents (molecular weight <1,000) differently from plasma proteins (e.g. fibrinogen, IgG, molecular weight >150,000). Transgenic mice that lack claudin-5 have brain vessels that are impermeable to large plasma proteins but permit leakage of a small contrast agent molecule (58). Such imperfect tight junctions in elderly human brains might explain the conflict between MRI data, i.e. on one hand suggesting an association of plasma leakage with SVD (11, 12), and on the other hand, neuropathological data from our study and others (16, 20) showing no such association.
Other Clinical Variables
We observed an association of DGM fibrinogen with history of hypertension, which might reflect long-term use of antihypertensive medications. We also found an association of DWM fibrinogen with clinical history of dementia. This may reflect a neurotoxic action of fibrinogen in nerve cells, axons and myelin, as demonstrated by in vivo studies (47, 55).
We saw a modest association of cellular fibrinogen in DWM with increasing CAA. This is in accordance with other evidence for a link between amyloid-related disease and BBB integrity (19, 28, 29). Experiments using transgenic mice clearly indicate a role for the AD-associated proteins APP and ApoE in BBB function (46, 59, 60). In studies of AD patients, the proportion of CAA-positive vessels in cortical grey matter strongly correlated with the presence of intramural fibrinogen (28) and the plasma protein prothrombin was detected in neurons, vessel walls and in perivascular tissue (19, 29). Pericyte-dependent signaling appears to be a key factor linking ApoE status with BBB function (45, 46), and pericyte degeneration was strongly associated with BBB abnormalities in AD patient brains (30).
The strengths of this study are 1) a well-powered cohort detailed in-life clinical assessment; 2) the use of 2 independent, robust markers (fibrinogen and IgG); and 3) the use of 2 independent measures of fibrinogen labeling (categorical CELL scoring by a blinded neuropathologist, and automated AF). The study has several caveats. First, the cohort size is limited. Second, the cohort is a well-medicated, middle-income group, possibly unrepresentative of populations where risk factors are less well controlled. Third, we have not addressed the membrane transporters that contribute to BBB function by exporting potential toxins from brain endothelial cells into plasma (1, 27).
In conclusion, some degree of extravascular fibrinogen and IgG, assumed to reflect plasma extravasation, was a frequent finding in DGM and DWM of older people without AD. Measures of fibrinogen labeling were not associated with SVD.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge tissue donors and their families for use of donated tissue. We thank our colleagues in Oxford Project to Investigate Memory and Ageing (OPTIMA); Thomas Willis Oxford Brain Collection; St George’s Healthcare NHS Trust Cellular Pathology Service; Institute for Memory Impairments and Neurological Disorders, University of California, Irvine. We acknowledge the Oxford Brain Bank.
This study received financial support from St George’s Hospital Charity (AHH), The Neuroscience Research Foundation (AHH), National Institute for Health Research (HSM) and NIHR via Oxford Biomedical Research Centre (MME) and Alzheimer’s Society UK (HSM, AHH). HSM is supported by an NIHR Senior Investigator award. Funding for the UCI-ADRC is provided by NIH/NIA Grant P50 AG016573. The Oxford Brain Bank is supported by the Medical Research Council (MRC), Brains for Dementia Research (BDR) and the NIHR Oxford Biomedical Research Centre.
Footnotes
The authors declare that they have no conflict of interest.
REFERENCES
- 1.Abbott NJ, Patabendige AA, Dolman DE, et al. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Cserr HF, DePasquale M, Patlak CS. Regulation of brain water and electrolytes during acute hyperosmolality in rats. Am J Physiol. 1987;253:F522–F29. doi: 10.1152/ajprenal.1987.253.3.F522. [DOI] [PubMed] [Google Scholar]
- 4.Fraser PA, Dallas AD. Permeability of disrupted cerebral microvessels in the frog. J Physiol. 1993;461:619–32. doi: 10.1113/jphysiol.1993.sp019532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuntz M, Mysiorek C, Petrault O, et al. Stroke-induced brain parenchymal injury drives blood-brain barrier early leakage kinetics: a combined in vivo/in vitro study. J Cereb Blood Flow Metab. 2014;34:95–107. doi: 10.1038/jcbfm.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Argaw AT, Asp L, Zhang J, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–68. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Argaw AT, Zhang Y, Snyder BJ, et al. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol. 2006;177:5574–84. doi: 10.4049/jimmunol.177.8.5574. [DOI] [PubMed] [Google Scholar]
- 8.Seo JH, Miyamoto N, Hayakawa K, et al. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J Clin Invest. 2013;123:782–6. doi: 10.1172/JCI65863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114:459–69. doi: 10.1007/s00401-007-0274-x. [DOI] [PubMed] [Google Scholar]
- 10.Starr JM, Wardlaw J, Ferguson K, et al. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2003;74:70–6. doi: 10.1136/jnnp.74.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taheri S, Gasparovic C, Huisa BN, et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. 2011;42:2158–63. doi: 10.1161/STROKEAHA.110.611731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topakian R, Barrick TR, Howe FA, et al. Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatry. 2010;81:192–7. doi: 10.1136/jnnp.2009.172072. [DOI] [PubMed] [Google Scholar]
- 13.Simpson JE, Ince PG, Higham CE, et al. Microglial activation in white matter lesions and nonlesional white matter of ageing brains. Neuropathol Appl Neurobiol. 2007;33:670–83. doi: 10.1111/j.1365-2990.2007.00890.x. [DOI] [PubMed] [Google Scholar]
- 14.Tomimoto H, Akiguchi I, Suenaga T, et al. Alterations of the blood-brain barrier and glial cells in white-matter lesions in cerebrovascular and Alzheimer’s disease patients. Stroke. 1996;27:2069–74. doi: 10.1161/01.str.27.11.2069. [DOI] [PubMed] [Google Scholar]
- 15.Utter S, Tamboli IY, Walter J, et al. Cerebral small vessel disease-induced apolipoprotein E leakage is associated with Alzheimer disease and the accumulation of amyloid beta-protein in perivascular astrocytes. J Neuropathol Exp Neurol. 2008;67:842–56. doi: 10.1097/NEN.0b013e3181836a71. [DOI] [PubMed] [Google Scholar]
- 16.Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology. 2008;71:804–11. doi: 10.1212/01.wnl.0000319691.50117.54. [DOI] [PubMed] [Google Scholar]
- 17.Alafuzoff I, Adolfsson R, Grundke-Iqbal I, et al. Perivascular deposits of serum proteins in cerebral cortex in vascular dementia. Acta Neuropathol. 1985;66:292–8. doi: 10.1007/BF00690961. [DOI] [PubMed] [Google Scholar]
- 18.Lammie GA, Brannan F, Wardlaw JM. Incomplete lacunar infarction (Type Ib lacunes) Acta Neuropathol (Berl) 1998;96:163–71. doi: 10.1007/s004010050877. [DOI] [PubMed] [Google Scholar]
- 19.Zipser BD, Johanson CE, Gonzalez L, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging. 2007;28:977–86. doi: 10.1016/j.neurobiolaging.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 20.Viggars AP, Wharton SB, Simpson JE, et al. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci Lett. 2011;505:25–30. doi: 10.1016/j.neulet.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 21.Kirk J, Plumb J, Mirakhur M, et al. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol. 2003;201:319–27. doi: 10.1002/path.1434. [DOI] [PubMed] [Google Scholar]
- 22.Leech S, Kirk J, Plumb J, et al. Persistent endothelial abnormalities and blood-brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol Appl Neurobiol. 2007;33:86–98. doi: 10.1111/j.1365-2990.2006.00781.x. [DOI] [PubMed] [Google Scholar]
- 23.van HJ, Brink BP, de Vries HE, et al. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol. 2007;66:321–8. doi: 10.1097/nen.0b013e318040b2de. [DOI] [PubMed] [Google Scholar]
- 24.Gveric D, Herrera B, Petzold A, et al. Impaired fibrinolysis in multiple sclerosis: a role for tissue plasminogen activator inhibitors. Brain. 2003;126:1590–8. doi: 10.1093/brain/awg167. [DOI] [PubMed] [Google Scholar]
- 25.Dallasta LM, Pisarov LA, Esplen JE, et al. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155:1915–27. doi: 10.1016/S0002-9440(10)65511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown H, Hien TT, Day N, et al. Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol Appl Neurobiol. 1999;25:331–40. doi: 10.1046/j.1365-2990.1999.00188.x. [DOI] [PubMed] [Google Scholar]
- 27.Liu JY, Thom M, Catarino CB, et al. Neuropathology of the blood-brain barrier and pharmaco-resistance in human epilepsy. Brain. 2012;135:3115–33. doi: 10.1093/brain/aws147. [DOI] [PubMed] [Google Scholar]
- 28.Hultman K, Strickland S, Norris EH. The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J Cereb Blood Flow Metab. 2013;33:1251–8. doi: 10.1038/jcbfm.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berzin TM, Zipser BD, Rafii MS, et al. Agrin and microvascular damage in Alzheimer’s disease. Neurobiol Aging. 2000;21:349–55. doi: 10.1016/s0197-4580(00)00121-4. [DOI] [PubMed] [Google Scholar]
- 30.Sengillo JD, Winkler EA, Walker CT, et al. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013;23:303–10. doi: 10.1111/bpa.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomimoto H, Akiguchi I, Wakita H, et al. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 1997;94:146–52. doi: 10.1007/s004010050686. [DOI] [PubMed] [Google Scholar]
- 32.Fisher CM. The arterial lesions underlying lacunes. Acta Neuropathol. 1968;12:1–15. doi: 10.1007/BF00685305. [DOI] [PubMed] [Google Scholar]
- 33.Lammie GA. Hypertensive cerebral small vessel disease and stroke. Brain Pathol. 2002;12:358–70. doi: 10.1111/j.1750-3639.2002.tb00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt R, Schmidt H, Haybaeck J, et al. Heterogeneity in age-related white matter changes. Acta Neuropathol. 2011;122:171–85. doi: 10.1007/s00401-011-0851-x. [DOI] [PubMed] [Google Scholar]
- 36.Giwa MO, Williams J, Elderfield K, et al. Neuropathologic evidence of endothelial changes in cerebral small vessel disease. Neurology. 2012;78:167–74. doi: 10.1212/WNL.0b013e3182407968. [DOI] [PubMed] [Google Scholar]
- 37.Wardlaw JM, Sandercock PA, Dennis MS, et al. Is breakdown of the blood-brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke. 2003;34:806–12. doi: 10.1161/01.STR.0000058480.77236.B3. [DOI] [PubMed] [Google Scholar]
- 38.Wardlaw JM, Doubal FN, Valdes-Hernandez M, et al. Blood-brain barrier permeability and long-term clinical and imaging outcomes in cerebral small vessel disease. Stroke. 2013;44:525–7. doi: 10.1161/STROKEAHA.112.669994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smallwood A, Oulhaj A, Joachim C, et al. Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: a pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol Appl Neurobiol. 2012;38:337–43. doi: 10.1111/j.1365-2990.2011.01221.x. [DOI] [PubMed] [Google Scholar]
- 40.Mendes Ribeiro HK, Barnetson LP, Hogervorst E, et al. A new visual rating scale for white matter low attenuation on CT. Eur Neurol. 2001;45:140–4. doi: 10.1159/000052112. [DOI] [PubMed] [Google Scholar]
- 41.Hogervorst E, Ribeiro HM, Molyneux A, et al. Plasma homocysteine levels, cerebrovascular risk factors, and cerebral white matter changes (leukoaraiosis) in patients with Alzheimer disease. Arch Neurol. 2002;59:787–93. doi: 10.1001/archneur.59.5.787. [DOI] [PubMed] [Google Scholar]
- 42.Hainsworth AH, Allsopp RC, Jim A, et al. Death-associated protein kinase (DAPK1) in cerebral cortex of late-onset Alzheimer’s disease patients and aged controls. Neuropathol Appl Neurobiol. 2010;36:17–24. doi: 10.1111/j.1365-2990.2009.01035.x. [DOI] [PubMed] [Google Scholar]
- 43.Esiri MM, Wilcock GK, Morris JH. Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry. 1997;63:749–53. doi: 10.1136/jnnp.63.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chalmers K, Wilcock GK, Love S. APOE epsilon 4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol Appl Neurobiol. 2003;29:231–8. doi: 10.1046/j.1365-2990.2003.00457.x. [DOI] [PubMed] [Google Scholar]
- 45.Armulik A, Genove G, Mae M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 46.Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–6. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davalos D, Ryu JK, Merlini M, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Sem Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 49.Winblad B, Andreasen N, Minthon L, et al. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;11:597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- 50.Golde TE, Das P, Levites Y. Quantitative and mechanistic studies of Abeta immunotherapy. CNS Neurol Disord Drug Targets. 2009;8:31–49. doi: 10.2174/187152709787601830. [DOI] [PubMed] [Google Scholar]
- 51.Boche D, Denham N, Holmes C, et al. Neuropathology after active Abeta42 immunotherapy: implications for Alzheimer’s disease pathogenesis. Acta Neuropathol. 2010;120:369–84. doi: 10.1007/s00401-010-0719-5. [DOI] [PubMed] [Google Scholar]
- 52.Greenberg SM, Al-Shahi SR, Biessels GJ, et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol. 2014;13:419–28. doi: 10.1016/S1474-4422(14)70003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abuqayyas L, Balthasar JP. Investigation of the role of FcgammaR and FcRn in mAb distribution to the brain. Mol Pharm. 2013;10:1505–13. doi: 10.1021/mp300214k. [DOI] [PubMed] [Google Scholar]
- 54.Tabrizi P, Wang L, Seeds N, et al. Tissue plasminogen activator (tPA) deficiency exacerbates cerebrovascular fibrin deposition and brain injury in a murine stroke model: studies in tPA-deficient mice and wild-type mice on a matched genetic background. Arterioscler Thromb Vasc Biol. 1999;19:2801–6. doi: 10.1161/01.atv.19.11.2801. [DOI] [PubMed] [Google Scholar]
- 55.Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med. 2007;204:1999–2008. doi: 10.1084/jem.20070304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–52. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 57.Wardlaw JM, Doubal F, Armitage P, et al. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. 2009;65:194–202. doi: 10.1002/ana.21549. [DOI] [PubMed] [Google Scholar]
- 58.Nitta T, Hata M, Gotoh S, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–60. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishitsuji K, Hosono T, Nakamura T, et al. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 2011;286:17536–42. doi: 10.1074/jbc.M111.225532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hartz AM, Bauer B, Soldner EL, et al. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke. 2012;43:514–23. doi: 10.1161/STROKEAHA.111.627562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.