

Abstract
Our laboratory is investigating ivermectin (IVM) and other members of the avermectin family as new pharmaco-therapeutics to prevent and/or treat alcohol use disorders (AUDs). Prior work found that IVM significantly reduced ethanol intake in mice and that this effect likely reflects IVM’s ability to modulate ligand-gated ion channels. We hypothesized that structural modifications that enhance IVM’s effects on key receptors and/or increase its brain concentration should improve its anti-alcohol efficacy. We tested this hypothesis by comparing the abilities of IVM and two other avermectins, abamectin (ABM) and selamectin (SEL), to reduce ethanol intake in mice, to alter modulation of GABA ARs and P2X4Rs expressed in Xenopus oocytes and to increase their ability to penetrate the brain. IVM and ABM significantly reduced ethanol intake and antagonized the inhibitory effects of ethanol on P2X4R function. In contrast, SEL did not affect either measure, despite achieving higher brain concentrations than IVM and ABM. All three potentiated GABAA receptor function. These findings suggest that chemical structure and effects on receptor function play key roles in the ability of avermectins to reduce ethanol intake and that these factors are more important than brain penetration alone. The direct relationship between the effect of these avermectins on P2X4R function and ethanol intake suggest that the ability to antagonize ethanol-mediated inhibition of P2X4R function may be a good predictor of the potential of an avermectin to reduce ethanol intake and support the use of avermectins as a platform for developing novel drugs to prevent and/or treat AUDs.
Keywords: Ivermectin, Medications, Development, Alcoholism Therapy, P2X4 Receptor, GABAA Receptor
INTRODUCTION
Alcohol use disorders (AUDs) rank third on the list of preventable causes of morbidity and mortality in the United States, affecting over 18 million people, causing over 100,000 deaths annually (Bouchery et al., 2011; Grant et al., 2004; Johnson, 2010) and costing in excess of $220 billion (Bouchery et al., 2011). This exceeds the costs of other leading preventable causes of death such as cigarette smoking and physical inactivity (Naimi, 2011). Presently, the only pharmacotherapeutic agents approved by the United States Food and Drug Administration (FDA) for the treatment of AUDs are disulfiram (Antabuse®), naltrexone (Revia® and Vivitrol®), and acamprosate (Campral®), with Vivitrol® being an extended-release injectable formulation of naltrexone (Harris et al., 2010; Litten et al., 2012). These drugs attempt to deter alcohol intake by blocking its metabolism or by targeting the neurochemical and neuropeptide systems in the downstream cascades leading to craving and dependence (Colombo et al., 2007; Gewiss et al., 1991; Johnson, 2010; Litten et al., 2012; Steensland et al., 2007). However, their success rate even when combined with psychotherapy, has been limited with approximately 70% of patients relapsing back into heavy drinking within one year (Johnson, 2008; Litten et al., 2012). Thus, the development of new drugs that will more effectively treat AUDs is of paramount importance.
Our team is investigating ivermectin (IVM) as a potential platform for developing novel agents for preventing or treating AUDs. IVM is a FDA approved drug that is currently used worldwide as a broad-spectrum antiparasitic agent (Crump and Omura, 2011; Omura, 2008). We recently demonstrated that IVM significantly reduces ethanol intake in both male and female mice across several models of self-administration (Yardley et al., 2012). Doses of IVM that significantly reduced ethanol intake also produced significant, dose-dependent anxiolytic responses in these animals without exhibiting any addiction potential (Bortolato et al., 2013). Moreover, IVM did not cause significant changes in pain sensitivity, motor competency or memory (Bortolato et al., 2013) and did not cause any obvious signs of toxicity (Bortolato et al., 2013; Yardley et al., 2012). Overall, these findings indicate that IVM reduces ethanol intake and has an excellent safety profile with good tolerability; thus pointing to this agent and potentially other related avermectins as novel therapeutic agents for the prevention and/or treatment of AUDs.
The mechanism(s) by which IVM reduces ethanol intake is/are not known. The current therapeutic application of IVM as an antihelmentic is attributed to action on a non-mammalian, glutamate-gated inhibitory chloride channel (Crump and Omura, 2011; Cully et al., 1994; Dent et al., 1997; Vassilatis et al., 1997). Thus, action on this channel cannot contribute to its anti- alcohol effect. On the other hand, IVM does potentiate mammalian ligand-gated ion channels, including gamma-aminobutyric acid A (GABA A) and glycine receptors (Dawson et al., 2000; Krusek and Zemkova, 1994; Shan et al., 2001), and has been shown in rodents to have anticonvulsant and anxiolytic properties linked to its action on GABAA receptors (GABAARs) (Dawson et al., 2000; Spinosa et al., 2002). More recent studies indicate that IVM acts on several other ligand-gated ion channel proteins in the mammalian central nervous system (CNS) (Sung et al., 2009) including nicotinic acetylcholine (Krause et al., 1998; Sattelle et al., 2009) and P2X4 receptors (P2X4Rs) (Khakh et al., 1999). We recently reported that IVM antagonizes ethanol inhibition of P2X4Rs expressed in Xenopus oocytes (Asatryan et al., 2010; Popova et al., 2013), suggesting that P2X4Rs may play an important role in the anti-alcohol intake effects of IVM. In support of the hypothesis that P2X4Rs are important in the anti-alcohol intake effects of IVM, preliminary investigations in our laboratory found that IVM did not significantly reduce ethanol intake in P2X4 knockout (KO) mice, but did reduce alcohol intake in wildtype (WT) controls.
In summary, available evidence indicates that IVM can reduce alcohol intake. Given that IVM is already approved for use in humans, IVM has the potential for rapid repurposing as a novel treatment for AUDs. The anti-alcohol actions of IVM likely reflect its ability to modulate one or more ligand-gated ion channels in the brain, but this hypothesis has yet to be tested. IVM, due to its lipophilic nature, should pass the blood brain barrier (BBB), but does not readily achieve high brain concentration, ostensibly due to its high efflux by P-glycoprotein (P-gp). Therefore, structural modifications that reduce its P-gp substrate recognition should increase brain concentration (Lespine et al., 2007; Menez et al., 2012) and should positively impact its ability to reduce ethanol intake (Yardley et al., 2012). Likewise, structural changes that alter its interaction with targeted brain receptors should also impact its efficacy in this regard. The present study investigates these possibilities by comparing the effects of IVM with two IVM- related macrocyclic lactones, abamectin (ABM) and selamectin (SEL), for their abilities to reduce alcohol intake in mice and to alter modulation of GABAARs and P2X4Rs expressed in Xenopus oocytes.
MATERIALS AND METHODS
Drugs
In vitro 10 mM stock solutions of IVM (powder from Sigma, St. Louis, MO), ABM (powder from Sigma (Supelco), St. Louis, MO) were dissolved in DMSO and kept at −20°C until use (See Fig. 1 for chemical structures). The highest DMSO concentration in the final solution was 0.1 %. SEL was kindly provided by Pfizer Pharmaceuticals (Groton, CT) as a 1% solution suspended in propylene glycol. In vivo: drugs were administered via daily intraperitoneal (IP) injections. Noromectin (10 mg/ml in 60% propylene glycol (Norbrook Inc, Lenexa, KS) was used for IVM injections. Drugs were diluted using a 0.9% sodium chloride solution (saline) to a concentration that would allow for an injection volume of 0.01 ml/g of body weight. Ethanol (190 proof USP, Sigma, St. Lois, MO) was used in in vitro studies. For drinking studies, Gold Shield Alcohol (200 proof USP solution, Gold Shield Chemical Company, Hayward, CA) was diluted in water to achieve a 10% v/v solution (10E).
Fig 1. Structures of IVM, ABM and SEL.
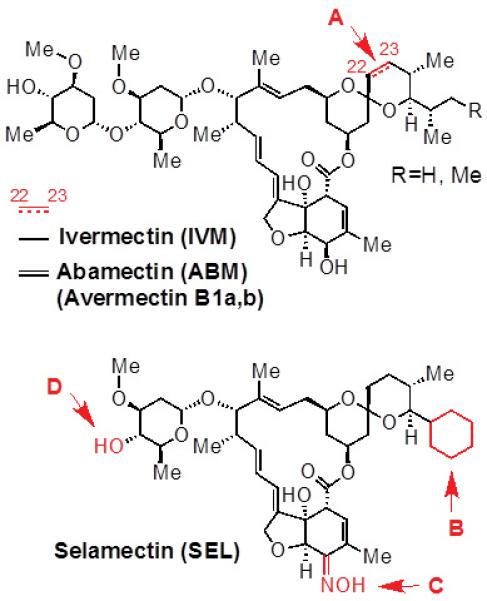
Both IVM and ABM feature a spiroketal moiety with a mixture of isopropyl/isobutyl substituents. (A) ABM differs from IVM with the presence of an unsaturated double bond at the C22-23 position. SEL has three structural differences from IVM and ABM: (B) it has a cyclohexyl ring in place of the isopropyl/isobutyl substituent on IVM and ABM; (C) it has a ketoxime substituent in the place of the hydroxyl group of the tetrahydro- benzofuran unit on IVM and ABM; and (D) it lacks the second carbohydrate unit that is present in both IVM and ABM.
In Vivo Studies
Animals
Studies were performed on C57BL/6J male mice that were 8 weeks old upon purchase (Jackson Laboratory, Bar Harbor, ME, USA). Mice were singly housed in polycarbonate/polysulfone cages at a 12 h light/dark cycle with lights off at 12:30PM. The holding room was maintained at approximately 22°C. Mice had been tested for the effects of IVM on ethanol intake using the drinking in the dark paradigm for approximately 2 months before testing in the present study. Prior to the onset of the present experiment, mice were acclimated to 24-h two-bottle choice ethanol paradigm as described below for five days. All procedures in this study were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and all efforts were made to minimize animal suffering. The USC Institutional Animal Care and Use Committee approved the protocols.
24-h access two-bottle choice paradigm
Individually housed mice had 24-h access to two inverted bottles with metal sippers placed on the cage tops with one tube containing tap water and the other a 10% v/v solution in tap water (10E). Alcohol determinations followed previously described procedures (Yardley et al., 2012).
Ethanol intake studies with avermectins
Following 5 days of acclimation to the two-bottle choice paradigm, mice were injected daily with saline until ethanol intake on the 2-bottle choice stabilized (+/− 10% variability from the mean dose of the last 3 days). Then, mice were assigned to drug treatment groups and were injected with 5 mg/kg of IVM, ABM or SEL according to a within subjects design. Mice were injected with saline on subsequent days until drinking once again stabilized.
Analysis of brain concentrations of IVM, ABM and SEL using liquid chromatography with tandem mass spectrometry (LC-MS/MS)
Plasma and brain samples were collected from separate groups of animals at 0.25, 1, 4, 8, 10 and 32 hours (n=2/time point) after administration of 5 mg/kg (i.p.) of IVM, ABM or SEL. To each 100 μL plasma or 150 mg of brain, 2 μg IVM was added as internal standard for samples containing either ABM or SEL. For samples containing IVM, 2 μg ABM was added as internal standards. Plasma samples were mixed with 2 mL of acetonitrile, centrifuged at 10,000 rpm for 5 min with the organic layer collected and air dried. To brain samples, 1 scoop of 1 mm zirconium bead and 1 mL of acetonitrile were added, and vigorously homogenized using the bullet blender for 5 min. Samples were then centrifuged at 10,000 rpm for 5 min, where the supernatant was collected. The above steps were repeated with another 1mL of fresh acetonitrile, and the combined 2 mL supernatant were dried by using a steady stream of filtered and dried air. Both the evaporated residues of plasma and brain samples were reconstituted in 100 μL of acetonitrile : water (90:10 v/v).
The LC-MS method was validated using a calibration curve of known standard solutions with the lower detection limit found to be 5 ng/mL. To each 100 μL plasma or 150 mg of brain, 2 μg of IVM was added as internal standard. The samples were extracted as described above. A 40 μL aliquot was injected into the Agilent 1100 HPLC System linked to an AB Sciex API 3000 turboion spray mass spectrometer. The analytes were separated using an ACE C18 column with an isocratic mobile phase consisting of acetonitrile/0.1% formic acid : water/0.1% formic acid (90 v : 10 v). The amount of IVM, ABM and SEL was quantified using the mass spectrometer set in positive mode with multiple reaction monitoring using the parent to transition ions of 888.8 → 551.5, 890.1 → 449.6 and 764.0→338.0 respectively.
In Vitro Studies
Preparation of Xenopus oocytes
Xenopus oocytes isolation and maintenance followed procedures described previously (Asatryan et al., 2010; Davies et al., 2005).
cRNA synthesis, cRNA and cDNA injections
The cDNAs of rat P2X4R (GenBank accession No. X87763) and of rat α1, β2 and γ2 subunits of GABAAR were sub-cloned into pcDNA3 vector (Invitrogen, Carlsbad, CA). The DNA containing P2X4R gene was then linearized and transcribed using the mMESSAGE mMACHINE kit (Ambion, Austin, TX) to result in cRNA, which was stored at −70°C until injection. Twenty four hours post isolation the oocytes were injected with 10 or 20 ng cRNA or 1 ng of DNA mixture of GABAAR subunits at 1:1:1 ratio using Nanoject II Nanoliter injection system (Drummond Scientific, Broomall, PA). The oocytes were incubated at 17°C and used in electrophysiological recordings for 3-7 days after injections.
Whole cell voltage clamp recordings
Two electrode voltage clamp recordings were performed using the Warner instrument model OC-725C oocyte clamp (Hamden, CT) following previously described procedures (Davies et al., 2005; Davies et al., 2002). The oocytes were voltage clamped at −70 mV and the currents were recorded on a strip-chart recorder (Barnstead/Thermolyne, IA).
Experimental procedures
P2X4Rs
Oocytes were continuously perfused at a rate of 3-4 ml/min with extracellular modified Ringers buffer containing (in mM) 110 NaCl, 2.5 KCl, 10 HEPES and 1.8 BaCl2, pH 7.5, using a peristaltic pump (Rainin Instrument, Oakland, CA). Ca2+ in the solution was replaced with Ba2+ to prevent the activation of Ca2+-dependent Cl− channels (Khakh et al., 1999). All experiments were performed at room temperature (20-23°C).
To induce currents, submaximal concentrations (i.e. EC10) of adenosine 5’-triphosphate (ATP, Sigma, St. Lois, MO) were used. Normally, P2X4Rs EC10 is at 1 μM ATP. Using EC10 has been previously shown to maximise the effects of ethanol while causing minimal receptor desensitisation (Davies et al., 2005; Davies et al., 2002). Ethanol and drugs were applied after stable responses to EC10 ATP were obtained. A washout period of 5 min was allowed between each application to allow for re-sensitisation of the receptor (Asatryan et al., 2010; Davies et al., 2005; Davies et al., 2002; Popova et al., 2013).
Effects of ethanol (50, 100 or 200 mM) or IVM, ABM and SEL (0.5 - 30 μM) were tested alone and in combination during co-application with ATP for 20 seconds. ATP currents were measured before and after each drug application in order to confirm the existence of a stable baseline response. Pilot studies determined that the drugs did not have an effect on the membrane potential of uninjected cells, nor did the drugs produce currents when applied in the absence of agonist.
Experiments on GABAARs
Oocytes were perfused at a rate of 3-4 ml/min at room temperature with modified Bart’s saline containing in mM (83 NaCl, 1 KCl, 10 HEPES, 0.82 MgSO4, 2.4 NaHCO3, 0.91 CaCl2, and 0.33 Ca(NO3)2 , pH 7.5). Similar to the approach for P2X4Rs, EC10 concentration of GABA was used to test the effects of the drugs, i.e. IVM, ABM and SEL. A washout period of 5 min was allowed between each GABA application.
IVM, ABM and SEL were applied with GABA EC10 after a stable response to GABA EC10 was obtained. Each oocyte was tested for one concentration of avermectins since these drugs caused irreversible effects that were not washable. The second consecutive response to the application of the drug was always larger; therefore, both responses in the presence of the drug were averaged for data analysis.
Data Analyses
In vivo studies
Ethanol dose (g/kg) and ethanol preference ratio (mls ethanol/total mls) were calculated for each drug. The dependent variables included 10E intake (g/kg), 10E preference (%), water (ml) and total fluid intake (ml). Two-tailed t-tests were used to assess the effects of drug treatment groups (IVM, SEL, ABM) versus the respective saline injected pre-drug control groups for each dependent variable. The significance level was set at p ≤ 0.05.
In vitro studies
Data were obtained from batches of oocytes from at least 3 different frogs and are expressed as mean ± SEM for an indicated number of tests. The results are presented as the percentage change in agonist EC10 activated currents after normalising these with the response of the EC10 alone. To assess concentration response relationships, data were fitted to a concentration-response curve by using the following logistic equation: I = Imax * [drug]/([drug] +(EC50)drug), where I is the percentage of the maximum obtainable response (Imax), EC50 is the drug concentration producing a half-maximal response. Bar graphs were used to compare the effects of ethanol with and without IVM, ABM or SEL on P2X4Rs. GraphPAD Prism software (San Diego, CA) was used for data analysis and curve fitting. Statistical analysis was performed using unpaired t-tests with significance set at p ≤ 0.05.
Pharmacokinetic Analysis
The pharmacokinetics (PK) of IVM, SEL and ABM in plasma and brain were analysed using a non-compartmental PK modeling. Serial blood and tissue IVM, SEL and ABM quantification was used to calculate PK parameters such as maximum drug concentration (Cmax), time to achieve maximal drug exposure (Tmax), half-life, elimination constant and area under the curve (AUC).
RESULTS
Behavioural studies
IVM and ABM, but not SEL, reduced alcohol intake and preference in C57BL/6J mice
We tested the effects of acute administration of IVM, SEL, and ABM using a 24-h access two-bottle choice alcohol paradigm (10E versus tap water) in C57BL/6J mice. This model was selected based on our initial investigation that tested the effects of IVM on ethanol intake and preference (Yardley et al., 2012). We used 5 mg/kg of IVM and analogs based on our prior study where doses ranging from 2.5 to 10.0 mg/kg provided significant reductions in drinking levels without any significant behavioral toxicity (Yardley et al., 2012).
Ethanol intake for the saline controls the day before injection of IVM, SEL and ABM were comparable (Fig 2A). IVM and ABM significantly reduced 10E intake versus their respective controls. In contrast, SEL did not have a significant effect on ethanol intake. For all drugs, 10E intake returned to comparable pre drug intake levels one-day post drug injection (data not shown).
Fig 2. IVM, SEL, ABM (5 mg/kg) administration in male C57BL/6J mice using a 24-h access two-bottle choice paradigm.
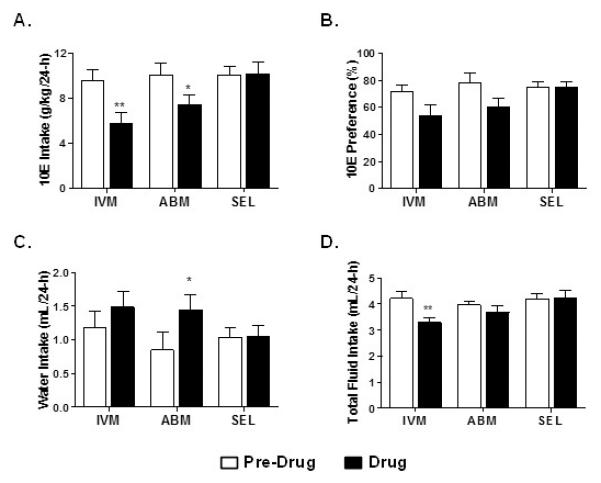
Bars represent levels from the saline control the day prior to drug injection (Pre Drug) and the day of the drug injection (Drug) for A) 10E intake, B) preference for 10E, C) water intake, and D) total fluid intake. Values represent the mean +/− SEM for 7-8 mice per dose group. *P<0.05, **P < 0.01 versus respective pre drug condition.
Similarly, IVM and ABM reduced preference for 10E versus saline injection (Fig. 2B). SEL had no effect on this parameter. In addition, ABM significantly increased water intake (Fig. 2C). There was a trend to increase water intake for IVM (p<0.06), whereas SEL had no effect on this measure (Fig 2C). Lastly, only IVM decreased total fluid intake (Fig. 2D).
Pharmacokinetics of IVM, ABM and SEL
To determine whether relative differences in avermectin penetration across the BBB influenced the brain concentration of IVM, ABM and SEL and might explain the differences in the respective effects of these agents on ethanol intake found in the in vivo studies described above, we measured both plasma and brain concentrations at the designed time points of 0.25, 1, 4, 8, 10 and 32 hours after i.p administration of 5 mg/kg of each. The findings are shown in Table 1. Brain AUCs for IVM, ABM and SEL were 1.81 ng*hr/mL, 40.1 ng*hr/mL and 69.3 ng*hr/mL respectively, where SEL and ABM had the highest brain concentrations. Interestingly, ABM and SEL AUCs were 22- and 38-times higher than those for IVM.
Table 1.
Pharmacokinetic parameters of avermectins. The plasma exposure of the three avermectins was similar as determined by mean AUC0-t. Despite similar plasma exposures, there were marked differences in brain disposition in this study, where brain AUC0-t for SEL and ABM was 38- and 22-fold higher than IVM.
| IVM | ABM | SEL | |
|---|---|---|---|
| Plasma PK Parameter | |||
| Cmax (ng/mL) | 232 | 351 | 370 |
| Tmax (hrs) | 10.00 | 0.25 | 1.00 |
| Half-life (hrs) | 12.67 | 17.40 | 21.40 |
| AUC0-t (ng*hr/mL) | 4970 | 4049 | 4276 |
| Brain PK Parameter | |||
| Cmax (ng/mg of tissue) | 0.31 | 2.32 | 6.21 |
| Tmax (hrs) | 10.0 | 10.0 | 4.0 |
| Half-life (hrs) | 19.0 | 8.8 | 19.4 |
| AUC0-t (ng*hr/mg of tissue) | 1.81 | 40.10 | 69.30 |
In vitro studies
Effect of avermectin analogs on P2X4R and GABAAR function
In order to determine the effect of avermectin analogs on receptor function, we performed several experiments testing IVM, ABM and SEL on two ion channels that are linked to IVM’s behavioral effects, P2X4Rs and GABAARs (Bortolato et al., 2013; Yardley et al., 2012). Previous findings demonstrated that IVM (0.5 – 10 μM) potentiated ATP-gated currents in P2X4Rs expressed in oocytes (Asatryan et al., 2010; Khakh et al., 1999; Priel and Silberberg, 2004). Therefore, to test the effects of ABM and SEL on P2X4Rs, we elected to use a similar concentration range (0.5 – 10 μM) that was used in our prior IVM study (Asatryan et al., 2010). We compared the ability of ABM and SEL versus IVM to modulate ATP-gated currents in P2X4Rs.
ABM (0.5 – 10 μM) significantly potentiated ATP-induced currents in P2X4Rs in a concentration-dependent manner (Fig 3A). The degree of ABM potentiation was significantly greater than that of IVM at concentrations ≥ 3 μM; i.e. reaching 100-fold greater activity at 10 μM (Fig. 3A). However, at lower concentrations, i.e. 0.5 and 1 μM, the degree of ABM and IVM potentiation did not significantly differ (Fig. 3B). ABM, at higher concentrations (3 and 10 μM), directly induced P2X4R currents in the absence of ATP and this effect was concentration- dependent (Fig 3C). In addition, there was a residual effect on post ATP currents (Fig. 3C, final tracings in each row) suggesting that ABM was slow to washout.
Fig 3. IVM, ABM and SEL modulation of P2X4R and GABAAR activity.
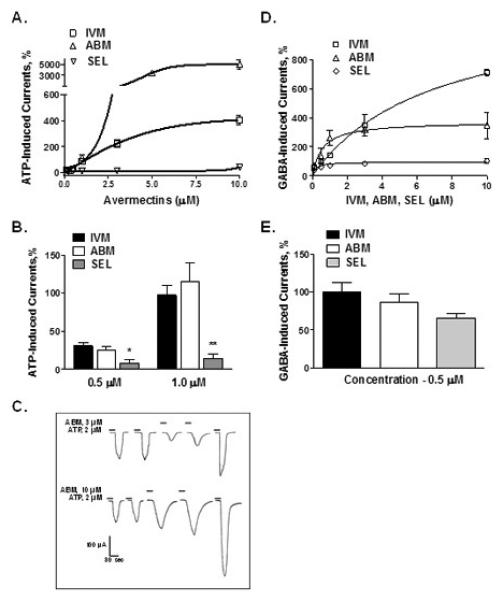
(A) Concentration-response curves of co-application of avermectins (0. 5 -10 μM) with EC10 ATP demonstrate that IVM and ABM are potent positive modulators whereas SEL is a weak modulator of P2X4R activity. (B) ABM potentiation was significantly greater than that of IVM at concentrations ≥ 3 μM but not at lower 0.5 and 1 μM concentrations. (C) ABM directly induces currents in P2X4Rs at higher concentrations (3 and 10 μM) in a concentration-dependent manner. (D) All avermectins significantly potentiated GABAAR function. (E) At low 0.5 μM concentration, the effect of SEL was smaller compared to that of IVM but did not reach statistical significance. Data are presented as mean ± SEM of 4-12 oocytes per data point.
In contrast to IVM and ABM, SEL poorly potentiated ATP-gated currents in P2X4Rs (Fig. 3A) and had markedly lower responses than either IVM or ABM across all concentrations tested (0.5 - 10 μM) with the response of SEL at 10 μM being similar to IVM and ABM at 0.5 μM. Therefore, we tested a higher concentration of SEL to see if we could elicit responses more comparable to IVM and ABM. We found that 30 μM SEL induced a significantly higher degree of potentiation compared to 10 μM SEL (78.8 ± 24.2 vs 42.4 ± 12.7 % of control, P < 0.05). However, the SEL response at 30 μM was still 5-fold less in magnitude compared to the degree of potentiation induced by 10 μM IVM (IVM 405 ± 36 % vs SEL 78.8 ± 24.2 %).
We also tested the effects of IVM, ABM and SEL on GABAARs. For this study we used α1β2γ2 GABAARs due to their predominant expression in the CNS. As illustrated (Fig. 3D), IVM and ABM significantly potentiated GABAAR function. The effects of ABM and IVM were similar in the concentration range of 0.5 - 3 μM. At 10 μM IVM, the extent of IVM potentiation was significantly greater compared to the effect of ABM. The greater degree of potentiation by 10 μM IVM may be due to IVM acting as a partial agonist for GABAARs. The degree of potentiation of GABA-induced currents by SEL on GABAAR function tested at 0.5 μM did not significantly differ from IVM and ABM. However, the effects of SEL were significantly less than IVM and ABM at concentrations ≥ 1.0 μM (Fig. 3E).
IVM and ABM, but not SEL, antagonize the effects of ethanol on P2X4Rs
Our previous work found that 0.5 μM IVM significantly antagonized the effects of ethanol caused by intoxicating (25 mM or 0.1 %) and higher (up to 100 mM) ethanol concentrations (Asatryan et al., 2010). The current study extended this work by testing concentrations of ABM and SEL that induced a similar degree of potentiation of P2X4R function as that of 0.5 μM IVM (i.e. 0.5 μM for ABM and 10 μM for SEL). We selected 100 mM ethanol since it caused a robust inhibition of ATP-induced currents.
ABM (0.5 μM) significantly reduced 100 mM ethanol inhibition and the degree of antagonism by ABM was similar to that of IVM (Fig. 4A). On the other hand, we found that a 20X higher concentration of SEL was required in order for SEL to reach a similar degree of potentiation of P2X4R function as that of IVM and ABM. Notably, even at this higher concentration, SEL did not significantly antagonize the effects of ethanol on P2X4R function (Fig. 4A).
Fig 4. Effects of IVM, ABM and SEL on ethanol inhibition of P2X4R function.
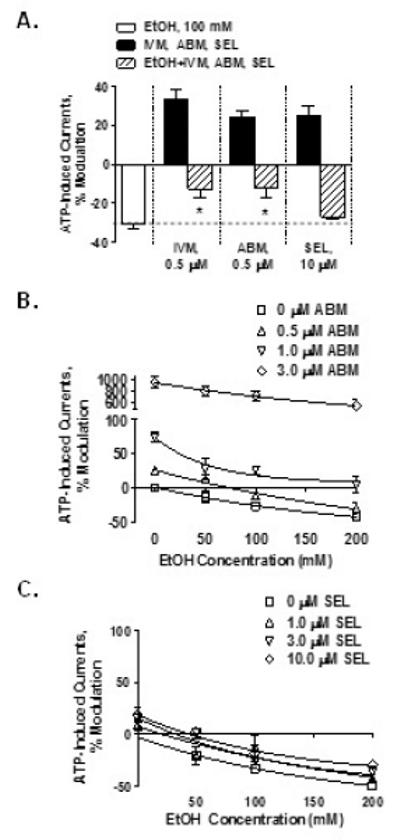
(A) IVM and ABM at 0.5 μM antagonize the 100 mM ethanol inhibition. SEL at 10 μM is not able to antagonize the effect of 100 mM ethanol. There was a right-shift in the ethanol concentration-response curves for ABM (B) but not for SEL (C). Data are presented as mean ± SEM of 5-17 oocytes per experimental point. * P < 0.05 compared to the ATP + 100 mM ethanol.
We extended this investigation to include concentration-response studies for ABM and SEL antagonism of ethanol using methods similar to our recent IVM studies (Asatryan et al., 2010; Popova et al., 2013). This was accomplished by testing a concentration range (0.5 – 3 μM) of ABM and SEL on the effects of ethanol (50, 100 and 200 mM) on P2X4Rs. ABM (Fig 4B), but not SEL (Fig 4C), antagonized ethanol inhibition of P2X4R function as indicated by a parallel right-shift in the ethanol concentration-response curves.
DISCUSSION
The present study provides insights into the mechanisms and important structural features of IVM responsible for its ability to reduce ethanol intake that can inform future development of more effective agents. Using three related, but structurally distinct avermectins (IVM, ABM, and SEL) the findings indicate that 1) the structure and 2) ability to modulate P2X4Rs and antagonize ethanol effects in P2X4Rs are important determinants for predicting the ability of avermectins to reduce ethanol intake.
Using an in vivo continuous access two-bottle choice drinking paradigm, the present study found that IVM significantly reduced ethanol consumption. IVM also reduced total fluid intake, however this probably reflected the decrease in ethanol intake since IVM did not affect water intake. These findings are in good agreement with our previous work (Yardley et al., 2012) and extend this investigation, showing that ABM also significantly reduced ethanol intake, but to a lesser extent than did IVM. In contrast, SEL did not significantly reduce ethanol consumption.
The disposition of the avermectins in plasma and brain were evaluated by LC-MS/MS to determine whether brain disposition might play a role in determining the efficacy of IVM, ABM, and SEL. The results indicate that there was minimal difference between IVM, ABM, and SEL in their plasma concentrations, as defined by the area under the curve (AUC). In contrast, the brain disposition differed considerably between the three avermectins. SEL had the highest brain concentration which was followed by ABM and then IVM. Thus, the degree of BBB penetration and resultant brain concentration of the respective drugs alone cannot explain the differences in the ability of these avermectins to reduce alcohol intake. Rather, these differences reflect their ability to modulate or offset underlying neurochemical effects of ethanol.
In agreement with this contention, we identified substantial differences between the abilities of avermectins to modulate P2X4R function and to antagonize the inhibitory effects of ethanol on P2X4Rs in vitro. Specifically, IVM and ABM significantly potentiated P2X4R function and antagonized the inhibitory effects of ethanol at therapeutically relevant concentrations. In contrast, SEL, even at much higher concentrations, showed only minimal activity to potentiate P2X4R function and to antagonize the effects of ethanol, in vitro. The right-shift in the ethanol concentration-response with ABM supports a competitive mechanism for the ethanol antagonism. Notably, the findings with ABM are similar to our previous findings with IVM (Asatryan et al., 2010; Popova et al., 2013). On the other hand, the absence of significant reductions in ethanol response or a right shift for the concentration-response curves for SEL further supports the notion that SEL lacks the ability to antagonize the inhibitory effects of ethanol on P2X4Rs.
SEL appeared to be equally potent to IVM and ABM in its ability to positively modulate GABAARs when tested at concentrations up to 1 μM. On the other hand, at concentrations ≥ 1.0 μM, SEL was less effective in potentiating GABAAR function as compared to IVM and ABM. Taken together, the findings suggest that the significant differences in structure between IVM and ABM, compared to SEL, play an important role in the differences in the compounds ability to positively modulate P2X4R and to a lesser extent, GABAAR function.
IVM belongs to a class of lipophilic, water insoluble compounds, the avermectins, that are produced by the soil microorganisms Streptomyces avermitilis (for review see Crump and Omura, 2011; Omura, 2008). The structural basis of IVM as an anti-parasitic agent has been linked to the hexahydro-benzofuran site, but not the spiroketal group of the molecule (Michael et al., 2001). In addition, the size of the carbohydrate side chain is suggested to be important in the interaction with the P-gp and should influence the ability of P-gp to remove avermectins that penetrate the BBB (Lespine et al., 2007). ABM and IVM are structurally similar; differing only by a double bond at C22-23 (Fig. 1). ABM is a mixture of the fermentation natural products avermectin B1a and B1b containing a double bond at C22-23 of the spiroketal unit, while IVM is the product of selective chemical hydrogenation of ABM at C22-23. SEL is a synthetic analog that is structurally different from both ABM and IVM. SEL, in addition to having a saturated spiroketal moiety that is similar to IVM, also has a cyclohexyl ring in place of the isopropyl/isobutyl substituent on IVM and ABM. The absence of the C22-C23 double bond in IVM alters the conformation of the spiroketal moiety and modifies its metabolism in comparison with ABM (Halley et al., 1992). However, given the comparable ability of both IVM and ABM to antagonize the inhibitory effects of ethanol in P2X4Rs more effectively than SEL, it seems that their spiroketal differences are not the primary determinant for this effect.
There are two other major structural differences between IVM and ABM, as compared to SEL. First, SEL features an unsaturated ketoxime substituent in the place of the simple allylic hydroxyl group that is found on IVM and ABM. Second, SEL contains only one carbohydrate moiety rather than two as in IVM and ABM (Fig. 1). The latter feature is likely responsible for reduced interaction of SEL with P-gp (Lespine et al., 2007) and as a result the higher brain concentrations seen with SEL versus IVM and ABM. On the other hand, despite higher brain concentrations, SEL did not reduce ethanol intake. Together, these findings suggest three possibilities: 1) the presence of the two carbohydrates is required for the anti-alcohol effects of IVM and ABM; 2) the presence of the ketoxime in the structure of SEL is sufficient to eliminate this effect or 3) both of these structural differences are important in determining the anti-alcohol efficacy of avermectins.
The identified structural modifications in SEL (Fig. 1) may be important pharmacophore sites required for the greater anti-alcohol potency of IVM and ABM. In support of this notion, recent studies identified an overlapping putative binding pocket for IVM and ethanol, an alcohol-IVM pocket, in P2X4Rs (Asatryan et al., 2010; Popova et al., 2013). In this context, we propose that the second sugar moiety that is present in both IVM and ABM, but lacking in SEL is important for the affinity of IVM and ABM for this alcohol/IVM pocket that is key for the efficacy of avermectins in blocking the actions of ethanol on P2X4Rs and reducing alcohol intake. The presence/absence of this structural element may also play an important role for the interaction of IVM and ABM with other receptors systems such as GABAARs. Future studies will begin to address these issues by investigating structure-function interactions including molecular modeling and extensive docking of avermectins at the identified putative binding site.
Taken together, the findings suggest that chemical structure and effects on receptor function play key roles in the ability of avermectins to reduce ethanol intake and that these factors are more important than brain penetration alone. The direct relationship between the effect of these avermectins on P2X4R function and ethanol intake suggest that the ability to antagonize ethanol-mediated inhibition of P2X4R function may be a good predictor of the potential of an avermectin to reduce ethanol intake and support the use of avermectins as a platform for developing novel drugs to prevent and/or treat AUDs.
ACKNOWLEDGEMENTS
L. Asatryan and M. M. Yardley contributed equally to this work as co-first authors. This work was conducted as partial fulfilment of the requirements for the PhD degree in Molecular Pharmacology and Toxicology, University of Southern California (M.M.Y.). We would like to thank Miriam Fine, Mark Ambroso and Ellen Martin for technical and laboratorial assistance.
This work was supported in part by the American Foundation for Pharmaceutical Education Fellowship (M.M.Y.) and by Research grants SC CTSI (NIH/NCRR/NCATS -- TL1TR000132 [M.M.Y.]) and UL1TR000130 (D.L.D.), and by NIAAA/NIH KO1 AA017243 (L.A.), AA022448 (D.L.D.), and AA020980 (J.R.T.), and with support from the USC Office of Research Advancement Collaboration Grant "USC Center for Discovery and Drug Development" (S.G.L and N.A.P.) and the USC School of Pharmacy.
Footnotes
The authors declare no conflict of interest and are entirely responsible for the scientific content of the paper.
REFERENCES
- Asatryan L, Popova M, Perkins DI, Trudell JR, Alkana RL, Davies DL. Ivermectin antagonizes ethanol inhibition in P2X4 receptors. J Pharmacol Exp Ther. 2010;334:720–728. doi: 10.1124/jpet.110.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolato M, Yardley M, Khoja S, Godar SC, Asatryan L, Finn DA, Alkana RL, Louie SG, Davies DL. Pharmacological insights into the role of P2X4 receptors in behavioral regulation: lessons from ivermectin. Int J Neuropsychopharmacol. 2013;16(5):1059–1070. doi: 10.1017/S1461145712000909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchery EE, Harwood HJ, Sacks JJ, Simon CJ, Brewer RD. Economic Costs of Excessive Alcohol Consumption in the U.S., 2006. Am J Prevent Med. 2011;41(5):516–524. doi: 10.1016/j.amepre.2011.06.045. [DOI] [PubMed] [Google Scholar]
- Colombo G, Orr A, Lai P, Cabras C, Maccioni P, Rubio M, Gessa GL, Carai MA. The cannabinoid CB1 receptor antagonist, rimonabant, as a promising pharmacotherapy for alcohol dependence: preclinical evidence. Mol Neurobiol. 2007;36(1):102–112. doi: 10.1007/s12035-007-0017-y. [DOI] [PubMed] [Google Scholar]
- Crump A, Omura S. Ivermectin, 'wonder drug' from Japan: the human use perspective. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(2):13–28. doi: 10.2183/pjab.87.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully DF, Vassilatis DK, Liu KK, Paress PS, Van der Ploeg LHT, Schaeffer JM, Arena JP. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nat. 1994;371(6499):707–711. doi: 10.1038/371707a0. [DOI] [PubMed] [Google Scholar]
- Davies DL, Kochegarov AA, Kuo ST, Kulkarni AA, Woodward JJ, King BF, Alkana RL. Ethanol differentially affects ATP-gated P2X(3) and P2X(4) receptor subtypes expressed in Xenopus oocytes. Neuropharmacol. 2005;49:243–253. doi: 10.1016/j.neuropharm.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Davies DL, Machu TK, Guo Y, Alkana RL. Ethanol sensitivity in ATP-gated P2X receptors is subunit dependent. Alcohol Clin Exp Res. 2002;26:773–778. [PubMed] [Google Scholar]
- Dawson GR, Wafford KA, Smith A, Marshall GR, Bayley PJ, Schaeffer JM, Meinke PT, McKernan RM. Anticonvulsant and adverse effects of avermectin analogs in mice are mediated through the gamma-aminobutyric acid A receptor. J Pharmacol Exp Ther. 2000;295(3):1051–1060. [PubMed] [Google Scholar]
- Dent JA, Davis MW, Avery L. avr-15 encodes a chloride channel subunit that mediates inhibitory glutamatergic neurotransmission and ivermectin sensitivity in Caenorhabditis elegans. EMBO J. 1997;16(19):5867–5879. doi: 10.1093/emboj/16.19.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewiss M, Heidbreder C, Opsomer L, Durbin P, De Witte P. Acamprosate and diazepam differentially modulate alcohol-induced behavioural and cortical alterations in rats following chronic inhalation of ethanol vapour. Alcohol Alcohol. 1991;26:129–137. doi: 10.1093/oxfordjournals.alcalc.a045093. [DOI] [PubMed] [Google Scholar]
- Grant BF, Dawson DA, Stinson FS, Chou P, Dufour MC, Pickering RP. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991-1992 and 2001-2002. Drug Alcohol Depend. 2004;74(3):223–234. doi: 10.1016/j.drugalcdep.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Halley BA, Narasimhan NI, Venkataraman K, Taub R, Erwin MLG, Andrew NW, Wislocki PG. Ivermectin and Abamectin Metabolism: Differences and Similarities. Vol. 503. American Chemical Society; Washington, DC: 1992. pp. 203–216. [Google Scholar]
- Harris AH, Kivlahan DR, Bowe T, Humphreys KN. Pharmacotherapy of alcohol use disorders in the Veterans Health Administration. Psychiatr Serv. 2010;61(4):392–398. doi: 10.1176/ps.2010.61.4.392. [DOI] [PubMed] [Google Scholar]
- Johnson B. Medication treatment of different types of alcoholism. Am J Psychiatry. 2010;167:630–639. doi: 10.1176/appi.ajp.2010.08101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA. Update on neuropharmacological treatments for alcoholism: Scientific basis and clinical findings. Biochem Pharmacol. 2008;75:34–56. doi: 10.1016/j.bcp.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Proctor WR, Dunwiddie TV, Labarca C, Lester HA. Allosteric control of gating and kinetics at P2X4 receptor channels. J Neurosci. 1999;19:7289–7299. doi: 10.1523/JNEUROSCI.19-17-07289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause RM, Buisson B, Bertrand S, Corringer PJ, Galzi JL, Changeux JP, Bertrand D. Ivermectin: A positive allosteric effector of the alpha 7 neuronal nicotinic acetylcholine receptor. Mol Pharmacol. 1998;53(2):283–294. doi: 10.1124/mol.53.2.283. [DOI] [PubMed] [Google Scholar]
- Krusek J, Zemkova H. Effect of ivermectin on gamma-aminobutyric acid-induced chloride currents in mouse hippocampal embryonic neurones. Eur J Pharmacol. 1994;259(2):121–128. doi: 10.1016/0014-2999(94)90500-2. [DOI] [PubMed] [Google Scholar]
- Lespine A, Martin S, Dupuy J, Roulet A, Pineau T, Orlowski S, Alvinerie M. Interaction of macrocyclic lactones with P-glycoprotein: Structure-affinity relationship. Eur J Pharm Sci. 2007;30:84–94. doi: 10.1016/j.ejps.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Litten RZ, Egli M, Heilig M, Cui C, Fertig JB, Ryan ML, Falk DE, Moss H, Huebner R, Noronha A. Medications development to treat alcohol dependence: a vision for the next decade. Addict Biol. 2012;17(3):513–527. doi: 10.1111/j.1369-1600.2012.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menez C, Sutra JF, Prichard R, Lespine A. Relative neurotoxicity of ivermectin and moxidectin in Mdr1ab (−/−) mice and effects on mammalian GABA(A) channel activity. PLoS Negl Trop Dis. 2012;6(11):e1883. doi: 10.1371/journal.pntd.0001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael B, Meinke PT, Shoop W. Comparison of ivermectin, doramectin, selamectin, and eleven intermediates in a nematode larval development assay. J Parasitol. 2001;87(3):692–696. doi: 10.1645/0022-3395(2001)087[0692:COIDSA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Naimi TS. The cost of alcohol and its corresponding taxes in the U.S.: a massive public subsidy of excessive drinking and alcohol industries. Am J Prevent Med. 2011;41(5):546–547. doi: 10.1016/j.amepre.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Omura S. Ivermectin: 25 years and still going strong. Int J Antimicrobial Agents. 2008;31:91–98. doi: 10.1016/j.ijantimicag.2007.08.023. [DOI] [PubMed] [Google Scholar]
- Popova M, Trudell J, Li K, Alkana R, Davies D, Asatryan L. Tryptophan 46 is a site for ethanol and ivermectin action in P2X4 receptors. Purinergic Signal. 2013 doi: 10.1007/s11302-013-9373-4. e-pub ahead of print 2 July 2013. doi: 1007/s11302-013-9373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Silberberg SD. Mechanism of Ivermectin Facilitation of Human P2X4 Receptor Channels. J Gen Phys. 2004;123(3):281–293. doi: 10.1085/jgp.200308986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattelle DB, Buckingham SD, Akamatsu M, Matsuda K, Pienaar I, Jones AK, Sattelle BM, Almond A, Blundell CD. Comparative pharmacology and computational modelling yield insights into allosteric modulation of human alpha7 nicotinic acetylcholine receptors. Biochem Pharmacol. 2009;78(7):836–843. doi: 10.1016/j.bcp.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Shan Q, Haddrill JL, Lynch JW. Ivermectin, an unconventional agonist of the glycine receptor chloride channel. J Biol Chem. 2001;276(16):12556–12564. doi: 10.1074/jbc.M011264200. [DOI] [PubMed] [Google Scholar]
- Spinosa HS, Stilck SRAN, Bernardi MM. Possible anxiolytic effects of ivermectin in rats. Vet Res Commun. 2002;26:309–321. doi: 10.1023/a:1016094726033. [DOI] [PubMed] [Google Scholar]
- Steensland P, Simms JA, Holgate J, Richards JK, Bartlett SE. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, selectively decreases ethanol consumption and seeking. Proc Natl Acad Sci U S A. 2007;104(30):12518–12523. doi: 10.1073/pnas.0705368104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YF, Huang CT, Fan CK, Lin CH, Lin SP. Avermectin intoxication with coma, myoclonus, and polyneuropathy. Clin Toxicol. 2009;47(7):686–688. doi: 10.1080/15563650903070901. [DOI] [PubMed] [Google Scholar]
- Vassilatis DK, Arena JP, Plasterk RH, Wilkinson HA, Schaeffer JM, Cully DF, Van de Ploeg LH. Genetic and biochemical evidence for a novel avermectin-sensitive chloride channel in Caenorhabditis elegans: Isolation and characterization. J Biol Chem. 1997;272(52):33167–33174. doi: 10.1074/jbc.272.52.33167. [DOI] [PubMed] [Google Scholar]
- Yardley M, Wyatt L, Khoja S, Asatryan L, Ramaker MJ, Finn DA, Alkana RL, Huynh N, Louie SG, Petasis NA, Bortolato M, Davies DL. Ivermectin reduces alcohol intake and preference in mice. Neuropharmacol. 2012;63(2):190–201. doi: 10.1016/j.neuropharm.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]