

Significance
Platelets are cell fragments in the blood that initiate clot formation at the site of bleeding. Although the biological aspects of this process have been well characterized, whether platelets can detect and physiologically respond to the mechanical aspects of its local environment is unclear. Here, we show that platelets sense the stiffness of the underlying clot substrate, and increasing substrate stiffness increases platelet adhesion and spreading. Importantly, adhesion on stiffer substrates leads to higher levels of platelet activation. Mechanistically, we determined that Rac1, actin, and myosin activity mediate this process. This newfound capability of how platelets adjust their degree of activation based on the mechanical properties of their environment provides new insight into how clots are formed.
Keywords: mechanotransduction, cell mechanics, platelet cytoskeleton, biomaterials
Abstract
As platelets aggregate and activate at the site of vascular injury to stem bleeding, they are subjected to a myriad of biochemical and biophysical signals and cues. As clot formation ensues, platelets interact with polymerizing fibrin scaffolds, exposing platelets to a large range of mechanical microenvironments. Here, we show for the first time (to our knowledge) that platelets, which are anucleate cellular fragments, sense microenvironmental mechanical properties, such as substrate stiffness, and transduce those cues into differential biological signals. Specifically, as platelets mechanosense the stiffness of the underlying fibrin/fibrinogen substrate, increasing substrate stiffness leads to increased platelet adhesion and spreading. Importantly, adhesion on stiffer substrates also leads to higher levels of platelet activation, as measured by integrin αIIbβ3 activation, α-granule secretion, and procoagulant activity. Mechanistically, we determined that Rac1 and actomyosin activity mediate substrate stiffness-dependent platelet adhesion, spreading, and activation to different degrees. This capability of platelets to mechanosense microenvironmental cues in a growing thrombus or hemostatic plug and then mechanotransduce those cues into differential levels of platelet adhesion, spreading, and activation provides biophysical insight into the underlying mechanisms of platelet aggregation and platelet activation heterogeneity during thrombus formation.
As the first responders at the site of vascular injury, platelets are subjected to a dynamic microenvironment during the process of hemostasis (1–5). Biochemically, platelets are exposed to diverse and rapidly changing gradients of soluble proteins and agonists such as von Willebrand factor, ADP, and thrombin, all of which drive platelet adhesion and activation (6, 7). During this process, platelet activation may take several forms including activation of platelet αIIbβ3 integrins, secretion of α- and dense granules, and membrane phosphatidylserine (PS) exposure leading to a procoagulant phenotype (8, 9). Biophysically, platelets also activate and aggregate in response to the hemodynamic and shear forces of the circulation (10, 11). As clot formation ensues, platelets then interact with polymerizing fibrin scaffolds, exposing platelets to a large range of mechanical microenvironments. Although the underlying biochemical signaling pathways that govern the fibrinogen-αIIbβ3–mediated processes have been well characterized, if and how the mechanical cues in the microenvironment affect platelet activation and physiology remain largely unknown. Indeed, as clot structure and mechanics are known to be heterogeneous within the same clot and more recent studies have demonstrated that platelet activation is also vastly heterogeneous within a growing thrombus (12–14), a systematic approach to investigate how platelet activation is affected by the mechanical microenvironment could lead to profound insights into platelet physiology.
To that end and to decouple the mechanical cues from biological and biochemical factors that may be involved, we immobilized fibrinogen on polyacrylamide (PA) gels of varied stiffnesses and investigated how platelets biologically respond to these differences in substrate stiffness. Here, we observed that the number of adherent platelets and the average spreading area of these platelets increased with increasing substrate stiffness. Importantly, we also found that PA gels stiffer than 5 kPa caused more platelet activation, namely increased integrin αIIbβ3 activation, P-selectin secretion, and PS exposure, compared with softer gels. Furthermore, through a series of pharmacologic inhibition studies, we explored the underlying mechanisms that govern mechanotransduction in platelets during adhesion, spreading, and subsequent activation.
Results
Differential Platelet Adhesion and Spreading on Immobilized Fibrinogen Is Mediated by Substrate Stiffness.
For these experiments, platelets adhered and spread onto PA gels of varied stiffness in which the gel surfaces were conjugated with a constant density of fibrinogen. These matrices enable exquisite control of matrix mechanics without concomitantly altering ligand presentation or density (15–17). More specifically, one can modulate the stiffness of these materials as an isolated variable by manipulating cross-linking density in ways that do not require addition of more matrix ligand (i.e., fibrinogen), which could independently alter platelet function and confound interpretation of results. As such, use of these fibrinogen-conjugated PA gels enables the decoupling of the mechanical from biochemical cues of the platelet microenvironment (18–20). PA gels of different stiffnesses were fabricated as previously reported (21), and their stiffnesses, which are defined by elastic moduli, ranged from 0.25 to 100 kPa. PA gel stiffness was modulated by adjusting the cross-linking density of the PA network. Fibrinogen was then covalently conjugated to the PA gel surface through a reaction between free amine groups on fibrinogen and prebound NHS groups on PA gels via UV grafting of Sulfo-SANPAH, as reported by others (22). The fibrinogen density was confirmed to be constant on the PA gels of different stiffnesses (Fig. S1). Therefore, this platform allows us to focus only on varying substrate stiffness, while maintaining constant fibrinogen density on the PA gel surface (Fig. 1A).
Fig. 1.

Differential platelet adhesion and spreading on immobilized fibrinogen is mediated by substrate stiffness. (A) Experimental setup illustrating how human platelets were incubated on polyacrylamide (PA) gels of varied stiffnesses but conjugated with the same fibrinogen concentration on the top surface. This creates a microenvironment in which the mechanical properties are uncoupled from the biochemical properties. (B) Confocal microscopy images of adherent platelets, stained with a fluorescent membrane dye, on PA gels with stiffnesses ranging from 0.25 to 100 kPa and glass. (C) The number of platelets adhered on PA gels of different stiffness and glass. (D) The spread surface area of adherent platelets on PA gels of different stiffness and glass. (E) Cumulative frequency of spread platelet surface area on substrates of different stiffnesses. (Scale bar: 10 μm.) (P < 0.05; n = 3 experiments; error bars indicate SD.)
With this setup, we found that the number of adherent platelets on the PA gel surfaces after incubation increased with increasing substrate stiffness, reaching a plateau (∼10,000 platelets per mm2) on 50-kPa PA gels and stiffer. In fact, the number of adherent platelets on 50- and 100-kPa gels is similar to the number of platelets adhered to fibrinogen-coated glass surfaces, which have a stiffness of >50 GPa (23), with no statistical differences (Fig. 1 B and C). The average spread area of adherent platelets on gels of different stiffnesses also exhibited a similar trend as that of platelet adhesion and reached a similar plateau (∼23 μm2) on 50-kPa gels and stiffer (Fig. 1 B, D, and E). According to this trend, we used three stiffnesses, 0.5, 5, and 50 kPa, for the remainder of our experiments. In addition, for all of our studies, platelets were incubated for 2 h on fibrinogen-conjugated PA gels of different elastic moduli, ensuring that platelets, which may adhere more slowly on softer gels, have sufficient time to spread and activate.
As a previous study demonstrated that different concentrations of fibrinogen affects platelet adhesion and spreading (24), we conjugated fibrinogen at a lower (3 µg/mL) and a higher concentration (100 µg/mL) on PA gels of different elastic moduli ranging from 0.25 to 100 kPa. Although platelet adhesion and spreading were diminished overall at the higher fibrinogen concentration, which is consistent with the aforementioned study (24), the substrate stiffness-mediated effects on platelet adhesion persisted (Fig. S2). This indicates that fibrinogen concentration and substrate stiffness affect platelet adhesion independently. In addition, we also determined that substrate stiffness-mediated platelet adhesion persisted under physiologic flow conditions at a shear rate of 100 s−1 (Fig. S3).
Platelet activation upon adhesion onto immobilized fibrinogen can cause secretion of agonists, such as ADP and thromboxane A2, which in turn can lead to paracrine activation of neighboring platelets. To rule out that the difference of adhesion and spreading of adherent platelets on gels of different stiffnesses is caused only by agonist secretion, we treated platelets with apyrase, an ADP-degrading enzyme, and acetylsalicylic acid, a thromboxane A2 inhibitor, during incubation. Although apyrase and acetylsalicylic acid decreased the number of adherent platelets on gels of all stiffnesses, it did not change the trend of increasing platelet adhesion or spreading with increasing substrate stiffness (Fig. S4 A and B). Interestingly, exposure to 10 μM ADP significantly increased platelet adhesion on the softest 0.5-kPa gels to ∼7,000 platelets per mm2, and that on gels of 5 kPa to ∼9,500 platelets per mm2, whereas it did not affect platelet adhesion on gels of 50 kPa, thus “shifting” the plateau of platelet adhesion to a lower gel stiffness of 5 kPa (Fig. S4A). This increased platelet adhesion is likely due to the inside-out activation of integrin αIIbβ3 by exogenous ADP (7). In addition, ADP exposure significantly increased the average spreading of adherent platelets on both 5- and 50-kPa gels (∼28 μm2), whereas ADP-exposed platelets remained unspread on 0.5 kPa (∼9 μm2) (Fig. S4B). This also rules out the possibility that the comparably lower adhesion and spreading of platelets on soft substrates is due only to slower activation of integrin αIIbβ3 on soft substrates. Taken together, platelet mechanosensing during adhesion and spreading is not mediated by the secretion of agonists ADP and thromboxane A2.
Platelet Mechanosensing During Adhesion Is Mediated by Rac-1 Activity, Whereas Mechanosensing During Platelet Spreading Is Mediated by Myosin Activity and Actin Polymerization.
To investigate the underlying mechanisms that govern platelet mechanosensing during adhesion and spreading, we exposed platelets to a series of pharmacological cytoskeletal inhibitors. Specifically, we found that GTPase Rac-1 inhibition with NSC23766 eliminated the substrate stiffness-mediated effect on both platelet adhesion and spreading on PA gels of all stiffnesses in a dose-dependent manner (Fig. 2 A and B, and Fig. S5). Indeed, when platelets were exposed to 300 μM NSC23766, platelet adhesion and spreading on both 5- and 50-kPa gels decreased to the levels of those observed on gels of 0.5 kPa.
Fig. 2.

Rac1 mediates platelet mechanosensing during adhesion, whereas Rac1, myosin, and actin polymerization mediate platelet mechanosensing during spreading. (A) Rac1 inhibition attenuates platelet mechanosensing during adhesion on fibrinogen-conjugated PA gels in a dose-dependent fashion. (B) Likewise, Rac1 inhibition also attenuates platelet mechanosensing during spreading on fibrinogen-conjugated PA gels in a dose-dependent fashion. (C) The number of platelets treated with vehicle (DMSO) or other cytoskeletal inhibitors after adhering onto PA gels of different stiffnesses. Inhibition of ROCK, MLCK, or actin polymerization does not alter the substrate stiffness-mediated effects on platelet adhesion. (D) On the other hand, inhibition of ROCK, MLCK, or actin polymerization all attenuate the substrate stiffness-mediated effects on platelet adhesion. (*P < 0.05; n = 3 experiments; error bars indicate SD.)
In nucleated adherent cells, mechanotransduction is actomyosin mediated and affects cellular processes such as motility and spreading (25, 26). In platelets, actin polymerization and myosin activity is also known to mediate shape change after activation (27, 28). Therefore, in separate experiments, we pretreated platelets with two inhibitors of myosin light chain phosphorylation, ML-7 and Y-27632, which inhibit Ca2+/calmodulin-dependent myosin light chain kinase (MLCK) and Rho kinase (ROCK), respectively, as well as latrunculin A, which inhibits actin polymerization. Regarding adhesion, MLCK, ROCK, and actin polymerization inhibition did not affect the overall trend of increasing platelet adhesion with increasing substrate stiffness (Fig. 2C and Fig. S6). However, in terms of platelet spreading, the substrate stiffness-mediated effects were eliminated with MLCK and actin polymerization inhibition but not with ROCK inhibition (Fig. 2D and Fig. S6). This is consistent with a recent report showing that platelet adhesion and spreading on immobilized fibrinogen involve different signaling pathways (29). These results indicate that, during spreading, platelet mechanosensing involves myosin activity and actin polymerization.
As Src family kinases (SFKs), including Lyn and Src, activate upstream of actin polymerization and play a major role in integrin αIIbβ3-related outside-in signaling (30, 31), we also explored the effect of SFK inhibitor PP2 on platelet mechanosensing. Similar to latrunculin A and ML-7, PP2 did not block the substrate stiffness-mediated effects on platelet adhesion, but did block substrate stiffness-mediated effects on platelet spreading (Fig. S7). In addition, as focal adhesion kinase (FAK) interacts with myosin contractility and is involved in mechanotransduction in nucleated cells, we also explored whether FAK regulates platelet mechanosensing using a FAK inhibitor but did not observe any detectable effect (Fig. S8).
Substrate Stiffness Mediates Platelet Activation.
As platelet adhesion and spreading triggers downstream platelet activation and signaling, we then investigated how substrate stiffness mediates platelet activation. Platelet adhesion to fibrinogen leads to integrin αIIbβ3 activation on the platelet surface via outside-in signaling (32, 33). This, in turn, leads to further activation and platelet secretion of α- and dense granules. As granule contents are released into circulation, membrane proteins such as P-selectin translocate from the granule to the plasma membrane (34). During the latter stages of activation, a subset of platelets, known as procoagulant platelets, expose PS on their surface, which promotes thrombin generation and amplifies clot formation (35). Therefore, we used integrin αIIbβ3 activation, P-selectin surface expression, and PS exposure as markers of platelet activation (Fig. 3).
Fig. 3.

Platelet activation is mediated by substrate stiffness. (A) Increased substrate stiffness increases activation of the αIIbβ3 platelet integrin as measured by PAC-1–FITC immunostaining via confocal microscopy. When normalized to spread platelet surface area, the average intensity is higher for the fibrinogen-conjugated 5- and 50-kPa gels compared with 0.5-kPa gels. (B) Inhibition of Rac1, SFK, MLCK, and actin polymerization attenuates the substrate stiffness-mediated αIIbβ3 activation of adherent platelets as measured by the average intensity of PAC-1–FITC staining. (C) Increased substrate stiffness increases P-selectin expression on adherent platelets as measured by P-selectin immunostaining (red: fluorescent membrane dye; green: P-selectin immunostaining) via confocal microscopy. (D) Inhibition of Rac1, SFK, ROCK, and MLCK differentially attenuate the substrate stiffness-mediated P-selectin expression on adherent platelets. (E) PS exposure was found only on platelets adhered onto stiffer gels of 5 and 50 kPa as measured as measured by Annexin V staining via confocal microscopy. (F) Inhibition of Rac1, SFK, MLCK, and actin polymerization abolished the substrate stiffness-mediated PS exposure on adherent platelets. (P < 0.05; n = 3 experiments; error bars indicate SD.) (Scale bar: 10 μm.)
Using fluorescently conjugated PAC1, an antibody that specifically binds to activated αIIbβ3 integrins (36), we quantified PAC-1–FITC intensity using confocal microscopy. Although all adherent platelets positively stained, the level of integrin αIIbβ3 activation differed among platelets adhered on PA gels of different substrate stiffnesses. The average intensity of adherent platelets on 0.5-kPa gels (∼4,000 A.U./platelets) was significantly lower than that of platelets on 5- and 50-kPa gels (∼11,000 A.U./platelets) (Fig. 3A). All adherent platelets on soft gels of 0.5 kPa remained less spread with less integrin αIIbβ3 activation, even when normalized for platelet spreading area (Fig. 3A). We also observed that the few unspread platelets on stiffer gels also exhibited less overall normalized integrin αIIbβ3 activation. This suggests that integrin αIIbβ3 activation is closely regulated by platelet mechanosensing during spreading and is further confirmed when inhibition of platelet spreading via latrunculin A, PP2, ML-7, and NSC23766 decreased the average PAC-1–FITC intensity of adherent platelets on both 5- and 50-kPa gels to the same level as platelets on 0.5-kPa gels (Fig. 3B).
P-selectin immunostaining, a marker for α-granule secretion (34, 37), on nonpermeabilized platelets revealed that substrate stiffness also affected platelet P-selectin expression. The ratio of P-selectin–expressing platelets to total adherent platelets on 0.5-kPa gels was ∼18% and is significantly lower than that of platelets adhered onto stiffer 5- and 50-kPa gels, which was ∼55% (Fig. 3C). Several studies have suggested that platelet α-granule secretion and P-selectin translocation to the platelet surface occurs during shape change (34). Our results are consistent with this—as platelet spreading increased on stiffer substrates, P-selectin expression increased as well (Fig. 3C). Inhibition of spreading using NSC23766, ML-7, and PP2 decreased the ratio of P-selectin–expressing platelets on 5- and 50-kPa gels to the same level as that on the softest 0.5-kPa gels (Fig. 3D). However, the role of actin polymerization on P-selectin expression of adherent platelet is more complex. Although treatment with latrunculin A completely inhibited platelet spreading on stiffer gels, it did not block P-selectin expression on adherent platelets on gels of different stiffnesses. Low latrunculin concentrations have been reported to actually increase platelet P-selectin expression possibly due to inhibition of actin polymerization, thereby increasing cell membrane fusion and P-selectin exocytosis, which may also be the case in our experiments (38).
We lastly tested how substrate stiffness affected procoagulant activity of activated platelets by measuring the percentage of PS-exposed adherent platelets via Annexin V staining. Our data revealed no detectable PS-exposed platelets on 0.5-kPa gels. However, on 5- and 50-kPa gels, ∼15–20% of adherent platelets exposed PS, again indicating that higher substrate stiffness leads to more platelet activation (Fig. 3E). Moreover, PS exposure was only found on spread platelets on both 5- and 50-kPa gels. In addition, with inhibition of actin polymerization and myosin contractility, PS exposure was completely inhibited on platelets adhered on the stiffer 5- and 50-kPa gels (Fig. 3F). Taken together, these results indicate that the procoagulant activity of platelets is actomyosin mediated and is coupled with the mechanotransductive signals that occur during spreading.
Discussion
Upon vascular injury, platelets are known to activate and respond to the hemodynamic and biochemical cues of the microenvironment, which ultimately leads to the formation of a hemostatic plug or thrombus. Here, we report for the first time (to our knowledge) that simply modulating substrate stiffness, as a pure and isolated variable that is precisely and quantitatively defined by the elastic modulus, directly changes the level of platelet adhesion, spreading, and activation. As the elastic modulus is an intrinsic physical property of a material that is independent of biological and biochemical factors, measurements of this purely mechanical variable can theoretically be used in concert with biological and biochemical characterization techniques to help predict how platelets will physiologically respond to different in vivo microenvironments and engineered biomaterials. The substrate stiffnesses used in this study span the physiologic elastic moduli of substrates that platelets might encounter in vivo during clot formation, including fibrinogen-coated vessel walls (39, 40), fibrin networks (13, 41), and individual fibrin fibers (13, 42). Our findings have significant implications for our understanding of hemostasis and thrombosis, establishing that the elastic modulus of the developing clot, in and of itself, is sufficient to predictably mediate platelet adhesion, spreading, and activation.
Regarding the underlying mechanisms of our observations, platelet mechanosensing of the underlying substrate likely initiates as the αIIbβ3 integrin engages with fibrinogen/fibrin (Fig. 4). As the platelet initiates contact, resistance of the substrate, which is dictated by its stiffness, balances the force generated by the inertial movement of the engaged platelet. Softer substrates will deform, exert less resistant force, and engage the integrin in a low-affinity state, which will cause relatively less αIIbβ3 integrin activation and platelet adhesion. Interestingly, this decreased outside-in αIIbβ3 integrin activation can be bypassed via platelet activation by exogenous ADP. Platelet activation with ADP leads to inside-out αIIbβ3 integrin activation, therefore significantly increasing platelet adhesion on the softer substrates (Fig. S4). Larger resistant forces from stiffer substrates will likely switch αIIbβ3 integrins to the activated, extended state, increasing affinity, prolonging the lifetime of binding, and triggering stronger outside-in signaling than softer substrates (Fig. 4). Our data also indicate that, further downstream, Rac1 also mediates platelet mechanosensing of substrate stiffness. In platelets, Rac1 acts downstream of integrin αIIbβ3 outside-in signaling to regulate lamellipodia formation, platelet aggregation, and granule secretion (43, 44), and can activate Rap1b, which is involved in inside-out integrin αIIbβ3 activation (45). As Rac1 inhibition decreases platelet adhesion and activation on stiffer gels in a dose-dependent manner, it is likely that this integrin–Rac1–Rap1b circuit provides the mechanosensing machinery during platelet adhesion, further αIIbβ3 activation, and integrin clustering when platelets adhered on stiffer substrates, thus increasing adhesion as substrate stiffness increases. To balance the higher resistant forces of stiffer substrates, platelet actomyosin contractility increases, leading to further platelet spreading and activation, including granule secretion and PS exposure. However, as cytoskeletal inhibitors are not completely specific, there is a possibility that other signaling pathways may be involved as well.
Fig. 4.
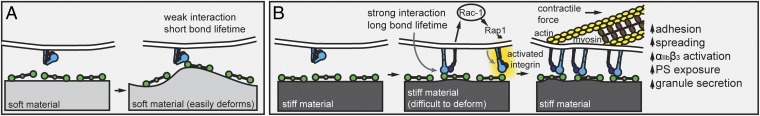
Proposed mechanisms of how substrate stiffness affects platelet activation. Platelet mechanosensing of the microenvironment, which includes heterogeneous fibrin polymer networks of differing mechanical properties, likely starts with the initial adhesive event, during which the resistant substrate force balances the inertial movement of the platelet. (A) A platelet engaging with a relatively soft substrate results in a weak αIIbβ3–fibrinogen interaction and short bond lifetime. (B) A stiffer substrate, however, would provide more resistant force leading to more platelet adhesion and outside-in αIIbβ3 signaling. This in turn, would generate a high acto-myosin–mediated internal balancing force, resulting in more platelet spreading on stiffer substrate. Downstream signals will then further trigger platelet activation, thus up-regulating more αIIbβ3 activation, granule secretion, and PS exposure.
Our observations are also pertinent from the perspectives of cellular mechanics and mechanotransduction, further establishing the platelet as a unique cellular entity. A major focus in cell biology is currently devoted to investigating the role of the nucleus, nuclear lamina, and nucleoskeleton in cellular mechanosensing (46–48). As the platelet is anucleate and its cytoskeletal machinery is relatively simpler than that of other nucleated cells (49), our data show that mechanosensing can occur and plays important roles even when the cellular structural building blocks are fairly basic. Therefore, we have discovered that functionally important mechanosensing does not depend on the presence of the nucleus or connections between the nucleoskeleton and cytoskeleton. In addition, our work establishes the platelet as a (relatively) simplified, experimental model to study the basic fundamentals of cellular mechanics and mechanotransduction.
The 2D nature of our data is an inherent limitation of our experimental setup as in vivo thrombi and clots are obviously 3D. To that end, we formed 3D fibrin networks with different fibrinogen concentrations, which directly affect fibrin stiffness (13), and observed that platelets in the stiffer fibrin networks exhibited stronger PS staining than platelets in softer fibrin networks (Fig. S9 A–D). Although these experiments demonstrate that altering the architecture, and therefore stiffness, of the fibrin network directly affects platelet activation, use of 3D fibrin matrices is associated with several potentially confounding factors. Indeed, varying fibrinogen concentration upon fibrin polymerization not only alters the stiffness of the network but also changes ligand density, fibrin porosity, fibrin fiber length and thickness, and fibrin branching (13, 41). Therefore, the 2D and 3D approaches complement each other in that the 2D PA studies enable rigorous and systematic investigation of how matrix stiffness influences platelet physiology, and the 3D fibrin studies provide some confirmation that predictions from these 2D studies are also observed in a matrix platform that more closely resembles in vivo clots.
Our findings may also provide insight into platelet aggregation and help explain the heterogeneity of platelet activation in a growing thrombus, which has clearly been documented in recent work (14, 50). During thrombus formation and platelet aggregation, platelets interact and bind via monomeric fibrinogen, which functions as the bridging molecule between adjacent platelets (51). Whereas biological agonists such as thrombin, ADP, and epinephrine are known to activate platelet aggregation, our data suggest that the mechanical microenvironment plays a potential role in this process as well.
Our results also add a previously unidentified dimension to the literature investigating platelet interactions with fibrinogen surfaces. Several groups have observed that multilayered fibrinogen decreases platelet adhesion (52) and that the fibrinogen density significantly affects platelet adhesion (24). Our findings suggest that fibrinogen concentration and substrate stiffness are independent factors that affect platelet adhesion and spreading and appear to affect platelet behavior synergistically, although the relationship between the effects may not be linear. Consistent with the data published by Jirousková et al. (24), platelet spreading on glass surfaces coated with lower fibrinogen concentrations (3 μg/mL) was significantly increased compared with that with higher fibrinogen concentrations (100 μg/mL) (Fig. S2). At low fibrinogen concentrations, platelet spreading increased as substrate stiffness increased (Fig. 1). In contrast, at high fibrinogen concentrations, platelet spreading was significantly diminished at all stiffness conditions including glass, although the overall substrate stiffness-mediated trend persisted (Fig. S2). Therefore, both substrate stiffness and fibrinogen density affect platelet spreading. Mechanistically, it has been suggested that lower concentrations of immobilized fibrinogen expose more αIIbβ3 binding sites due to the horizontal orientation of fibrinogen molecules on the surface and, therefore, cause increased platelet spreading (24, 53). On the other hand, higher concentrations of immobilized fibrinogen may expose only the γ-chain C-terminal αIIbβ3 binding sites due to the vertical orientation of fibrinogen, which is likely caused by molecular “crowding” on the surface. This, in turn, could lead to decreased αIIbβ3 binding and decreased platelet spreading overall. In greater context, this phenomenon likely works in concert with substrate stiffness, as spreading platelets engage immobilized fibrinogen with differing resistance forces due to differences in substrate stiffness. Therefore, platelet spreading is likely most pronounced at relatively low fibrinogen concentrations, which maximizes exposure of αIIbβ3 recognition sites, and at relatively high substrate stiffness, which maximizes αIIbβ3 affinity and outside-in signaling.
Leveraging a technique that specifically varies only the mechanical properties of the surface while maintaining the fibrinogen chemistry and density constant, we observed that substrate stiffness, in and of itself, significantly alters platelet adhesion, spreading, and activation as well. This mechanical microenvironmental parameter of platelet physiology also has profound implications for improving the biocompatibility of engineered biomaterials. Simply modifying the stiffness of catheters and medical implants could enable bioengineers to significantly decrease platelet deposition and clot formation on those surfaces, which is currently a major cause of device failure and patient morbidity.
Materials and Methods
Glass slides (12 mm in diameter) were silanized by surface activation with oxygen plasma for 1 min followed by treatment with 10% (vol/vol) (3-aminopropyl)-trimethoxysilane (Sigma) in 95% ethanol solution at 60 °C for 1 h. Silanized glasses were then treated with 0.5% glutaraldehyde (Sigma) for 30 min. Prepolymer solutions with different ratios of acrylamide to bis-acrylamide (Sigma) were cross-linked on the treated glass surface with 0.1% ammonium persulfate and tetramethylethylenediamine to fabricate PA gels with different stiffnesses. PA gels were then soaked in Sulfo-SANPAH (Pierce)-Hepes buffer solution (0.25 mg/mL; pH 8.25) and exposed to UV light (365 nm) for 10 min. The PA gels were washed three times with Hepes buffer (pH 8.25) to remove Sulfo-SANPAH residues and then incubated in 3 μg/mL fibrinogen solution at 37 °C overnight. The fibrinogen density was confirmed to be constant on the PA gels of different stiffnesses (Fig. S1). Platelet adhesion, spreading, and activation were analyzed after pretreated or nontreated platelets were incubated on the PA gels for 2 h. Detailed explanations of materials and methods can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
Finanical support for this work was provided by National Science Foundation CAREER Award 1150235 (to W.A.L.); National Science Foundation Grant CBET-0939511 (to G.B.); an American Heart Association Innovative Research Grant (to W.A.L.); National Institutes of Health Grants U54HL112309 (to W.A.L.), R01HL121264 (to W.A.L.), and PN2EY018244 (to G.B. and W.A.L.); and American Heart Association Postdoctoral Fellowships (to A.C.B. and D.R.M.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a Prearranged Editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322917111/-/DCSupplemental.
References
- 1.Jackson SP. Arterial thrombosis—insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–1436. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- 2.Brass LF, Wannemacher KM, Ma P, Stalker TJ. Regulating thrombus growth and stability to achieve an optimal response to injury. J Thromb Haemost. 2011;9(Suppl 1):66–75. doi: 10.1111/j.1538-7836.2011.04364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coller BS, Shattil SJ. The GPIIb/IIIa (integrin alphaIIbbeta3) odyssey: A technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112(8):3011–3025. doi: 10.1182/blood-2008-06-077891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8(11):1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 5.Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: The platelet paradigm. Blood. 1998;91(8):2645–2657. [PubMed] [Google Scholar]
- 6.Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res. 2007;120(Suppl 1):S5–S9. doi: 10.1016/j.thromres.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. 2006;99(12):1293–1304. doi: 10.1161/01.RES.0000251742.71301.16. [DOI] [PubMed] [Google Scholar]
- 8.Heemskerk JWM, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. 2002;88(2):186–193. [PubMed] [Google Scholar]
- 9.Schoenwaelder SM, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114(3):663–666. doi: 10.1182/blood-2009-01-200345. [DOI] [PubMed] [Google Scholar]
- 10.Sheriff J, Bluestein D, Girdhar G, Jesty J. High-shear stress sensitizes platelets to subsequent low-shear conditions. Ann Biomed Eng. 2010;38(4):1442–1450. doi: 10.1007/s10439-010-9936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996;88(5):1525–1541. [PubMed] [Google Scholar]
- 12.Campbell RA, Overmyer KA, Selzman CH, Sheridan BC, Wolberg AS. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114(23):4886–4896. doi: 10.1182/blood-2009-06-228940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan EA, Mockros LF, Weisel JW, Lorand L. Structural origins of fibrin clot rheology. Biophys J. 1999;77(5):2813–2826. doi: 10.1016/S0006-3495(99)77113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stalker TJ, et al. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121(10):1875–1885. doi: 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol. 2005;204(1):198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- 16.Yeung T, et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60(1):24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 17.Sen S, Ng WP, Kumar S. Contractility dominates adhesive ligand density in regulating cellular de-adhesion and retraction kinetics. Ann Biomed Eng. 2011;39(4):1163–1173. doi: 10.1007/s10439-010-0195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam WA, et al. Extracellular matrix rigidity modulates neuroblastoma cell differentiation and N-myc expression. Mol Cancer. 2010;9:35. doi: 10.1186/1476-4598-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci USA. 2012;109(26):10334–10339. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009;69(10):4167–4174. doi: 10.1158/0008-5472.CAN-08-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tse JR, Engler AJ. Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb1016s47. Chapter 10:Unit 10.16. [DOI] [PubMed] [Google Scholar]
- 22.Pelham RJ, Wang YL. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA. 1997;94(25):13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inaba S, Fujino S, Morinaga K. Young's modulus and compositional parameters of oxide glasses. J Am Ceram Soc. 1999;82(12):3501–3507. [Google Scholar]
- 24.Jirousková M, Jaiswal JK, Coller BS. Ligand density dramatically affects integrin alpha IIb beta 3-mediated platelet signaling and spreading. Blood. 2007;109(12):5260–5269. doi: 10.1182/blood-2006-10-054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2(12):a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark K, Langeslag M, Figdor CG, van Leeuwen FN. Myosin II and mechanotransduction: A balancing act. Trends Cell Biol. 2007;17(4):178–186. doi: 10.1016/j.tcb.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Kiss E, et al. Integrin-linked kinase phosphorylates the myosin phosphatase target subunit at the inhibitory site in platelet cytoskeleton. Biochem J. 2002;365(Pt 1):79–87. doi: 10.1042/BJ20011295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox JEB. The platelet cytoskeleton. Thromb Haemost. 1993;70(6):884–893. [PubMed] [Google Scholar]
- 29.Shen B, et al. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. 2013;503(7474):131–135. doi: 10.1038/nature12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obergfell A, et al. Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol. 2002;157(2):265–275. doi: 10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao J, Zoller KE, Ginsberg MH, Brugge JS, Shattil SJ. Regulation of the pp72syk protein tyrosine kinase by platelet integrin alpha IIb beta 3. EMBO J. 1997;16(21):6414–6425. doi: 10.1093/emboj/16.21.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30(12):2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibbins JM. Platelet adhesion signalling and the regulation of thrombus formation. J Cell Sci. 2004;117(Pt 16):3415–3425. doi: 10.1242/jcs.01325. [DOI] [PubMed] [Google Scholar]
- 34.Furie B, Furie BC, Flaumenhaft R. A journey with platelet P-selectin: The molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost. 2001;86(1):214–221. [PubMed] [Google Scholar]
- 35.Sims PJ, Wiedmer T. Unraveling the mysteries of phospholipid scrambling. Thromb Haemost. 2001;86(1):266–275. [PubMed] [Google Scholar]
- 36.Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J Biol Chem. 1985;260(20):11107–11114. [PubMed] [Google Scholar]
- 37.Merten M, Thiagarajan P. P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation. 2000;102(16):1931–1936. doi: 10.1161/01.cir.102.16.1931. [DOI] [PubMed] [Google Scholar]
- 38.Flaumenhaft R, et al. The actin cytoskeleton differentially regulates platelet alpha-granule and dense-granule secretion. Blood. 2005;105(10):3879–3887. doi: 10.1182/blood-2004-04-1392. [DOI] [PubMed] [Google Scholar]
- 39.Massberg S, et al. Fibrinogen deposition at the postischemic vessel wall promotes platelet adhesion during ischemia-reperfusion in vivo. Blood. 1999;94(11):3829–3838. [PubMed] [Google Scholar]
- 40.Stroka KM, Vaitkus JA, Aranda-Espinoza H. Endothelial cells undergo morphological, biomechanical, and dynamic changes in response to tumor necrosis factor-α. Eur Biophys J. 2012;41(11):939–947. doi: 10.1007/s00249-012-0851-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weisel JW. The mechanical properties of fibrin for basic scientists and clinicians. Biophys Chem. 2004;112(2-3):267–276. doi: 10.1016/j.bpc.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 42.Brown AEX, Litvinov RI, Discher DE, Purohit PK, Weisel JW. Multiscale mechanics of fibrin polymer: Gel stretching with protein unfolding and loss of water. Science. 2009;325(5941):741–744. doi: 10.1126/science.1172484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aslan JE, McCarty OJT. Rho GTPases in platelet function. J Thromb Haemost. 2013;11(1):35–46. doi: 10.1111/jth.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCarty OJT, et al. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280(47):39474–39484. doi: 10.1074/jbc.M504672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stefanini L, et al. Rap1-Rac1 circuits potentiate platelet activation. Arterioscler Thromb Vasc Biol. 2012;32(2):434–441. doi: 10.1161/ATVBAHA.111.239194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertrand AT, et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J Cell Sci. 2014;127(Pt 13):2873–2884. doi: 10.1242/jcs.144907. [DOI] [PubMed] [Google Scholar]
- 47.Swift J, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341(6149):1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin JW, et al. Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc Natl Acad Sci USA. 2013;110(47):18892–18897. doi: 10.1073/pnas.1304996110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitsios JV, et al. What is vinculin needed for in platelets? J Thromb Haemost. 2010;8(10):2294–2304. doi: 10.1111/j.1538-7836.2010.03998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Munnix ICA, Cosemans JMEM, Auger JM, Heemskerk JWM. Platelet response heterogeneity in thrombus formation. Thromb Haemost. 2009;102(6):1149–1156. doi: 10.1160/TH09-05-0289. [DOI] [PubMed] [Google Scholar]
- 51.Brass LF, Zhu L, Stalker TJ. Minding the gaps to promote thrombus growth and stability. J Clin Invest. 2005;115(12):3385–3392. doi: 10.1172/JCI26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Podolnikova NP, et al. Control of integrin alphaIIb beta3 outside-in signaling and platelet adhesion by sensing the physical properties of fibrin(ogen) substrates. Biochemistry. 2010;49(1):68–77. doi: 10.1021/bi9016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dyr JE, et al. Molecular arrangement of adsorbed fibrinogen molecules characterized by specific monoclonal antibodies and a surface plasmon resonance sensor. Sens Actuators B Chem. 1998;51(1-3):268–272. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.