

Abstract
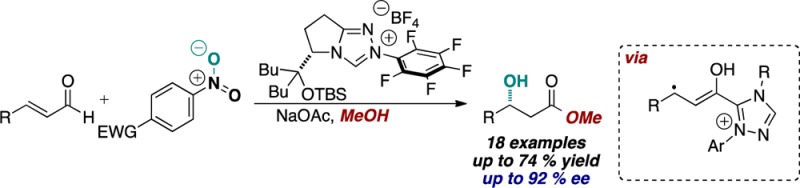
A novel oxidative N-heterocyclic carbene-catalyzed reaction pathway has been discovered. Alkyl and aryl enals undergo β-hydroxylation via oxygen atom transfer from electron-deficient nitrobenzenes, followed by trapping of the resultant acyl azolium by the solvent. The proposed mechanism involves a single electron transfer event to initiate the reaction followed by radical recombination. This represents a profound mechanistic departure from the established two-electron disconnects in NHC catalysis.
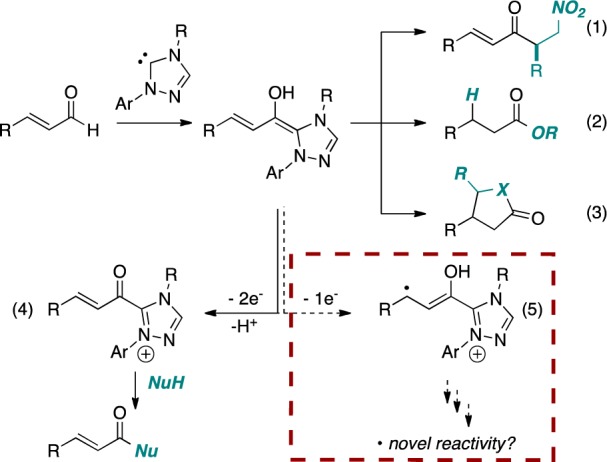
Over the past decade, N-heterocyclic carbene (NHC) catalysis has received considerable attention from the synthetic community.1 The advent of NHC catalysis embodied acyl anion reactivity exemplified by the benzoin and Stetter reactions.2 The presence of a nearby leaving group or a reducible functionality opened the door to NHC-based redox catalysis.3 The use of α,β-unsaturated aldehydes as substrates for NHC catalysis is particularly illustrative (Figure 1): they have been demonstrated to undergo the asymmetric Stetter reaction (eq 1),4 β-protonation/esterification by the redox pathway (eq 2),5 or trapping with an exogenous aldehyde,6 imine,7 or nitroalkene8 from the homoenolate-type position (eq 3). Direct oxidation of enals has also been accomplished under NHC catalysis (eq 4).9 All of these previously described NHC-catalyzed reactions presumably operate via a two-electron manifold.10 We hypothesized that it may be possible to access a new single-electron pathway through the judicious choice of a single-electron oxidant (eq 5), which would enable an entirely new class of reactions.11 Herein we disclose our results.
Figure 1.
Background.
In our previous studies of the aza-Breslow intermediates, we found that they exhibit a well-behaved, readily reversible oxidation at −0.49 eV, suggestive of a single-electron cycle.12 Using this as an approximation for the reactivity of the oxo-Breslow intermediate, we screened a number of potential one-electron oxidants (Table 1). While exploring electron-deficient pyridine N-oxides, we were surprised to find that the use of 4-nitropyridine N-oxide (2a) as an oxidant with a carbene catalyst and cinnamaldehyde in methanol afforded the corresponding β-hydroxy ester 3a in 40% yield. The remainder of the mass balance is unsaturated methyl cinnamate (4a).
Table 1. Oxidant Screen.
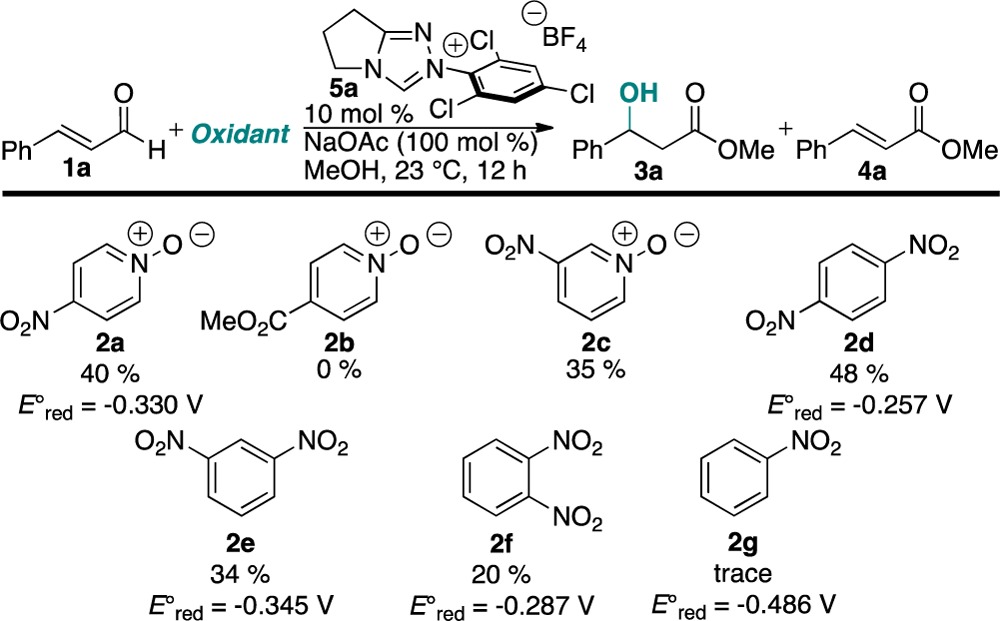
Initially, we hypothesized that oxygen transfer arose from the N-oxy substituent of 2a. After screening a variety of pyridine N-oxides, we found that only nitro-containing N-oxides provided the product. We then explored nitrobenzenes and found that a variety of sufficiently electron-deficient nitrobenzenes are capable of effecting the β-hydroxylation. Nitrosoarenes and over-reduced products were observed in the reaction mixture, implicating the nitro group in the oxygen transfer step.13,14 The reduction potential of the nitrobenzene derivative plays a critical role in the reactivity, with potentials greater than −0.49 V being required for oxidation of the Breslow intermediate.15 We believe that this explains the low conversion with nitrobenzene (Ered° = −0.486 V).16
The scope of the reaction proved remarkably broad (Table 2), with the reaction being tolerant of aryl, heteroaryl, and alkyl substitution at the β position of the enal. Strongly electron-deficient systems lead to lower yields, with increased amounts of β-protonation byproducts. Aliphatic substrates behave particularly well under these conditions, affording some of the highest yields. Indeed, the chemistry is even tolerant of β,β-disubstitution, affording tertiary alcohols as products in reasonable yields.
Table 2. Scope of the Racemic Reactiona,b.
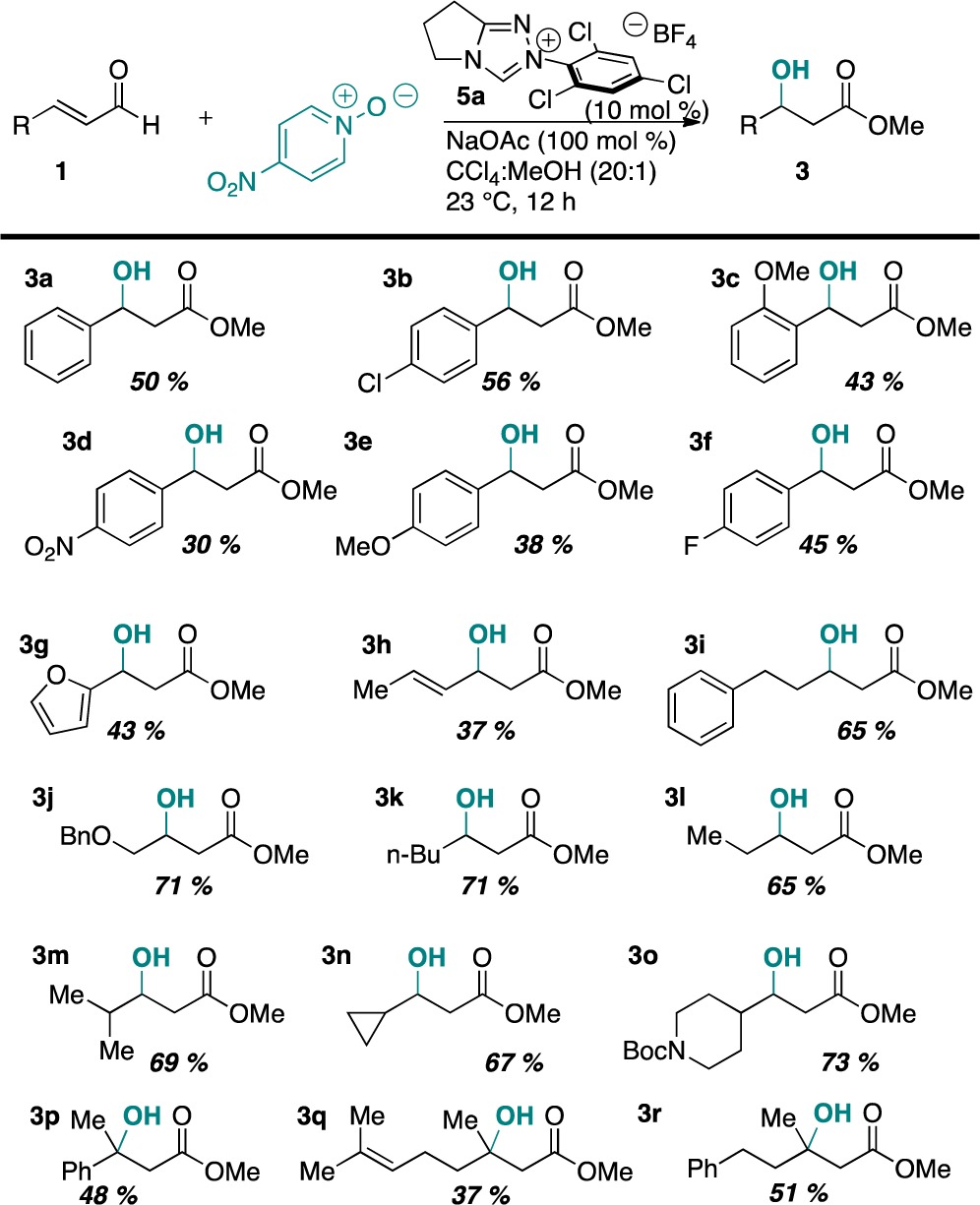
Reactions were conducted with 1.0 equiv of 1 and 1.5 equiv of 2.
Isolated yields after chromatography are shown.
We then turned our focus to the asymmetric variant of the reaction (Table 3). Through a chiral catalyst screen, we found that our previously developed O-silyl-protected NHC scaffold 5g provides the product with the highest selectivity.17 The use of mixed solvents provides an increase in enantioselectivity. A 20:1 mixture of carbon tetrachloride and methanol proved optimal, delivering β-hydroxy ester 3a in 45% yield with 92% ee. The nitrobenzene oxidant also plays a role in the selectivity, with 4-nitropyridine N-oxide providing the best results.
Table 3. Optimization of the Asymmetric Varianta.
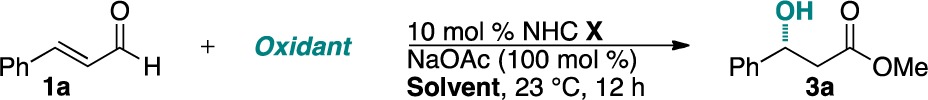
| entry | NHC | oxidant | solvent | yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | 5b | 2a | MeOH | 39 | –25 |
| 2 | 5c | 2a | MeOH | 36 | –7 |
| 3 | 5d | 2a | MeOH | 15 | 66 |
| 4 | 5e | 2a | MeOH | 22 | 0 |
| 5 | 5f | 2a | MeOH | 42 | –8 |
| 6 | 5g | 2a | MeOH | 51 | 31 |
| 7 | 5g | 2a | DCM/MeOH (20:1) | 36 | 79 |
| 8 | 5g | 2a | toluene/MeOH (20:1) | 30 | 87 |
| 9 | 5g | 2a | trifluorotoluene/MeOH (20:1) | 44 | 88 |
| 10 | 5g | 2a | CCl4/MeOH(20:1) | 45 | 92 |
| 11 | 5g | 2d | CCl4/MeOH (20:1) | 34 | 84 |
| 12 | 5g | 2e | CCl4/MeOH (20:1) | 32 | 76 |
| 13 | 5g | 2f | CCl4/MeOH (20:1) | 12 | 16 |
Reactions were conducted with 1.0 equiv of 1a and 1.5 equiv of 2.
Isolated yields after chromatography.
Enantiomeric excess was determined by HPLC analysis on a chiral stationary phase.
The scope of the asymmetric reaction was then explored (Table 4). Both aryl and aliphatic enals are tolerated. In most cases, the remainder of the mass balance is the corresponding α,β-unsaturated methyl ester. Control experiments confirmed that unsaturated esters do not form from elimination of the β-hydroxyl group. We were pleased to find that electron-rich and electron-deficient enals participate in the reaction. The reaction of 4-nitrocinnamaldehyde provided 3d in 20% yield, presumably as a result of rapid β-protonation that outcompetes oxidation. In all other cases, no β-protonation was observed. A variety of aliphatic enals participate in the reaction. Notably, enals 1j and 1o bearing a potential leaving group at the γ-position and a protected amine, respectively, are competent substrates.
Table 4. Scope of the Asymmetric Reactiona.
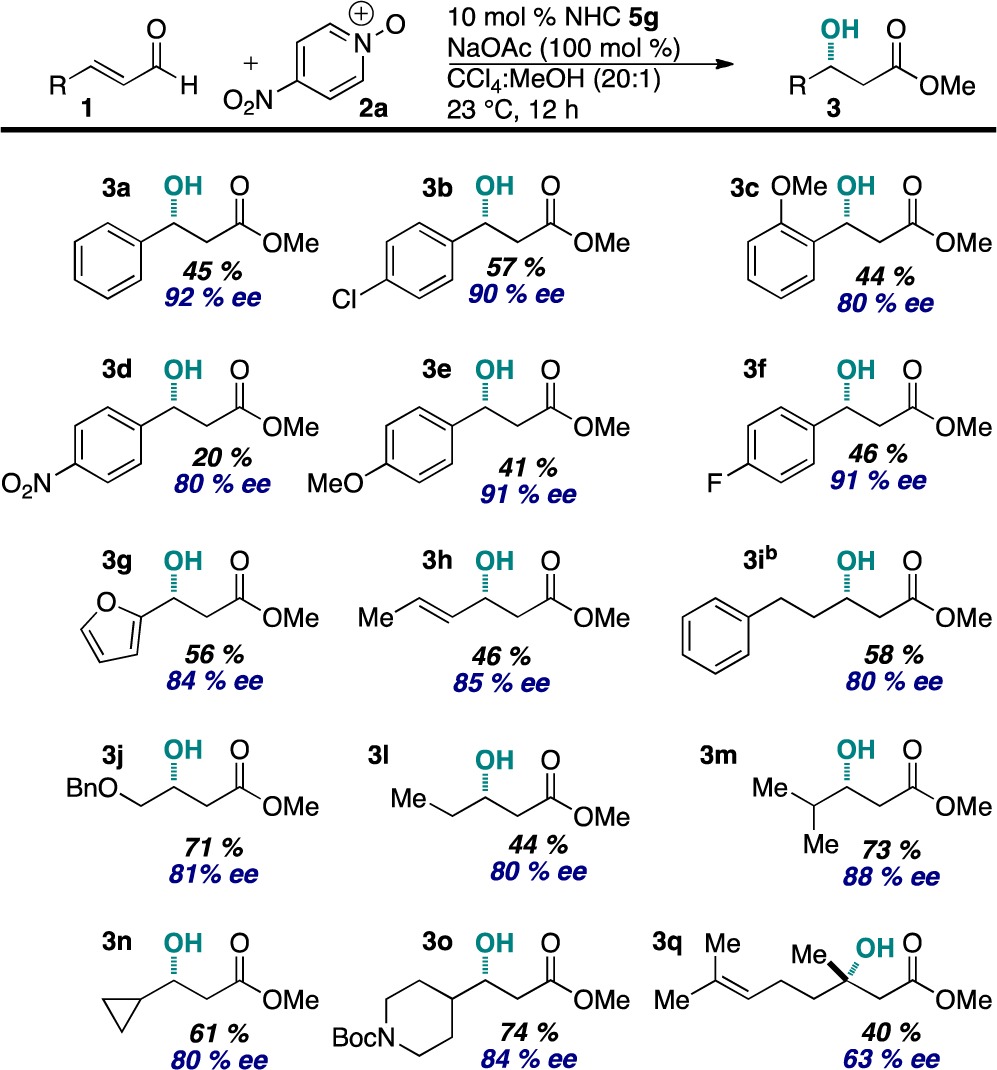
See footnotes a–c in Table 3.
The reaction was conducted in PhCF3/MeOH (20:1).
Mechanistically, we believe that this reaction proceeds first by formation of the Breslow intermediate II (Scheme 1), which undergoes a subsequent single-electron oxidation by the nitrobenzene to furnish Breslow-centered radical cation IIIa and nitrobenzene-derived radical anion IIIb. These radicals can then combine through a homoenolate-centered radical and an oxygen-centered radical to form intermediate IV, which subsequently collapses to expel an NHC-bound alkoxide and nitrosobenzene V. The NHC-bound alkoxide then reacts with methanol to liberate the catalyst and generate β-hydroxy ester VI. The α,β-unsaturated methyl ester VIII may form via deprotonation of intermediate III followed by a second abstraction of a single electron to furnish α,β-unsaturated acyl azolium VII, which is then intercepted with methanol.18
Scheme 1. Proposed Catalytic Cycle.

We entertained a few alternative mechanistic hypotheses. We first wondered whether the reaction proceeds by an initial epoxidation of the enal followed by an NHC-catalyzed epoxide opening.3a,19 However, control experiments in the absence of the NHC catalyst led to no observable epoxidation, and the enal was fully recovered (Scheme 2, top). Another mechanism we considered involved initial two-electron oxidation of the Breslow intermediate to the α,β-unsaturated acyl azolium followed by 1,4-addition of hydroxide to furnish the β-hydroxy ester. We believe this pathway to be unlikely, as subjecting phenylpropiolaldehyde to a mixture of NHC, hydroxide, and methanol afforded no β-hydroxy ester (Scheme 2, bottom).20 We also consider this pathway to be unlikely because we never observed any amount of the β-methoxy product that one would expect from solvent addition to the α,β-unsaturated acyl azolium. Finally, we considered the possibility that the homoenolate effects a nucleophilic attack on the oxygen of the nitro group. The nitro group as an electrophilic source of oxygen is rare in the literature, reacting only with very strong nucleophiles such as the Grignard reagent in the Bartoli indole synthesis.21 Furthermore, there is strong evidence that this reaction proceeds via single electron transfer.22
Scheme 2. Control Experiments.

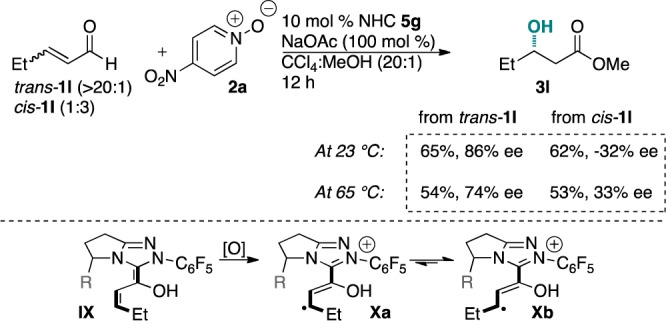
We then investigated the stereochemical course of the reaction when using cis- and trans-enals. If the reaction proceeds via a radical mechanism and the radical recombination is slower than bond rotation, the cis- and trans-enals should provide the same major enantiomer via interconversion of presumed intermediates Xa and Xb (Scheme 3). At ambient temperature, cis- and trans-olefin isomers gave opposite major enantiomers. However, at 65 °C we observed enantiomeric convergence, supporting the notion that the reaction proceeds via a discrete radical intermediate.23−25
Scheme 3. Stereochemical Probe.
In conclusion, we have discovered an NHC-catalyzed reaction that proceeds via a radical pathway. Crucial to the success of this reaction is the proper choice of oxidant. An asymmetric reaction has been developed utilizing a chiral NHC bearing a bulky silyl-protected tertiary alcohol, which delivers aldol adducts by a route complementary to established methods.
Acknowledgments
We thank NIGMS for generous support (GM72586) and Donald Gauthier (Merck Research Laboratories) for a generous gift of aminoindanol.
Supporting Information Available
Detailed experimental procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Moore J. L.; Rovis T. Top. Curr. Chem. 2010, 291, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vora H. U.; Rovis T. Aldrichimica Acta 2011, 44, 1. [PMC free article] [PubMed] [Google Scholar]; c Bugaut X.; Glorius F. Chem. Soc. Rev. 2012, 41, 3511. [DOI] [PubMed] [Google Scholar]
- For seminal references, see:; a Ciganek E. Synthesis 1995, 1311. [Google Scholar]; b Enders D.; Breuer K.; Runsink J.; Teles J. H. Helv. Chim. Acta 1996, 79, 1899. [Google Scholar]; c Kerr M. S.; Read de Alaniz J.; Rovis T. J. Am. Chem. Soc. 2002, 124, 10298. [DOI] [PubMed] [Google Scholar]
- a Chow K. Y.-K.; Bode J. W. J. Am. Chem. Soc. 2004, 126, 8126. [DOI] [PubMed] [Google Scholar]; b Reynolds N. T.; Read de Alaniz J.; Rovis T. J. Am. Chem. Soc. 2004, 126, 9518. [DOI] [PubMed] [Google Scholar]
- DiRocco D. A.; Rovis T. J. Am. Chem. Soc. 2011, 133, 10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chan A.; Scheidt K. A. Org. Lett. 2005, 7, 905. [DOI] [PubMed] [Google Scholar]; b Sohn S. S.; Bode J. W. Org. Lett. 2005, 7, 3873. [DOI] [PubMed] [Google Scholar]
- a Burstein C.; Glorius F. Angew. Chem., Int. Ed. 2004, 43, 6205. [DOI] [PubMed] [Google Scholar]; b Sohn S. S.; Rosen E. L.; Bode J. W. J. Am. Chem. Soc. 2004, 126, 14370. [DOI] [PubMed] [Google Scholar]
- a He M.; Bode J. W. Org. Lett. 2005, 7, 3131. [DOI] [PubMed] [Google Scholar]; b Raup D. E. A.; Cardinal-David B.; Holte D.; Scheidt K. A. Nat. Chem. 2010, 2, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhao X.; DiRocco D. A.; Rovis T. J. Am. Chem. Soc. 2011, 133, 12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nair V.; Sinu C. R.; Babu B. P.; Varghese V.; Jose A.; Suresh E. Org. Lett. 2009, 11, 5570. [DOI] [PubMed] [Google Scholar]; b Maji B.; Ji L.; Wang S.; Vedachalam S.; Rakesh G.; Liu X.-W. Angew. Chem., Int. Ed. 2012, 51, 8276. [DOI] [PubMed] [Google Scholar]; c White N. A.; DiRocco D. A.; Rovis T. J. Am. Chem. Soc. 2013, 135, 8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Maki B. E.; Scheidt K. A. Org. Lett. 2008, 10, 4331. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Guin J.; De Sarkar S.; Grimme S.; Studer A. Angew. Chem., Int. Ed. 2008, 47, 8727. [DOI] [PubMed] [Google Scholar]; c De Sarkar S.; Grimme S.; Studer A. J. Am. Chem. Soc. 2010, 132, 1190. [DOI] [PubMed] [Google Scholar]
- Studer has described an NHC-catalyzed, TEMPO-mediated oxidation of aldehydes that presumably proceeds via two successive single electron transfer events but still gives the product of net two-electron oxidation. See ref (9b).
- For the seminal report of asymmetric organocatalysis proceeding via one-electron pathways, see:Beeson T. D.; Mastracchio A.; Hong J.-B.; Ashton K.; MacMillan D. W. C. Science 2007, 316, 582–585. [PubMed] [Google Scholar]
- DiRocco D. A.; Oberg K. M.; Rovis T. J. Am. Chem. Soc. 2012, 134, 6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Informaton for details.
- The reaction is insensitive to the presence of oxygen, as strictly anaerobic (glovebox) and aerobic (O2 balloon) reaction conditions afforded similar yields.
- The reported reduction potentials were measured against the saturated calomel electrode. See:; a Wardman P. J. Phys. Chem. Ref. Data 1989, 18, 1637. [Google Scholar]; b Ziȩba-Mizgala A.; Puszko A.; Regiec A.; Kuduk-Jaworska J. Bioelectrochemistry 2005, 65, 113. [DOI] [PubMed] [Google Scholar]
- We had previously demonstrated the enantioselective dehydrogenative coupling of tetrahydroquinolines and aliphatic aldehydes using NHCs and photoredox catalysis. The oxidative quencher used in that chemistry was m-dinitrobenzene. See:DiRocco D. A.; Rovis T. J. Am. Chem. Soc. 2012, 134, 8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the development of this modified Ye catalyst, see ref (8c).
- A reaction conducted in the presence of the known radical inhibitor galvinoxyl led to a significantly reduced yield of product.
- Jiang H.; Gschwend B.; Albrecht Ł.; Jørgensen K. A. Org. Lett. 2010, 12, 5052. [DOI] [PubMed] [Google Scholar]
- a Kaeobamrung J.; Mahatthananchai J.; Zheng P.; Bode J. W. J. Am. Chem. Soc. 2010, 132, 8810. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cheng J.; Huang Z.; Chi Y. R. Angew. Chem., Int. Ed. 2013, 52, 8592. [DOI] [PubMed] [Google Scholar]
- Bartoli G.; Dalpozzo R.; Nardi M. Chem. Soc. Rev. 2014, 43, 4728. [DOI] [PubMed] [Google Scholar]
- Bosco M.; Dalpozzo R.; Bartoli G.; Palmieri G.; Petrini M. J. Chem. Soc., Perkin Trans. 2 1991, 657. [Google Scholar]
- A control experiment conducted with a stoichiometric amount of NHC catalyst delivered identical results at 65 °C, suggesting that the enantiomeric convergence is not due to a background cis- to trans-enal isomerization.
- The bond rotational barrier in the parent allyl radical has been determined to be 15.7 ± 1.0 kcal/mol. See:Korth H.-G.; Trill H.; Sustmann R. J. Am. Chem. Soc. 1981, 103, 4483. [Google Scholar]
- We also considered that the reaction might proceed via a charge transfer complex. NMR and UV–vis studies showed no ground-state aggregation between the aza-Breslow intermediate and 1,4-dinitrobenzene in CH2Cl2. Rigorous exclusion of light also did not alter the reaction yield.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.