

Abstract
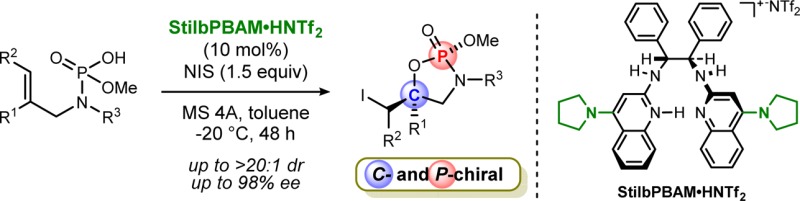
The first highly diastereo- and enantioselective additions of a halogen and phosphoramidic acid to unactivated alkenes have been developed, catalyzed by a chiral Brønsted acid. A unique feature of these additions is the opportunity for stereocontrol at two noncontiguous chiral centers, carbon and phosphorus, leading to cyclic P-chiral phosphoramidates. In addition to their inherent value, the phosphoramidates are precursors to enantioenriched epoxy allylamines.
Phosphoramidates and their derivatives are organophosphorous(V) compounds featuring a P–N bond and are used as pesticides in agriculture and prodrugs in therapeutic development, among other synthetic applications.1 The central chirality of the phosphorus group of a phosphoramidate has a strong impact on the biological activity of the drug;2,3 therefore, methods to stereoselectively construct phosphorus (P-) chiral centers are essential tools. The diastereoselective synthesis of phosphoramidate prodrugs was recently reported by using phosphoryl chloride with a chiral auxiliary, where the phosphorus group plays a role as the electrophilic reagent.3b The preparation of P-stereogenic compounds is still challenging,4,5 and to the best of our knowledge, there are no examples of enantioselective reactions in which the phosphoramidate configuration is governed by a chiral catalyst. In contrast to the use of phosphorus electrophiles, we reasoned that phosphoramidic acids might add as nucleophiles to a halogen-activated alkene. The halogen, in turn, could be activated by a hydrogen bond catalyst.6 This design raised the interesting challenge of not only controlling diastereotopic oxygen–carbon bond formation but also using a chiral Brønsted acid reagent as the master stereocontrol element since phosphoric acids and their derivatives are effective catalysts.7 We reasoned that the use of a polar ionic hydrogen-bond catalyst might offer the ion pairing necessary to achieve reagent-controlled stereoselection, and ultimately an innovative entry to P-chiral small molecules (Figure 1). Phosphoramidic acids have Brønsted acidic (O–H) and Brønsted basic (O) sites which offer potential interactions with a bifunctional Brønsted acid/base catalyst.
Figure 1.
Addition of phosphoramidic acids to alkenes using an electrophilic halogen source for the construction of carbon and phosphorus chiral centers.
Since 2010, enantioselective alkene functionalizations using an electrophilic halogen source by organocatalysis have emerged as an attractive method in asymmetric synthesis.8−10 Considerable efforts have been devoted to this area, especially to the development of enantioselective halolactonizations.9c We reported an enantioselective iodolactonization catalyzed by a chiral Brønsted acid, StilbPBAM·HNTf2 (1·HNTf2), which is composed of an uncharged chiral Brønsted base backbone with an achiral Brønsted acid.6 A variety of nucleophilic donors, e.g., carboxylic acids, amides, alcohols, and N-protected amines, have been employed for this class of reactions.9i,10 Organophosphorus acid derivatives are conspicuously absent from either chiral Brønsted acid or transition-metal-catalyzed alkene halo-oxygenation reactions, presumably due to the lower nucleophilicity of these acids relative to carboxylic acids.11 Conversely, this character may be responsible for their extensive success in catalysis, engaging substrates only transiently and in a noncovalent manner.7,12 Herein, we report the chiral Brønsted acid catalyzed diastereo- and enantioselective iodocyclizations of phosphoramidic acid 3 for the synthesis of C- and P-chiral cyclic phosphoramidates 4.13 Two noncontiguous chiral centers are constructed during the formation of 4, with high levels of absolute and relative stereocontrol.
At the outset of these studies, a control experiment was performed using phophoramidic acid 3aa, 1.5 equiv of N-iodosuccinimide (NIS), and molecular sieves (MS) 4A in toluene at −20 °C for 48 h (Table 1, entry 1). NIS cleanly afforded product 4aa in good yield (82%) but with low diastereoselection (2:1 dr). Insofar as the substrate is a Brønsted acid itself, capable of catalyzing the transformation, a series of experiments directed at formation of a chiral proton complex with a Bis(AMidine) ligand was performed; use of the chiral free base alone could form a phosphoramidic acid salt, whereas a separate strong Brønsted acid might favor a distinct chiral proton complex. This was investigated by probing the effect of the protonation state of StilbPBAM (1) on selectivity. Brønsted base 1 catalyzed the iodocyclization efficiently, affording 4aa in good yield with high diastereoselectivity and moderate enantioselectivity in spite of the background reaction proceeding under the same conditions (Table 1, entry 2). A higher level of enantioselection was observed with the preformed triflimidic acid complex (1·HNTf2) (Table 1, entry 3), and interestingly, a 1:2 mixture of free base (1) and HNTf2 gave the product in 94% ee (Table 1, entry 4). However, a 1:3 ratio of the 1/HNTf2 decreased the diastereo- and enantioselectivity (Table 1, entry 5) and illustrated the potential to inhibit the reaction relative to the catalyst-free case (Table 1, entry 1). In order to probe the importance of the bifunctional chiral acid’s basicity,14 we employed chiral base 2, having no pyrrolidine substituents, in an otherwise identical reaction. The reaction of 2 led to a considerable reduction in the stereoselectivity (Table 1, entry 6). In addition, both 2·HNTf2 and 2·(HNTf2)2 gave lower levels of diastereo- and enantioselection than those of 1 (Table 1, entries 7 and 8). These results indicate that the pyrrolidine unit is critical to achieve high levels of selectivity. Furthermore, iodine (I2) was tested as another halogen source, but both the chemical yield and stereoselectivity of 4aa dramatically dropped (Table 1, entry 9).15
Table 1. Iodocyclization Catalyzed by a Chiral Brønsted Acid: Initial Studya.
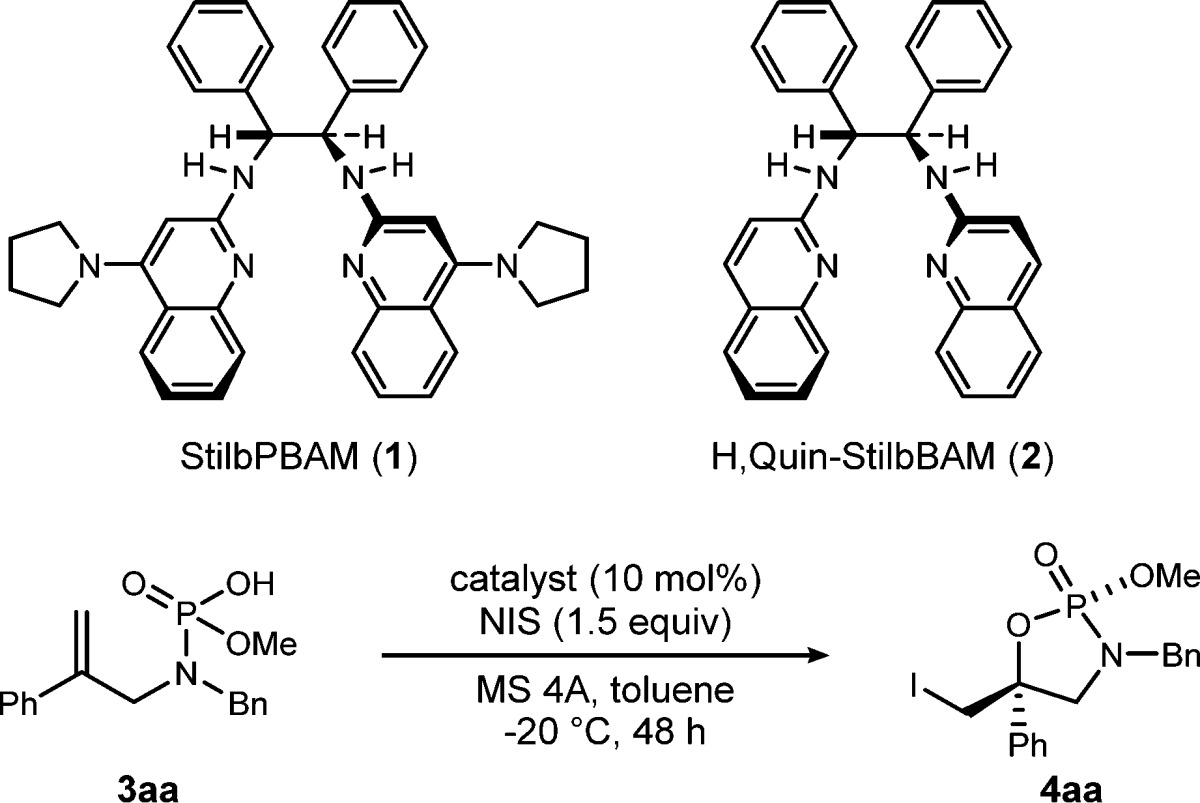
| entry | catalyst | yieldb | drc | eed |
|---|---|---|---|---|
| 1 | none | 82 | 2:1 | – |
| 2 | 1 | 86 | >20:1 | 83 |
| 3 | 1·HNTf2 | 85 | >20:1 | 96 |
| 4 | 1·(HNTf2)2 | 85 | >20:1 | 94 |
| 5 | 1·(HNTf2)3 | 70 | 3:1 | 15 |
| 6 | 2 | 62 | 4:1 | 32 |
| 7 | 2·HNTf2 | 70 | 4:1 | 46 |
| 8 | 2·(HNTf2)2 | 76 | 3:1 | 10 |
| 9e | 1·HNTf2 | 33 | 7:1 | 45 |
Unless otherwise noted, all reactions were carried out at 0.10 mmol scale using 1 equiv of phosphoramidic acid 3aa, 10 mol % catalyst, and 1.5 equiv of NIS in toluene (0.08 M) at −20 °C for 48 h. The relative and absolute configurations of 4aa have been determined by X-ray crystal structure analysis of 4ad; see Supporting Information for details.
Isolated yield of a mixture of diastereomers.
Determined by 1H NMR.
Determined by HPLC with a chiral stationary phase for the major diastereomer.
I2 was used instead of NIS.
The initial scope is summarized in Table 2. The investigation of substituents on the aromatic ring showed that both electron-donating groups16 and electron-withdrawing groups were tolerated to give the products in good yields with high levels of stereoselection (Table 2, entries 1–4). Secondary and primary alkyl groups also underwent the iodocyclization well, while lower diastereoselectivity (8:1 dr) was observed in the case of a sterically less hindered methyl group (Table 2, entries 5–7). Allylamine derivative 3ai was most challenging (Table 2, entry 8), and although the enantioselectivity for this case was promising at the 68/75% ee level, the transformation was not diastereoselective; overall, a substituent at the internal position of the alkene appears essential for relative stereocontrol. To expand the substrate scope and further challenge the catalyst, a trisubstituted alkene was examined. The reaction of 4aj having a dimethyl cis-substituted alkene afforded only two diastereomers with 6:1 dr and 93% ee, the same level of enantioselection as that for disubstituted alkenes (Table 2, entry 9). It should be noted that a trace amount of 6-endo-cyclized product was observed in the reaction using trisubstituted alkene 3aj. Phosphoramidic acid 3ba, bearing an N-4-methoxybenzyl (PMB) group, behaves similarly to 3aa (cf. Table 2, entry 10 and Table 1, entry 3). The relative and absolute configurations of phosphoramidate 4ad (R1 = 4-ClC6H4, R2 = H: Table 2, entry 3) was determined by X-ray crystallographic analysis (Figure 2). Intriguingly, a methoxy group on the phosphorus (P2) and a phenyl group on the quaternary carbon (C5) are located in the pseudo axial position of cyclic phosphoramidate 4ad.
Table 2. Substrate Scope of the Chiral Brønsted Acid Catalyzed Diastereo- and Enantioselective Iodocyclizationa.
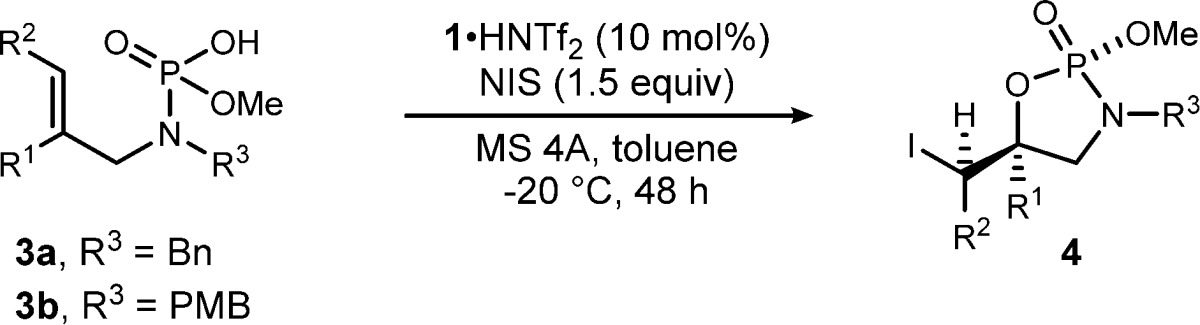
| entry | 3 | R1, R2 | yieldb | drc | eed |
|---|---|---|---|---|---|
| 1 | ab | 4-MeOC6H4, H | 83 | 18:1 | 94 |
| 2 | ac | 3-MeOC6H4, H | 71 | 18:1 | 93 |
| 3 | ad | 4-ClC6H4, H | 74 | >20:1 | 97 |
| 4 | ae | 2-FC6H4, H | 80 | >20:1 | 96 |
| 5 | af | Cy, H | 95 | >20:1 | 95 |
| 6 | ag | 3-butenyl, H | 92 | >20:1 | 98 |
| 7 | ah | Me, H | 91 | 8:1 | 91 |
| 8 | ai | H, H | 69 | 1:1 | 68/75e |
| 9 | aj | Me, Me | 92f | 6:1 | 93 |
| 10 | ba | Ph, H | 85 | >20:1 | 93 |
Unless otherwise noted, all reactions were carried out at 0.10–0.20 mmol scale using 1 equiv of the phosphoramidic acid, 10 mol % of 1·HNTf2, and 1.5 equiv of NIS in toluene (0.08 M) at −20 °C for 48 h.
Isolated yield of a mixture of diastereomers.
Determined by 1H NMR and/or HPLC.
Determined by HPLC with a chiral stationary phase for the major diastereomers.
Ee for each diastereomer.
Combined yield of 5-exo and 6-endo product.
Figure 2.
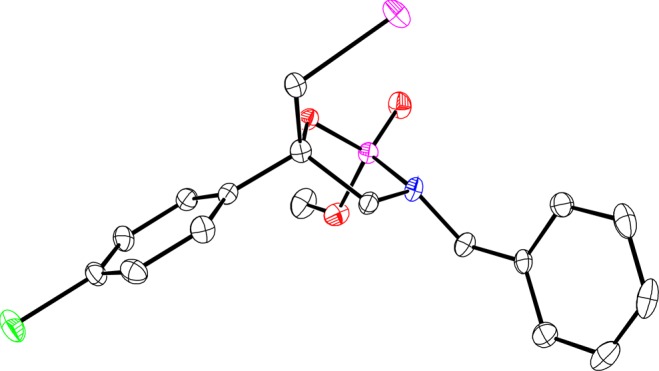
ORTEP drawing of cyclic phosphoramidate (2R,5S)-4ad.
The regioisomer and minor diastereomer could be removed by column chromatography, and as a result, 4aj was obtained in 63% yield (over 2 steps, from dimethyl phosphoramidate 5) with 92% ee (Scheme 1). Taking into account the electrophilic feature of cyclic organophosphorous compounds,11a iodophosphoramidate 4aj was treated with sodium methoxide to afford the corresponding cis-dimethyl-epoxide 6a. The present method could be regarded as a formal asymmetric epoxidation of allylamine derivatives, where only one example of an enantioselective epoxidation of allylamine derivatives has been reported.17 The protocol is also applicable to the synthesis of gem-disubstituted epoxy allylamines. Although partial epimerization at the P-chiral center was observed, cyclic phosphoramidate 4aa (>20:1 dr, 96% ee) was converted to enantioenriched epoxide 6b(18) as an acyclic chiral phosphoramidate in good yield with high enantioselectivity.1c,1d,3b
Scheme 1. Formal Asymmetric Epoxidation of Allylamine Derivatives via the Chiral Brønsted Acid Catalyzed Diastereo- and Enantioselective Iodocyclization Sequence.

In conclusion, the functionalization of unactivated alkenes with the use of NIS and phosphoramidic acids has been developed, leading to cyclic phosphoramidates in which both carbon and phosphorus configurations are forged with a high degree of absolute stereocontrol. Importantly, the present reaction enables a catalytic formation of P-chiral phosphoramidates using a tactic that could be applied more broadly to organophosphorous compounds. While stereodefined phosphoramidates are valuable in their own right, their treatment with a base leads to an epoxide and, vis-a-vis, epoxy allylamines in high enantiomeric excess. Noteworthy is the use of a (chiral) polar ionic bond to promote the enantioselective cyclization involving a polar covalent hydrogen bond. Efforts to identify new synergistic transformations using these two types of hydrogen bonds will be reported in due course.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM 084333).
Supporting Information Available
Experimental procedures and spectroscopic data for all new compounds, and X-ray data (cif) for 4ad. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Eto M.Organophosphorous Pesticides: Organic and Biological Chemistry; CRC: Cleveland, OH, 1974. [Google Scholar]; b Fest C.; Schmidt K. J.. The Chemistry of Organophosphorous Pesticides; Springer: Berlin, Heidelberg, NY, 1982. [Google Scholar]; c Mehellou Y.; Balzarini J.; McGuigan C. ChemMedChem 2009, 4, 1779–1791. [DOI] [PubMed] [Google Scholar]; d Sofia M. J.; Chang W.; Furman P. A.; Mosley R. T.; Ross B. S. J. Med. Chem. 2012, 55, 2481–2531. [DOI] [PubMed] [Google Scholar]; e Molt O.; Schrader T. Synthesis 2002, 2633–2670. [Google Scholar]
- a Lin K.; Zhou S.; Xu C.; Liu W. J. Agric. Food Chem. 2006, 54, 8134–8138. [DOI] [PubMed] [Google Scholar]; b Wang C.; Zhang N.; Li L.; Zhang Q.; Zhao M.; Liu W. Chirality 2010, 22, 612–617. [DOI] [PubMed] [Google Scholar]
- a Morales E. H. R.; Balzarini J.; Meier C. Chem.—Eur. J. 2011, 17, 1649–1659. [DOI] [PubMed] [Google Scholar]; b Román C. A.; Wasserthal P.; Balzarini J.; Meier C. Eur. J. Org. Chem. 2011, 4899–4909. [Google Scholar]; Related work, see:; c Wu S.-Y.; Hirashima A.; Eto M.; Yanagi K.; Nishioka E.; Moriguchi K. Agric. Biol. Chem. 1989, 53, 157–163. [Google Scholar]
- For selected reviews on P-stereogenic compounds, see:; a Wozniak L. A.; Okruszek A. Chem. Soc. Rev. 2003, 32, 158–169. [DOI] [PubMed] [Google Scholar]; b Glueck D. S. Synlett 2007, 2627–2634. [Google Scholar]; c Grabulosa A.; Granell J.; Muller G. Coord. Chem. Rev. 2007, 251, 25–90. [Google Scholar]; d Harvey J. S.; Gouverneur V. Chem. Commun. 2010, 46, 7477–7485. [DOI] [PubMed] [Google Scholar]; e Kolodiazhnyi O. I. Tetrahedron: Asymmetry 2012, 23, 1–46. [Google Scholar]
- Recent reports:; a Han Z. S.; Goyal N.; Herbage M. A.; Sieber J. D.; Qu B.; Xu Y.; Li Z.; Reeves J. T.; Desrosiers J.-N.; Ma S.; Grinberg N.; Lee H.; Mangunuru H. P. R.; Zhang Y.; Krishnamurthy D.; Lu B. Z.; Song J. J.; Wang G.; Senanayake C. H. J. Am. Chem. Soc. 2013, 135, 2474–2477. [DOI] [PubMed] [Google Scholar]; b Ding B.; Zhang Z.; Xu Y.; Liu Y.; Sugiya M.; Imamoto T.; Zhang W. Org. Lett. 2013, 15, 5476–5479. [DOI] [PubMed] [Google Scholar]; c Nikitin K.; Rajendran K. V.; Müller-Bunz H.; Gilheany D. G. Angew. Chem., Int. Ed. 2014, 53, 1906–1909. [DOI] [PubMed] [Google Scholar]; d Huang Y.; Li Y.; Leung P.-H.; Hayashi T. J. Am. Chem. Soc. 2014, 136, 4865–4868. [DOI] [PubMed] [Google Scholar]
- Dobish M. C.; Johnston J. N. J. Am. Chem. Soc. 2012, 134, 6068–6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For seminal studies on phosphoric acid catalysis, see:; a Akiyama T.; Itoh J.; Yokota K.; Fuchibe K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. [DOI] [PubMed] [Google Scholar]; b Uraguchi D.; Terada M. J. Am. Chem. Soc. 2004, 126, 5356–5357. [DOI] [PubMed] [Google Scholar]; For selected reviews, see:; c Akiyama T. Chem. Rev. 2007, 107, 5744–5758. [DOI] [PubMed] [Google Scholar]; d Terada M. Synthesis 2010, 1929–1982. [Google Scholar]; e Rueping M.; Kuenkel A.; Atodiresei I. Chem. Soc. Rev. 2011, 40, 4539–4549. [DOI] [PubMed] [Google Scholar]; See also:; f Vellalath S.; Čorić I.; List B. Angew. Chem., Int. Ed. 2010, 49, 9749–9752. [DOI] [PubMed] [Google Scholar]; g Čorić I.; List B. Nature 2012, 483, 315–319. [DOI] [PubMed] [Google Scholar]
- Selected reports:; a Whitehead D. C.; Yousefi R.; Jaganathan A.; Borhan B. J. Am. Chem. Soc. 2010, 132, 3298–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang W.; Zheng S.; Liu N.; Werness J. B.; Guzei I. A.; Tang W. J. Am. Chem. Soc. 2010, 132, 3664–3665. [DOI] [PubMed] [Google Scholar]; c Cai Y.; Liu X.; Hui Y.; Jiang J.; Wang W.; Chen W.; Lin L.; Feng X. Angew. Chem., Int. Ed. 2010, 49, 6160–6164. [DOI] [PubMed] [Google Scholar]; d Zhou L.; Tan C. K.; Jiang X.; Chen F.; Yeung Y.-Y. J. Am. Chem. Soc. 2010, 132, 15474–15476. [DOI] [PubMed] [Google Scholar]; e Veitch G. E.; Jacobsen E. N. Angew. Chem., Int. Ed. 2010, 49, 7332–7335. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Murai K.; Matsushita T.; Nakamura A.; Fukushima S.; Shimura M.; Fujioka H. Angew. Chem., Int. Ed. 2010, 49, 9174–9177. [DOI] [PubMed] [Google Scholar]; g Paull D. H.; Fang C.; Donald J. R.; Pansick A. D.; Martin S. F. J. Am. Chem. Soc. 2012, 134, 11128–11131. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also:; h Wang M.; Gao L. X.; Mai W. P.; Xia A. X.; Wang F.; Zhang S. B. J. Org. Chem. 2004, 69, 2874–2876. [DOI] [PubMed] [Google Scholar]
- For reviews, see:; a Chen G.; Ma S. Angew. Chem., Int. Ed. 2010, 49, 8306–8308. [DOI] [PubMed] [Google Scholar]; b Tan C. K.; Zhou L.; Yeung Y.-Y. Synlett 2011, 1335–1339. [Google Scholar]; c Castellanos A.; Fletcher S. P. Chem.—Eur. J. 2011, 17, 5766–5776. [DOI] [PubMed] [Google Scholar]; d Hennecke U. Chem.—Asian J. 2012, 7, 456–465. [DOI] [PubMed] [Google Scholar]; e Denmark S. E.; Kuester W. E.; Burk M. T. Angew. Chem., Int. Ed. 2012, 51, 10938–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tan C. K.; Yeung Y.-Y. Chem. Commun. 2013, 49, 7985–7996. [DOI] [PubMed] [Google Scholar]; g Murai H.; Fujioka H. Heterocycles 2013, 87, 763–805. [Google Scholar]; h Nolsøe J. M. J.; Hansen T. V. Eur. J. Org. Chem. 2014, 3051–3065. [Google Scholar]; i Tripathi C. B.; Mukherjee S. Synlett 2014, 25, 163–169. [Google Scholar]
- a Zhang Y.; Xing H.; Xie W.; Wan X.; Lai Y.; Ma D. Adv. Synth. Catal. 2013, 355, 68–72. [Google Scholar]; b Brindle C. S.; Yeung C. S.; Jacobsen E. N. Chem. Sci. 2013, 4, 2100–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tripathi C. B.; Mukherjee S. Angew. Chem., Int. Ed. 2013, 52, 8450–8453. [DOI] [PubMed] [Google Scholar]; For other alkene halogenations, see:; d Gustafson J. L.; Lim D.; Miller S. J. Science 2010, 328, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chen Z.-M.; Zhang Q.-W.; Chen Z.-H.; Li H.; Tu Y.-Q.; Zhang F.-M.; Tian J.-M. J. Am. Chem. Soc. 2011, 133, 8818–8821. [DOI] [PubMed] [Google Scholar]; f Li H.; Zhang F.-M.; Tu Y.-Q.; Zhang Q.-W.; Chen Z.-M.; Chen Z.-H.; Li J. Chem. Sci. 2011, 2, 1839–1841. [Google Scholar]
- For selected examples of the synthesis of racemic cyclic organophosphorus compounds via iodocyclization, see:; a Bartlett P. A.; Jernstedt K. K. J. Am. Chem. Soc. 1977, 99, 4829–4830. [Google Scholar]; b Zhao Y.-F.; Yan S.-J.; Zhai C. J. Org. Chem. 1985, 50, 2136–2140. [Google Scholar]; c Ye M.-C.; Chai W.-G.; Li L.-P.; Zhao Y.-F. Tetrahedron Lett. 1987, 28, 2615–2618. [Google Scholar]; d Yokomatsu T.; Shioya Y.; Iwasawa H.; Shibuya S. Heterocycles 1997, 46, 463–472. [Google Scholar]; e André V.; Lahrache H.; Robin S.; Rousseau G. Tetrahedron 2007, 63, 10059–10066. [Google Scholar]; See also:; f Hatano M.; Horibe T.; Ishihara K. Angew. Chem., Int. Ed. 2013, 52, 4549–4553. [DOI] [PubMed] [Google Scholar]
- For asymmetric additions of a dithiophosphoric acid, see:Shapiro N. D.; Rauniyar V.; Hamilton G. L.; Wu J.; Toste F. D. Nature 2011, 470, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Coe P. L.; Jones A. S.; Walker R. T.; Balzarini J.; De Clercq E. J. Med. Chem. 1990, 33, 2368–2375. [DOI] [PubMed] [Google Scholar]
- Davis T. A.; Wilt J. C.; Johnston J. N. J. Am. Chem. Soc. 2010, 132, 2880–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NBS afforded the corresponding product in good yield and with modest enantioselection. It is significant that these reactions provided moderate diastereo- and enantioselectivity, even with a different halogen donor. To-date, a single system is equally effective with two types of halogen sources; see:Wang Y.-M.; Wu J.; Hoong C.; Rauniyar V.; Toste F. D. J. Am. Chem. Soc. 2012, 134, 12928–12931. [DOI] [PubMed] [Google Scholar]
- Product 4ab (R1 = 4-MeOC6H4, R2 = H) racemizes at room temperature (1:1 dr, 45% ee after 3 days). The enantioenriched product could be obtained by a quick workup after the reaction; see Supporting Information for details.
- Olivares-Romero J. L.; Li Z.; Yamamoto H. J. Am. Chem. Soc. 2012, 134, 5440–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The P-stereochemistry of 6b is postulated by an inversion mechanism, in accord with prior studies (see ref (4a) and (4c)).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.