

The compensatory increase in the mass of β‐cells in response to peripheral insulin resistance contributes to the prevention of diabetes. Glucokinase‐mediated glucose signaling and insulin receptor substrate (IRS)‐2‐mediated insulin signaling in the β‐cells play crucial roles in the proliferation of the β‐cells in this process1. When the β‐cell mass fails to increase to compensate for the increased insulin demand, type 2 diabetes becomes manifest. Loss of β‐cell functional activity results in the development of the absolute or relative insulin insufficiency observed in patients with type 1 and type 2 diabetes, respectively. On the basis of the pathophysiological mechanism, rescue from β‐cell failure is an essential target for appropriate diabetes therapy.
In the presence of diabetes, β‐cells are exposed to hyperglycemia, increased free fatty acids, advanced glycation end‐products, glycated serum substances, inflammatory cytokines, hypoxia and abnormal neural regulation. These factors coordinately lead to oxidative stress, endoplasmic reticulum (ER) stress, uncontrolled autophagy, decreased proliferative capacity and cell death of β‐cells. Oxidative stress is thought to be the primary mechanism underlying the β‐cell dysfunction induced by chronic exposure to hyperglycemia, namely, glucose toxicity. Oxidative stress is also linked to ER stress and inflammation in β‐cells. β‐Cells show extraordinary vulnerability to superoxide damage as a result of the low levels of expression of anti‐oxidant enzyme genes.
Musculoaponeurotic fibrosarcoma oncogene family A (MAFA) is one of the large Maf transcription factors, a subgroup of the basic leucine‐zipper family including MAFA, MAFB, c‐MAF and neural retina leucine zipper (NRL). These proteins are characterized by N‐terminal transactivation and C‐terminal basic leucine‐zipper deoxyribonucleic acid (DNA)‐binding domains. MAFA is heavily phosphorylated in vivo, which affects its stability, DNA‐binding capacity, transactivation and oncogenic potential. Dephosphorylation by phosphatases has been shown to inhibit the DNA‐binding properties of MAFA in vitro. Pancreatic duodenal homeobox 1 (PDX1), a member of the large family of homeodomain (HD)‐containing transcription factors, is associated with both type 2 diabetes and maturity‐onset diabetes of the young (MODY)‐4. PDX1 is expressed in the precursors in the endocrine and exocrine compartments of the pancreas, and plays essential roles in pancreas development, β‐cell differentiation and maintenance of mature β‐cell function by regulating several β‐cell‐related genes. MAFA binding to the C1 element in the insulin gene enhancer region is crucial for insulin gene transcription, together with PDX1 binding to the A3 element and neurogenic differentiation‐1 binding to the E1 element. The expression levels and DNA‐binding capacity of MAFA and PDX1 are reportedly decreased in the presence of oxidative stress induced by chronic high glucose exposure of β‐cells. Reduction of these transcription factors has been shown to be correlated with the loss of insulin gene expression. Anti‐oxidant treatment was shown to recover the failure, induced by chronic exposure to high glucose concentrations, of MAFA and PDX1 expressions, and of their DNA binding. In the presence of chronically high glucose levels, downregulation of MAFA occurred earlier than that of PDX1. These stress conditions affect PDX1 by altering its subcellular localization from the nucleus to the cytoplasm, but inhibit MAFA activity through messenger ribonucleic acid processing, stabilization and changing the cellular localization. Although the nuclear PDX1 level is unaffected under oxidative stress in db/db mice, MAFA is translocated to the cytoplasm, and p38 mitogen‐activated protein kinase (MAPK)‐mediated degradation is increased2. Transgenic β‐cell‐specific overexpression of glutathione peroxidase‐1 (Gpx1) rescues the β‐cell functions, islet β‐cell ratio, insulin granulation and nuclear MAFA content in db/db mice2. Prolonged exposure of the islets to fatty acids also inhibits insulin gene transcription by impairing MAFA expression and PDX1 nuclear localization. However, the temporal, ordinal, and selective control of these transcription factors and other molecules in the β‐cells during the onset and progression of diabetes remains unclear.
Recently, Guo et al.3 found that selective loss of MAFA, MAFB, NK transcription factor‐related, gene family 6, locus 1 (NKX6.1) and PDX1 contribute to β‐cell failure in the course of development of diabetes (Figure 1). They showed that MAFA and/or MAFB are early and very sensitive targets of oxidative stress in β‐cells. Furthermore, they showed that NKX6.1 and PDX1 are the subsequent targets in the β‐cells under conditions of oxidative stress. Under oxidative stress conditions induced by hydrogen peroxide (H2O2), MAFA were translocated from the nucleus to the cytoplasm and dephosphorylated, which was mediated by covalent dimer formation by the C‐terminal cysteines in the β‐cells. Treatment with H2O2 also evoked dephosphorylation and cytoplasmic translocation of NKX6.1. Concurrently, the binding activity of PDX1 to endogenous target gene promoter sequences was also attenuated. It was noteworthy that the expressions and DNA‐binding capacity of other key β‐cell transcriptional factors were unaffected. Oxidative stress was triggered progressively in the β cells in obese hyperglycemic db/db mice. In the db/db islets, the nuclear content of NKX6.1 was drastically diminished, which was rescued by β‐cell‐specific transgenic expression of Gpx1, as well as MAFA or PDX1. Interestingly, sequential analysis of the endocrine pancreas in the db/db mouse showed transient cell proliferation at 4 weeks, forkhead box O1 (FOXO1) nuclear translocation at 6 weeks, loss of MAFA and its target glucose transporter/Slc2a2 (GLUT2) at 8 weeks, and reduction of nuclear NKX6.1 at 8 weeks in the β‐cells. Furthermore, selective loss of MAFA, MAFB, NKX6.1 and PDX1 was shown in the islets of humans with type 2 diabetes. Although MAFB expression is lost in rodent β‐cells soon after birth and detected only in α‐cells, both α‐ and β‐cells in the human islets retain MAFB expression into adulthood. While MAFB is thought to play a role in the development of both α‐ and β‐cells, and its expression is also implicated in β‐cell adaptation during pregnancy in mice, the role of MAFB in human islets still remains unclear. MAFB expression was downregulated in the β‐cells of humans with type 2 diabetes, but not in the α‐cells3. However, as aforementioned, the physiological role of MAFB in adult human islet cells still remains unclear. This gene might provide new clues to understanding the dissimilarities between human and rodent β‐cells.
Figure 1.
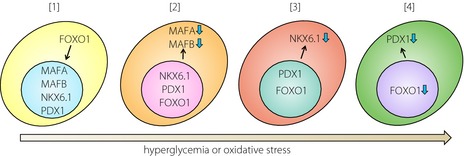
Schematic diagram of inactivation of β‐cell transcriptional factors in the development of diabetes. (a) Nuclear localization of forkhead box O1 (FOXO1) is the initial step in the maladaptation of β cells. (b) Sustained hyperglycemia or oxidative stress induces dephosphorylation, dimerization, translocation to the cytoplasm and loss of musculoaponeurotic fibrosarcoma oncogene family A (MAFA). (c) This behavior of might also be applicable to MAFB in human β‐cells. Reduction of nuclear NK transcription factor‐related, gene family 6, locus 1 (NKX6.1) is the next step of β‐cell failure. (d) Finally, the contents of pancreatic duodenal homeobox 1 (PDX1) and FOXO1 are diminished, causing further impairment of insulin production, reduction of the β‐cell mass, and overt diabetes.
Insulin signaling, including through the phosphoinositide‐3 kinases (PI3Ks), IRS‐2, pyruvate dehydrogenase kinase (PDK) through V‐akt murine thymoma viral oncogene homolog (AKT), and their downstream molecule FOXO1 promotes cell proliferation and survival of β‐cells. Translocation of FOXO1 from the cytosol to the nucleus was an earlier event than inactivation of MAFA, NKX6.1 and PDX1 during the course of development of β‐cell failure in db/db mice3. Both oxidative stress and ER stress induced translocation and retention of FOXO1 in the nucleus. β‐Cell‐specific deletion of FOXO1 aggravated glucose tolerance in db/db mice. These findings show that impairment of insulin signaling could cause β‐cell dysfunction through FOXO1 translocation, and subsequent inactivation of MAFA and PDX1. Downregulation of glucokinase activity triggered nuclear retention of FOXO1 through insufficient insulin signaling in the β‐cells of a mouse model of diet‐induced obesity (DIO)1. Meanwhile, glucokinase activation ameliorated ER stress‐induced β‐cell apoptosis, in part through downregulating the pro‐apoptotic protein, C/EBP homologous protein, in a MAPKs extracellular signal‐regulated kinase 1/2‐dependent manner4. These results are concordant with the view that MAFA regulates the expression of C/EBP homologous protein, mediated by (dependent on) extracellular signal‐regulated kinase 1/21/2, in glucose‐stimulated β‐cells5. Surprisingly, β‐cell‐specific FOXO1 deletion reportedly induced transdifferentiation from β‐cells to α‐cells under metabolic stress6. These FOXO1‐deficient β‐cells had lost the expressions of PDX1, MAFA and insulin, and instead expressed the progenitor markers neurogenin 3 (NEUROG3), v‐myc avian myelocytomatosis viral oncogene lung carcinoma‐derived homolog (MYCL), nanog homeobox (NANOG) and POU class 5 homeobox 1 (1 POU5F1). Meanwhile, according to one study, the expressions of NEUROG3, MYCL, NANOG and POU5F1 are unchanged in human diabetic islets3. Hence, the conversion of cell fate between α‐cells and β‐cells in human diabetic islets is still controversial. Cell fate determination between β‐cell apoptosis and β‐cell dedifferentiation is a major question that needs to be addressed in future studies in this area of research.
Accumulating evidence suggests that restoration of the specific aforementioned transcriptional factors could be an appropriate therapeutic strategy for preventing β‐cell failure in diabetes. The time might have come to contemplate the feasibility of application of this strategy in patients with diabetes. Further study is required to understand the mechanisms underlying the selective downregulation of transcriptional factors in the pancreatic β‐cells. In addition to the genetic background, epigenetic regulation by environmental cues could also be involved in this process. Above all, unraveling of the pathogenetic mechanisms of diabetes in human patients is required to open new avenues for the development of β‐cell‐protective interventions.
Acknowledgment
The authors have no conflict of interests to declare.
References
- 1.Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IRS‐2 are required for compensatory beta cell hyperplasia in response to high‐fat diet‐induced insulin resistance. J Clin Invest 2007; 117: 246–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harmon JS, Bogdani M, Parazzoli SD, et al. beta‐Cell‐specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009; 150: 4855–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo S, Dai C, Guo M, et al. Inactivation of specific beta cell transcription factors in type 2 diabetes. J Clin Invest 2013; 123: 3305–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shirakawa J, Togashi Y, Sakamoto E, et al. Glucokinase activation ameliorates ER stress‐induced apoptosis in pancreatic beta‐cells. Diabetes 2013; 62: 3448–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence MC, McGlynn K, Naziruddin B, et al. Differential regulation of CHOP‐10/GADD153 gene expression by MAPK signaling in pancreatic beta‐cells. Proc Natl Acad Sci USA 2007; 104: 11518–11525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talchai C, Xuan S, Lin HV, et al. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012; 150: 1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]