

Abstract
A receptor–ligand interaction can evoke a broad range of biological activities in different cell types depending on receptor identity and cell type-specific post-receptor signaling intermediates. Here, we show that the TNF family member LIGHT, known to act as a death-triggering factor in motoneurons through LT-βR, can also promote axon outgrowth and branching in motoneurons through the same receptor. LIGHT-induced axonal elongation and branching require ERK and caspase-9 pathways. This distinct response involves a compartment-specific activation of LIGHT signals, with somatic activation-inducing death, while axonal stimulation promotes axon elongation and branching in motoneurons. Following peripheral nerve damage, LIGHT increases at the lesion site through expression by invading B lymphocytes, and genetic deletion of Light significantly delays functional recovery. We propose that a central and peripheral activation of the LIGHT pathway elicits different functional responses in motoneurons.
Keywords: axon outgrowth, branching, cell death, LIGHT, motoneuron
Introduction
A same ligand can elicit diverse biological responses including degeneration, regeneration, proliferation, and cell migration under cell type-specific circumstances. This diversity relies on the commitment of distinct receptors, combinatorial co-receptor systems, or the recruitment of differential intracellular signaling intermediates that typifies a cell type 1–4. Identifying the cellular and molecular basis of this signaling pleiotropy is crucial to understand development and pathologies and in fine to design pertinent therapeutic interventions.
Activation of the tumor necrosis factor receptor superfamily (TNFRSF) member lymphotoxin beta receptor (LT-βR) by its ligand LIGHT (TNFSF14) triggers a death-signaling pathway in motoneurons 5. LIGHT has been proposed to contribute to the selective degeneration of motoneurons in the fatal paralytic disorder amyotrophic lateral sclerosis (ALS) 5, 6. Astrocytes expressing an ALS-causing mutation in the superoxide dismutase-1 (SOD1) gene are selectively toxic toward motoneurons 7. This astrocyte-mediated neurotoxicity depends on the production of interferon gamma (IFN-γ), which leads to an upregulation of LIGHT in motoneurons and commitment of a death program through LT-βR activation. Another function of LIGHT within the peripheral nervous system has been documented in nodose ganglion neurons 8 where activation of the herpes virus entry mediator (HVEM) receptor by LIGHT leads to reduced neurite length and number of branch points, without triggering neuronal death. This evidence led us to explore whether LIGHT could elicit different responses in motoneurons.
In the present report, we show that LIGHT promotes axon outgrowth and branching in motoneurons through the activation of LT-βR. The neuronal compartment on which LIGHT acts dictates this bivalent response: While an axonal stimulation elicits axon outgrowth and branching, a somatic stimulation triggers motoneuron death. During sciatic nerve injury, we found that LIGHT, expressed by infiltrating B lymphocytes, is required for proper reinnervation and functional motor recovery.
Results and Discussion
LIGHT promotes axon outgrowth and branching in motoneurons
As previously demonstrated, activation of LT-βR by recombinant soluble LIGHT (sLIGHT) triggers death of about 50% of purified embryonic motoneurons after 48 h of treatment (Supplementary Fig S1A) 5. To determine whether LIGHT elicits axon outgrowth and branching in motoneurons, we added sLIGHT to motoneuron cultures for 24 h to avoid biased analysis due to the loss of differentially vulnerable populations of motoneurons after longer exposure to the ligand. Indeed, after 24 h of treatment, sLIGHT did not significantly induce death of Hb9::GFP neurons (Fig 1A). However, sLIGHT treatment increased motoneuron axon length and branching (Fig 1A–C). The proportion of motoneurons that survive LIGHT death signals at 48 h also showed increased axon length and branching (Supplementary Fig S1B, C). Quantification of axonal length distribution indicated that LIGHT promotes a homogeneous shift toward longer axons (Fig 1D). Live cell time-lapse imaging showed that LIGHT increased by twofold the average axon elongation rate (Fig 1E, F). Consistently, motoneurons isolated from mice with a targeted deletion of Light 9 presented reduced axon length and arborization when compared to wild-type motoneurons (Fig 1G–I and Supplementary Fig S1D). We confirmed that the addition of exogenous sLIGHT to Light−/− motoneurons after 24 h of culture compensated for LIGHT deficiency (Supplementary Fig S1E, F).
Figure 1. LIGHT selectively promotes axon outgrowth and branching in motoneurons.
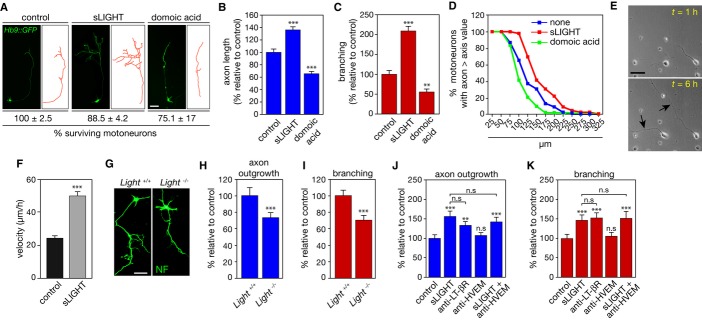
A–D Motoneurons were isolated from Hb9::GFP mice and cultured for 24 h before being treated or not with 100 ng/ml of soluble LIGHT (sLIGHT) or 10 mM domoic acid that served as a negative control. (A) Motoneuron survival was determined after 24 h of treatment and is expressed relative to non-treated cells. Measurements of axon length (B) and branching (C) were performed 24 h after sLIGHT or domoic acid treatment (traces in red (A)). (B, C) show one representation of three independent experiments. (D) The distribution of motoneurons with different axon lengths under indicated conditions is plotted as a cumulative histogram.
E Phase-contrast pictures of motoneurons obtained 1 and 6 h after sLIGHT addition (arrowheads indicate elongated axons). Scale bar, 50 μm.
F Bar graph of the average speed of axon outgrowth in the presence or not of sLIGHT.
G–I Motoneurons were isolated from Light+/+ and Light−/− embryos and 24 h after plating axon length (H) and branching (I) were determined. NF, neurofilament, scale bar, 50 μm.
J, K Motoneurons were treated (or not) with agonistic anti-LT-βR antibodies (100 ng/ml) or antagonistic anti-HVEM antibodies (100 ng/ml) in combination with sLIGHT (100 ng/ml). Twenty-four hours after, axon length (J) or branching (K) was determined. Graphs show one representation of three independent experiments.
Data information: Values are means ± SEM; **P < 0.01; ***P < 0.001; n.s, non-significant; ANOVA with Tukey–Kramer’s post hoc test (B, C, J, K) and unpaired two-tailed Student’s t-test (F, H, I).
Since both LT-βR and HVEM are expressed by motoneurons 5, we next determined whether the differential outcome of LIGHT signaling originates from a differential activation of LT-βR or HVEM. When added to the culture medium for 24 or 48 h, agonistic anti-LT-βR antibodies 5 increased both axon outgrowth and number of branch points as efficiently as sLIGHT (Fig 1J, K and Supplementary Fig S1G, H). However, the blockade of LIGHT-HVEM interaction by antagonistic anti-HVEM antibodies 8 did not affect motoneuron axons (Fig 1J, K and Supplementary Fig S1G, H). We further confirm that LT-βR agonist increased axon length and branching in Light-deficient motoneurons, whereas HVEM antagonist had no effect (Supplementary Fig S1I, J). The neurite length and growth rate of sensory neurons were not modified following LIGHT treatment (Supplementary Fig S1K–M), further reinforcing the cell-selective effects that LIGHT exerts on motoneurons.
The extracellular signal-related protein kinase (ERK) signaling pathway is known to operate in neurite outgrowth 3. We found that activation of LIGHT signaling, by sLIGHT and agonistic anti-LT-βR antibodies, led to a significant increase in phospho-ERK (p42 and p44 isoforms) content in motoneurons (Fig 2A–D). The pharmacological inhibition of ERK pathway by PD98059 or U0126 reduced the average axon length and number of branch points in the presence of sLIGHT, whereas it had no protective effect on LIGHT-induced motoneuron loss (Fig 2E–G). Unexpectedly, inhibition of caspase-9, which is involved in LIGHT-induced death 5, abolished LIGHT-promoted axon extension and branching in motoneurons. On the other hand, inhibition of caspase-3, which is not implicated in LIGHT-triggered death of motoneurons, did not interfere in the axon outgrowth and branching induced by LIGHT (Fig 2H, I). We did not observe that caspase-9 inhibition by LEHD blocked the phosphorylation of ERK in motoneurons after sLIGHT treatment (P-ERK/actin: sLIGHT, 2.8 ± 0.6 and sLIGHT with LEHD, 4.1 ± 0.2 arbitrary units, n = 3, non-significant, unpaired two-tailed t-test).
Figure 2. Inhibition of ERK and caspase-9 pathways blocks the effect of LIGHT on axon outgrowth and branching.

A–D Western blot analysis of phospho-ERK (P-ERK) and ERK in extracts from motoneurons treated or not with 100 ng/ml of sLIGHT (A) and agonistic anti-LT-βR antibodies (C) for 16 h. Actin served as a loading control. P-ERK intensities were normalized to actin (B, D) (n = 3, unpaired two-tailed Student’s t-test).
E–I Axon length (E, H) and branching (F, I) of motoneurons were determined following treatment with PD98059 (10 μM), U0126 (1 μM), LEHD (1 μM), DEVD (10 μM) in combination with sLIGHT. (G) Motoneurons were maintained in culture for 24 h and treated with sLIGHT, PD98059, and U0126. Survival was determined 48 h later.
Data information: Data are represented as means ± SEM, representative of three independent experiments, ANOVA with Tukey–Kramer’s post hoc test; n.s, non-significant, *P < 0.05, **P < 0.01
Regional activation of LIGHT-dependent signaling triggers opposite responses in motoneurons
Neurons are highly polarized cells that process and emit signals distally or locally through axonal and somatic compartments. To our concern, LT-βR is distributed along axon and soma in cultured motoneurons 5. We therefore hypothesized that the somatic and axonal compartments of neurons can process the same external cue toward distinct functional outcomes. To assess this possibility, we compartmentalized motoneuron cultures via microfluidic chambers (Fig 3A, B). We found a restricted expression of the dendritic marker microtubule-associated protein 2 (MAP2) in the somatic compartment after 120 h of culture 10, whereas a pan-axonal neurofilament marker (SMI312) immunostained axons in both compartments, since not all motoneurons cross into the axonal compartment (Fig 3C, D). When added to the somatic compartment, sLIGHT did not result in increased axonal length and branching in the axonal compartment. In contrast, treatment of the axonal compartment with sLIGHT elicited a significant axon outgrowth and branching (Fig 3E, F). Addition of sLIGHT to both somatic and axonal compartments, which recapitulates experiments in non-compartmentalized cell cultures, induced axonal elongation and branching. We next evaluated whether motoneuron death can be triggered by LIGHT following differential stimulation of motoneuron compartments. To take into account only the compartmentalized motoneurons, we counted the number of Hb9::GFP neurons that were retrogradely labeled after the addition of fluorochrome-conjugated wheat germ agglutinin (WGA) in the axonal compartment (Fig 3G). We found that somatic activation of the LIGHT pathway triggered motoneuron death, whereas axonal sLIGHT treatment had no effect on survival (Fig 3H). We confirmed that axonal, but not somatic, activation of LT-βR by agonistic anti-LT-βR antibodies elicited axonal elongation (Supplementary Fig S2A). Inversely, somatic, but not axonal, activation of LT-βR triggered motoneuron death (Supplementary Fig S2B). We also confirmed that axonal inhibition of caspase-9 and ERK pathways blocked the effect of LIGHT on axon elongation (Supplementary Fig S2C, D).
Figure 3. Somatic or axonal LIGHT signaling elicits opposite responses in motoneurons.
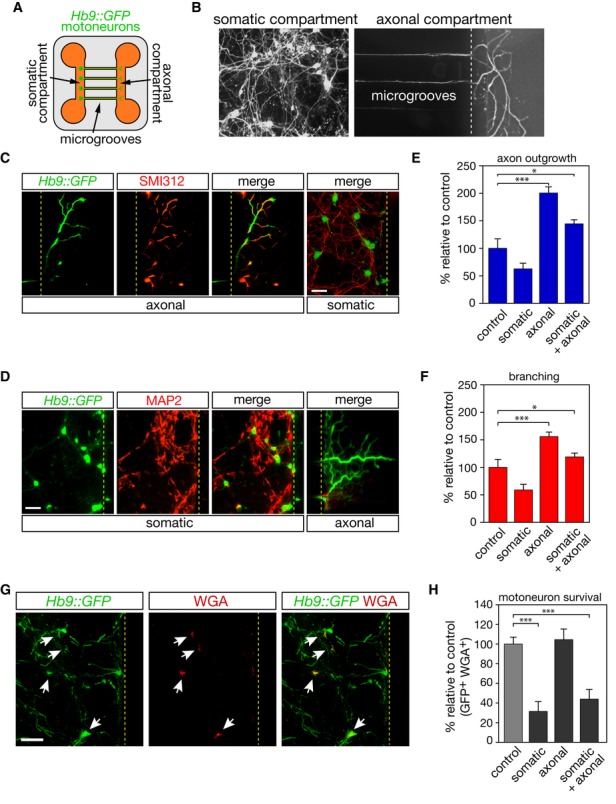
A Schematic representation of the microfluidic device.
B Motoneurons are plated into the somatic compartment and they extend their axons into the axonal compartment through 750-μm-long microgrooves.
C, D Hb9::GFP motoneurons were cultured for 120 h and immunostained using SMI312 (C) and MAP2(D). Scale bar, 50 μm.
E, F After 120 h of culture, sLIGHT (100 ng/ml) was added either in the somatic or in the axonal compartment, or both. Axonal length (E) and branching (F) were determined 48 h later.
G The retrograde transport of Alexa555-conjugated WGA allows tracing of motoneurons whose axons have crossed microchannels (white arrows).
H The survival of motoneurons whose axons have reached the axonal compartment is determined 48 h later by counting the number of GFP- and WGA-positive cells. Scale bar, 100 μm.
Data information: Histograms show mean values ± SEM in three independent experiments, ANOVA with Tukey–Kramer’s post hoc test; *P < 0.05, ***P < 0.001
LIGHT is upregulated at the site of nerve lesion
The death outcome of the somatic LIGHT signal is consistent with the increased levels of LIGHT in the spinal cord associated with motoneuron degeneration in ALS 5, 6. To confirm that upregulation of LIGHT in the spinal cord is associated with motoneuron death, we conducted a sciatic nerve axotomy that led to motoneuron death along with an upregulation of IFN-γ (Supplementary Fig S3A–D). While LT-βR levels were not modified, we found that LIGHT levels were significantly elevated in the axotomized spinal cord (Supplementary Fig S3E–H).
We then explored whether following a sciatic nerve damage that leads to motor axon regeneration but not motoneuron death, LIGHT levels might be differentially modified between the spinal cord and the periphery. Indeed, sciatic nerve lesion in adult mice did not induce motoneuron loss and led to increased levels of the neuronal regeneration marker growth-associated protein 43 (GAP-43) at the lesion site (Fig 4A, B and Supplementary Fig S3I, J). We did not observe any significant upregulation of LIGHT in the lumbar spinal cord or the tibialis anterior (TA) muscle. However, a robust increase in LIGHT was observed at the site of nerve lesion 10 days after crush (Fig 4C, D). We confirmed by quantitative RT-PCR and analysis of LIGHT immunoreactivity in motoneurons that mRNA and protein levels of LIGHT did not differ in the spinal cord after nerve lesion (Fig 4E–G). Expression of LT-βR was not affected by nerve lesion in spinal cord, sciatic nerve, and TA following injury (Fig 4H, I).
Figure 4. LIGHT is upregulated at the site of regeneration following nerve injury.
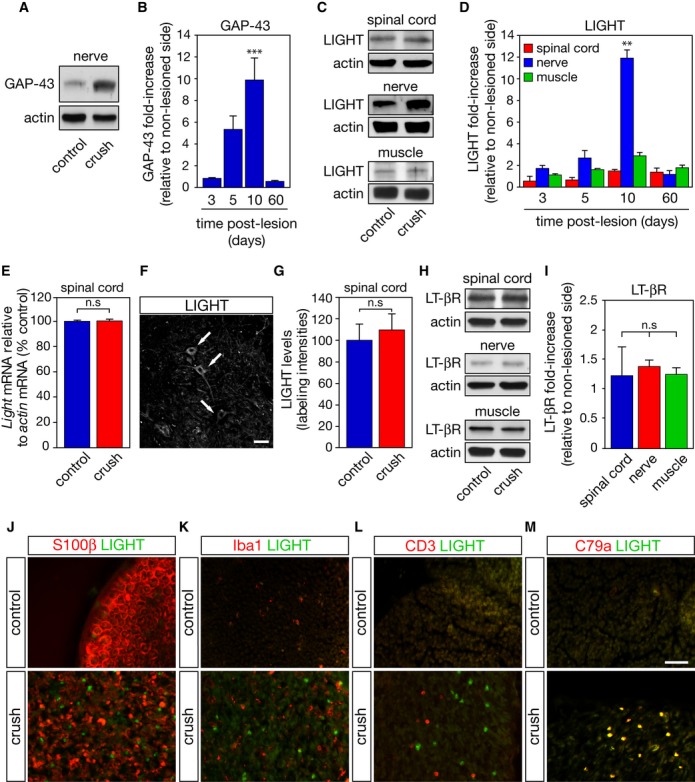
A, B Total levels of GAP-43 were monitored in the sciatic nerve by Western blot (A). GAP-43 band intensities were normalized to their corresponding actin band and expressed as the ratio of lesioned to non-lesion side values (B).
C A representative immunoblot of LIGHT signals in the spinal cord, sciatic nerve, and tibialis anterior muscle 10 days after the nerve lesion.
D At the indicated time, fold increase in LIGHT over sham-operated condition (control) was determined by densitometric analysis of immunoreactive bands, normalized to their respective actin signals.
E qRT-PCR analysis of Light mRNA expression in the spinal cord 10 days following sciatic nerve lesion.
F, G Ten days following a unilateral sciatic nerve crush, LIGHT immunoreactivity in motoneurons (F, arrows; scale bar, 25 μm) in both sham- and operated side was quantified (G).
H, I At the peak of LIGHT expression, LT-βR levels were determined as in (C) and (D).
J–M Ten days after a unilateral crush, sciatic distal nerve sections were co-immunostained for LIGHT, S100β (J), Iba1 (K), CD3 (L), and CD79a (M). Scale bar, 50 μm.
Data information: Results shown in (B, D, I) are the mean values ± SEM, n = 4, ANOVA with Tukey–Kramer’s post hoc test. Results shown in (E) and (G) are means ± SEM, n = 3, unpaired two-tailed Student’s t-test.
Following a crush, the lesioned peripheral nerve is progressively infiltrated by macrophages to eliminate myelin debris and facilitate axonal regeneration 11. In addition, T and B lymphocytes infiltrate the lesion site and have been proposed to contribute to the macrophage influx, myelin removal, and axonal regeneration 12. Interestingly, 10 days after nerve crush, LIGHT immunoreactive cells are observed in the distal nerve segment among the remaining Schwann cells debris, as identified by S100β (Fig 4J). Iba1+ macrophages and CD3+ T cells were found in the injured nerve segment but were distinct from LIGHT-immunopositive cells (Fig 4K, L). However, we found that CD79a+ B cells, which also invade the lesioned distal segment of the sciatic nerve, expressed LIGHT (Fig 4M).
Genetic ablation of Light retards regeneration of peripheral nerve following injury
To evaluate the in vivo role of LIGHT in peripheral nerve regeneration, we analyzed nerve regeneration in Light−/− mice. Light-deficient mice do not show any behavioral abnormalities and motoneuron numbers between Light+/+ and Light−/− mice did not differ 5. We also found that the neuromuscular junction (NMJ) innervation of TA from Light−/− mice did not differ from that of wild-type mice (Fig 5A,B). We then performed a unilateral sciatic nerve crush on wild-type and Light-deficient mice and followed their locomotor recovery. Nerve lesion resulted in a dramatic reduction in the sciatic function index (SFI), followed by recovery of locomotor activity within 2 weeks. However, Light-deficient mice showed a significant delay in recovery (Fig 5C). We quantified the frequency with which pre-synaptic terminals occupy the post-synaptic area in TA 7, 15 and 21 days after nerve crush in Light+/+ and Light−/− mice. Seven days after nerve damage, both wild-type and Light-deficient mice presented severe motor endplates denervation (Fig 5D,E). Reinnervation of NMJs in the TA of Light+/+ mice was almost complete as early as 15 days after nerve injury. However, reinnervation of NMJs was retarded in Light−/− mice at 15 and 21 days post-injury (Fig 5E).
Figure 5. LIGHT is required for axonal regeneration following nerve lesion.
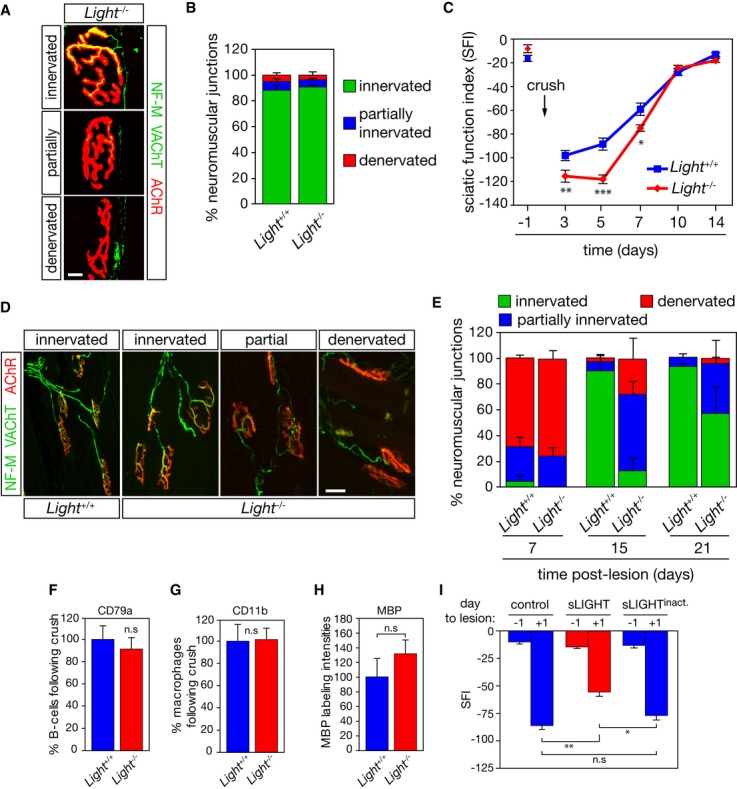
A Tibialis anterior muscle from 50-day-old Light+/+ (not shown) Light−/− mice was stained with anti-neurofilament (NF-M) and anti-vesicular acetylcholine transporter (VAChT) antibodies (in green) and fluorochrome-conjugated α-bungarotoxin (AChR, in red). Scale bar, 10 μm.
B The percentage of innervated, partially innervated and denervated motor endplates, based on the overlap of NF-M/VAChT and AChR area, was quantified (n = 3).
C Functional recovery was quantified by calculating the SFI before and on days 3, 5, 7, 10, and 14 post-nerve lesion in Light+/+ (n = 24) and Light−/− mice (n = 22).
D Representative confocal images illustrating complete to partial occupancy and vacancy of neuromuscular junctions following nerve lesion. Scale bar, 50 μm.
E The degree of neuromuscular junction occupancy was quantified after nerve injury in Light+/+ and Light−/− mice (n = 4).
F–H The number of CD79a+ (F), CD11b+ (G) and the intensity of MBP (H) immunoreactivity were quantified in the sciatic nerve of Light+/+ and Light−/− mice 10 days after crush (n = 3).
I At the time of a unilateral sciatic nerve lesion, mice were intramuscularly injected with PBS (control), sLIGHT and a heat-inactivated sLIGHT (250 ng/ml). The SFI was determined at indicated days to the lesion (n = 12 for each condition).
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. In (C, I) two-way repeated-measure ANOVA, Newman–Keuls post hoc test; in (F, G, H) unpaired two-tailed Student’s t-test.
B lymphocytes promote myelin clearance and macrophage influx at the lesion site 12. We determined the number of B cells (CD79a+), macrophages (CD11b+), and peripheral myelin levels in distal sciatic nerve after crush (Supplementary Fig S4A, B). We did not observe any significant difference in the number of CD79a+, CD11b+, and myelin basic protein (MBP) levels in the lesioned sciatic nerve of Light−/− compared to Light+/+ mice (Fig 5F–H).
To evaluate whether the effect of LIGHT was selective to motor axon regeneration, we next assessed sensory function recovery and did not observe a significant difference between Light+/+ and Light−/− mice following sciatic nerve damage (Supplementary Fig S4C). We then evaluated the effect of peripheral LIGHT administration on motor functions following sciatic nerve crush. We found that the intramuscular delivery of sLIGHT improved motor functions of mice compared to injection with saline or an inactive form of sLIGHT (Fig 5I).
A functional bivalence of death receptors has already been documented through FasL-Fas and TNF-α-TNFR interactions. Whereas activation of Fas by FasL triggers a death-signaling pathway in motoneurons 4 and has been proposed to contribute to ALS pathogenesis 13, the activation of Fas also promotes neurite outgrowth in sensory neurons, while not affecting their survival 3, 4. Although studies to further explore the potential effect of Fas engagement on motoneuron growth and branching are needed, the observed opposite responses may rely on the commitment of neuron type-dependent intracellular events. Another example of dual signaling is illustrated by the effect of TNF-α in embryonic motoneurons and post-natal sympathetic neurons. Interestingly, the opposite response elicited by TNF-α may not be governed by a cell-type feature but by the signaling orientation given by the membrane-integrated molecule acting as a receptor. Indeed, activation of TNFR signaling by soluble TNF-α triggers motoneuron death 14, while activation of the membrane-integrated form of TNF-α by TNFR1 induces a reverse signaling that promotes axon growth and branching in sympathetic neurons 15. Here, we implement the diversity of post-receptor signaling by showing that opposite neuronal responses can arise from the regional effect of the same ligand. At present, it remains to be determined what are the mechanisms involved in the dual response of motoneurons to LIGHT.
Both Fas- and TNF-α-induced neurite outgrowth of sensory and sympathetic neurons, respectively, require the activation of the ERK pathway 3, 15. However, contrary to what was observed in sensory neurons following Fas activation 3, we found that caspase signaling contributes to LIGHT-mediated axon outgrowth and branching. The non-apoptotic function of caspase signaling has already been documented for axon pathfinding and synapse maturation of olfactory sensory neurons 16. The caspase-9-mediated cleavage of Sema7A, a semaphorin family member that acts as a developmental axon guidance cue, might contribute to axon wiring in the olfactory system 16. Axon guidance molecules that play a role in the patterning and positioning of motoneurons during development also contribute to the regenerative ability of neurons 17. The caspase-mediated cleavage of axon-inhibitory proteins could therefore be envisaged as a molecular mechanism driving motor axon regeneration, plasticity, and functional recovery.
This work establishes that a same ligand can elicit, in a local-dependent manner, distinct responses within the same cell. This work further underscores the importance of therapeutic intervention site, whether a central or peripheral delivery of a therapeutic candidate must be considered, as well as the mode of therapeutic action, that is, the stimulation or the inhibition of the molecular target.
Materials and Methods
Complete details of the experimental procedures used in the present study can be found in the Supplementary Methods.
Motoneuron culture system and analysis
Motoneurons were isolated from embryonic day (E) 12.5 mice of indicated genotype as previously described 5. Microfluidic devices were prepared as described in 10. Axonal length and branching were determined in 50–100 neurons per condition from triplicate wells per experiment and from at least three independent experiments. Motoneuron survival was assessed by direct counting of GFP- or WGA-positive cells or by counting phase-bright neurons using morphological criteria. Time-lapse imaging was performed as previously described 18.
Immunostaining and Western blotting
Immunostaining of neurons, lumbar spinal cord, sciatic nerve, and muscle sections was done as described 5, 19. Preparation of tissue lysates, SDS–PAGE, immunoblotting, and quantification were performed as described 5.
Sciatic nerve lesion
The right sciatic nerve of anesthetized 50-day-old mice was exposed at its mid-thigh level and crushed for 30 s using forceps. For exploring the effect of LIGHT deficiency on nerve function, we calculated the sciatic function index based on walking track analysis as described 20.
Acknowledgments
We are thankful to E. Kremer for his critical reading of the manuscript; F. Mann, B. Schneider, and all members of the team for their helpful comments throughout the work. We thank K. Pfeffer and S. Scheu for the gift of Light-deficient mice. We thank G. Schiavo and G. Menendez for microfluidic molds and S. Nedelec for his helpful advices on microfluidic experiments. This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale (Inserm), Association Française contre les Myopathies, Association Française pour la Recherche sur la SLA, Languedoc-Roussillon Region, the Thierry Latran foundation, and EMBO long-term fellowship.
Author contributions
BO, AM, JA, MB, and CR conceived and designed the experiments; BO, AM, JA, EC, CS, CS, CS, SS. performed the experiments; BO, AM, JA, JV, MB, and CR analyzed the data; and BO, JA, MB, and CR wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://embor.embopress.org
References
- Chauvet S, Cohen S, Yoshida Y, Fekrane L, Livet J, Gayet O, Segu L, Buhot MC, Jessell TM, Henderson CE, et al. Gating of Sema3E/PlexinD1 signaling by neuropilin-1 switches axonal repulsion to attraction during brain development. Neuron. 2007;56:807–822. doi: 10.1016/j.neuron.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, Turner JR, Fu YX, Romero IL, Lengyel E, et al. CD95 promotes tumour growth. Nature. 2010;465:492–496. doi: 10.1038/nature09075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbarats J, Birge RB, Mimouni-Rongy M, Weinstein DE, Palerme JS, Newell MK. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat Cell Biol. 2003;5:118–125. doi: 10.1038/ncb916. [DOI] [PubMed] [Google Scholar]
- Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Aebischer J, Cassina P, Otsmane B, Moumen A, Seilhean D, Meininger V, Barbeito L, Pettmann B, Raoul C. IFNgamma triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ. 2011;18:754–768. doi: 10.1038/cdd.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebischer J, Moumen A, Sazdovitch V, Seilhean D, Meininger V, Raoul C. Elevated levels of IFNgamma and LIGHT in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Eur J Neurol. 2012;19:e745–e756. doi: 10.1111/j.1468-1331.2011.03623.x. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavalda N, Gutierrez H, Davies AM. Developmental regulation of sensory neurite growth by the tumor necrosis factor superfamily member LIGHT. J Neurosci. 2009;29:1599–1607. doi: 10.1523/JNEUROSCI.3566-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002;195:1613–1624. doi: 10.1084/jem.20020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry EJ, Ho C, David S. A role for Nogo receptor in macrophage clearance from injured peripheral nerve. Neuron. 2007;53:649–662. doi: 10.1016/j.neuron.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci USA. 2010;107:11993–11998. doi: 10.1073/pnas.1001948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Buhler E, Sadeghi C, Jacquier A, Aebischer P, Pettmann B, Henderson CE, Haase G. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc Natl Acad Sci USA. 2006;103:6007–6012. doi: 10.1073/pnas.0508774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolini G, Raoul C, Ferri A, Haenggeli C, Yamamoto Y, Salaün D, Henderson CE, Kato AC, Pettmann B, Hueber AO. Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J Neurosci. 2003;23:8526–8531. doi: 10.1523/JNEUROSCI.23-24-08526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisiswa L, Osorio C, Erice C, Vizard T, Wyatt S, Davies AM. TNFalpha reverse signaling promotes sympathetic axon growth and target innervation. Nat Neurosci. 2013;16:865–873. doi: 10.1038/nn.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa S, Hamada S, Kuida K, Yoshida H, Igaki T, Miura M. Maturation of the olfactory sensory neurons by Apaf-1/caspase-9-mediated caspase activity. Proc Natl Acad Sci USA. 2010;107:13366–13371. doi: 10.1073/pnas.0910488107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ER, Pasterkamp RJ, van den Berg LH. Axon guidance proteins: novel therapeutic targets for ALS? Prog Neurobiol. 2009;88:286–301. doi: 10.1016/j.pneurobio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Pieraut S, Laurent-Matha V, Sar C, Hubert T, Méchaly I, Hilaire C, Mersel M, Delpire E, Valmier J, Scamps F. NKCC1 phosphorylation stimulates neurite growth of injured adult sensory neurons. J Neurosci. 2007;27:6751–6759. doi: 10.1523/JNEUROSCI.1337-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard-Marissal N, Moumen A, Sunyach C, Pellegrino C, Dudley K, Henderson CE, Raoul C, Pettmann B. Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS. J Neurosci. 2012;32:4901–4912. doi: 10.1523/JNEUROSCI.5431-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, et al. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron. 2012;75:633–647. doi: 10.1016/j.neuron.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.