

Abstract

Ir catalysts supported by bidentate silyl ligands that contain P- or N-donors are shown to effect ortho borylations for a range of substituted aromatics. The substrate scope is broad, and the modular ligand synthesis allows for flexible catalyst design.
Directed ortho-metalation (DoM) is a powerful synthetic method that is widely applied by synthetic chemists.1 More recently, C–H borylation catalysts have been developed which are able to functionalize aromatic C–H bonds with regioselectivities that are often sterically determined, thus giving complementary selectivity to DoM.2 Because C–H borylations tolerate groups like esters, which can be incompatible with DoM, and do not require low temperatures to operate like DoM, there has been significant interest in ortho-directed borylations.3 Aside from an early report on ortho borylation of benzamide,4 the first examples of ortho borylations in Ir-catalyzed homogeneous systems involved silyl-directed borylation described by Hartwig and Boebel.5 This approach requires conversion of O–H and N–H bonds to O–SiR2H and N–SiR2H moieties. The Si–H bonds undergo formal σ bond metathesis with Ir–Bpin groups to generate Ir–Si bound intermediates that direct functionalization of C–H bonds ortho to O and N groups. Recently, Maleczka, Singleton, and Smith have proposed that hydrogen-bonding interactions between aniline N–H bonds and Bpin O atoms can direct ortho borylations of anilines.6
Other examples of ortho borylations rely on intrinsic substrate functionality. For example, Ishiyama and Miyaura have described ortho borylation of esters;7 Lassaletta has reported N-directed ortho borylations;8 Ito and Ishiyama have developed ortho borylations of ketones;9 and Clark has disclosed borylations of benzylamines and phosphines.10 Although detailed kinetic studies have not been reported, mechanisms where two coordination sites in the catalyst (structure 2) are available to the substrate are most plausible (Scheme 1). One site enables coordination of a directed metalating group (DMG), while the other is necessary to cleave the ortho C–H bond. Unfortunately, equilibria with thermodynamically favored 5-coordinate structures 3 likely reduces activity.
Scheme 1. Ortho Borylation Strategies with DMGs.

In contrast to the aforementioned homogeneous catalysts, Ir-catalyzed borylations using surface supported phosphine ligands, pioneered by Sawamura, demonstrate a broad scope for ortho-directed borylations.11 While the surface supported phosphine systems of Sawamura’s are highly active, their synthesis can be nontrivial, which makes modification of the phosphine structure challenging.12 Thus, we sought an appropriate ligand framework for homogeneous catalysis that could mimic these heterogeneous catalysts. Given that tridentate PSiP pincer ligands have been utilized in C–H borylation,13 we were hopeful that bidentate Si–P ligands could generate structures 4, where the silyl ligand replaces a spectating boryl. Silane metathesis with a boryl ligand would steer the phosphine to the metal center to generate intermediates (4) with accessible coordination sites. In contrast to other homogeneous approaches where electron-deficient ligands are used to achieve coordinative unsaturation, our strategy would create an electron-rich framework that facilitates C–H cleavage.14 If this approach proved to be viable, the ligand framework could be modified in substantive ways. For example, silanes with pendant N-donors (or other basic ligands) could be targeted.
To test this concept, we prepared ligand 5 (Scheme 1), using a protocol related to the synthesis of (2-diphenylphosphinophenyl)diphenylsilane.15 The synthesis is straightforward, modular, and scalable to gram quantities. With 5 in hand, we tested it for the C–H borylation of methylbenzoate. Table 1 shows the results compared to those obtained for catalysts generate from [Ir(OMe)(cod)]2 and P(3,5-bis(trifluoromethyl)phenyl)3 (PArF3) or triphenylarsine, which have both been used in directed borylations. As can be seen in entry 1, ligand 5 allows for full conversion of B2pin2 and some of the generated HBpin, while the PArF3 and AsPh3 give considerably lower conversions. In contrast, silica-SMAP ligated Ir catalysts are significantly more active.
Table 1. Comparison of Catalyst Efficiency for Ortho Borylation.
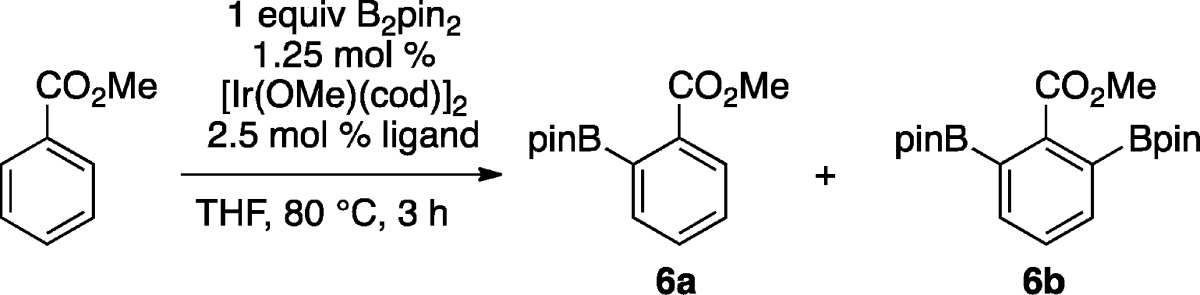
| entrya | ligand | %6ab | %6bb | % conversionc |
|---|---|---|---|---|
| 1 | 5 | 63 | 24 | 111d |
| 2 | PArF3 | 2 | 1 | 4 |
| 3 | AsPh3 | 44 | 3 | 50 |
| 4e | silica-SMAP | 101f | 4f | 109d |
Reactions run on 0.5 mmol scale in 0.5 mL THF.
% Conversions determined by GC integration.
% Conversion is calculated as %6a + 2(%6b) and is based on B2pin2 as limiting reagent.
Conversion exceeds 100% because some of the HBpin generated from B2pin2 participates in the borylation.
Data are from ref (11a). Reaction run at 25 °C with 1 mmol methylbenzoate, 0.5 mmol B2pin2, 0.5 mol % silica-SMAP-Ir(OMe)(cod) in 1.5 mL hexane.
Yields were determined by 1H NMR spectroscopy.
With promising preliminary data in hand we explored a number of substituted methyl benzoates (see Chart 1). The reactions were performed under identical conditions, and reaction times were not optimized. Yields range from fair to excellent with the lowest yields corresponding to methylbenzoates that were only substituted at the 4-position. For these substrates, significant amounts of 2,6-diborylated compounds form, which accounts for diminished yield of monoborylated products 6i and 6k. Some diborylation was also observed in the synthesis of 6e. It is noteworthy that lowering the catalyst loading to 0.5 mol % and reducing the temperature to 60 °C gave a 6% yield increase for 6k. In addition, diborylation is not observed for t-butylbenzoate in contrast to methylbenzoate. Lastly, the selectivity difference for ligand 5 relative to 4,4′-di(t-butyl)bipyridine (dtbpy) is highlighted in Scheme 2, where different regioisomers are obtained in the borylations of 2-fluoro-4-bromomethylbenzoate.
Chart 1. Ortho Borylation of Alkylbenzoatesa.

a Reactions were run with 1.0 mmol substrate, 1.0 mmol B2pin2, 0.025 mmol 5, and 0.0125 mmol [Ir(OMe)(cod)]2 in 1.0 mL THF. Yields are for isolated materials.
b 2.0 mmol substrate was used.
c 0.5 mol % [Ir(OMe)(cod)]2 was used.
d Reaction was run at 60 °C.
e 2.0 mmol B2pin2, 0.025 mmol [Ir(OMe)(cod)]2, and 0.050 mmol 5 were used.
f 0.5 mmol substrate was used.
g 1.25 mmol B2pin2 was used.
h 5% diborylated material was isolated.
i 17% diborylated material was isolated.
j 10% diborylated material was isolated.
Scheme 2. Ligand-Dependent Borylation Regioselectivity.

Given the results in Chart 1, we further explored the scope with respect to substrate and directing group, and the results are given in Table 2. Entry 1 shows that unsubstituted benzamides give excellent conversions to monoborylated substrates without resorting to using excess arene to minimize diborylation as is required for methyl benzoate. Entry 2 shows that the amide DMG trumps Cl direction. In fact, chlorobenzene does not give ortho-borylated product with 5, contrasting the high ortho selectivity for Ir-SMAP catalysts. In entry 3, diborylated product 8c was obtained. When 1 equiv B2pin2 was used with 7c, selectivity for borylation ortho to the amide vs the ester was 4.2:1. Entries 4 and 5 show that OMe can direct borylation to the ortho position, albeit in modest yield. This sets the chemistry of catalysts generated from ligand 5 apart from that of silica-SMAP, where borylation of 7d gives a 1:1 ratio of o:m + p borylated products. Entry 6 shows that esters are stronger DMGs that OMe, which is in line with SMAP supported catalysts. In entry 7, Weinreb amide 7g is converted cleanly to ortho-borylated product 8g. Borylation of 1-methylnaphthoate, 7i, gives a single isomer where functionalization of the β-C–H bond is favored over the γ-C–H position. Borylation of thiophene substrate 7h at 60 °C gave slightly better selectivity (14:1) for 3 vs 5-borylated products than that reported for the borylation with Ir-SMAP catalysts (9:1 at 80 °C). In contrast to the SMAP system, where the Ir catalysts are ineffective for borylation of 7j, borylation gives the products obtained by Lassaletta using hemilabile N,N ligands.8a Our reaction was performed at a lower temperature (60 vs 80 °C) and a shorter reaction time (2 vs 48 h). This was offset by a higher catalyst loading in our study (1.25 vs 0.5 mol %) and the formation of ∼15% diorthoborylated product. With 2.0 equiv B2pin2, this was the major product.
Table 2. Borylations with 5/[Ir(OMe)(cod)]2 Catalysta.
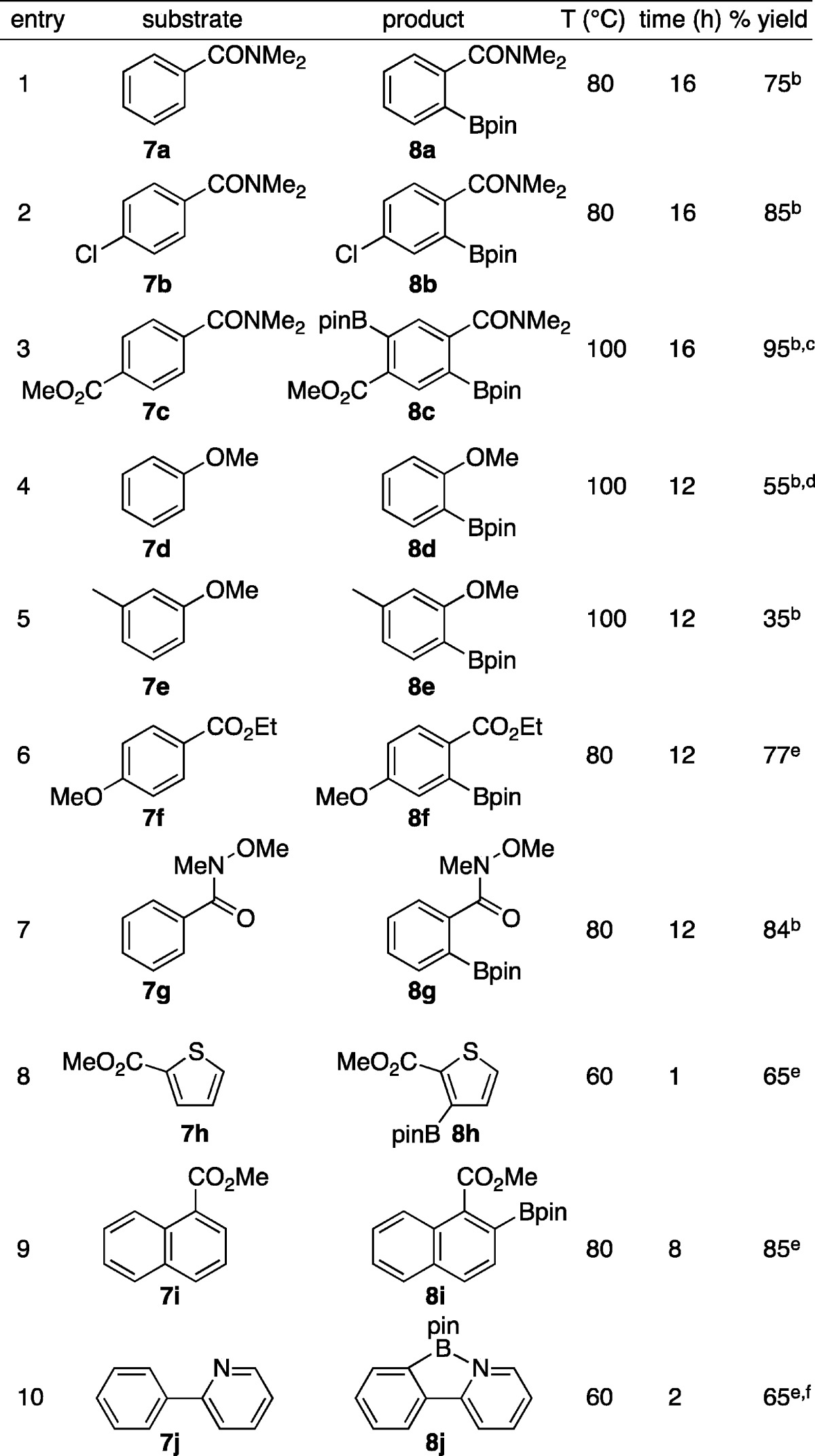
Typically reactions were run on 1 mmol scale with equimolar B2pin2 and substrate with 2.5 mol % 5 and 1.25 mol % [Ir(OMe)(cod)]2. Unless noted, yields are for isolated materials. See SI for details.
Reaction solvent was THF.
2.0 equiv B2pin2 was used.
GC-conversion with a 6:1:2 ratio of o:m + p:diborylated products.
Reaction solvent was n-hexane.
Approximately 15% of the crude mixture was the diorthoborylated compound (8j′ in the SI).
The generation of active catalyst from 5, B2pin2, and [Ir(OMe)(cod)]2 raises interesting questions with regard to how an Ir–Si bond is generated, if it is formed at all. Several literature reports describe the metathesis of M–OR and Si–H bonds to make M–H and Si–OR products. However, silane-directed borylations invoke M–B/Si–H metatheses to generate M–Si and B–H bonds. Thus, we considered that initial formation of Ir–Bpin intermediates and subsequent Ir–B/Si–H metathesis could account for formation of intermediates such as 4. Nevertheless, we performed a control experiment where 5 and [Ir(OMe)(cod)]2 were reacted. 31P and 1H NMR indicates rapid and quantitative conversion to compound 9 and methanol—an unprecedented reaction for a silane with a metal alkoxide (eq 1). This is critical for establishing a 1:1 ratio of 5:Ir, which is the key to creating sites for DMG coordination and C–H activation as posited for structure 4 in Scheme 1. The structure of 9 was confirmed by X-ray crystallography and is shown in Figure 1. Using the centroids of the cod bound carbons as coordinates, the sum of the angles about Ir is 370°, which is close to the value for square-planar IrI. The Ir–C and C–C distances for the carbons trans to Si are significantly longer than those for carbons trans to P, which is consistent with strong donation from Si to Ir.
Figure 1.
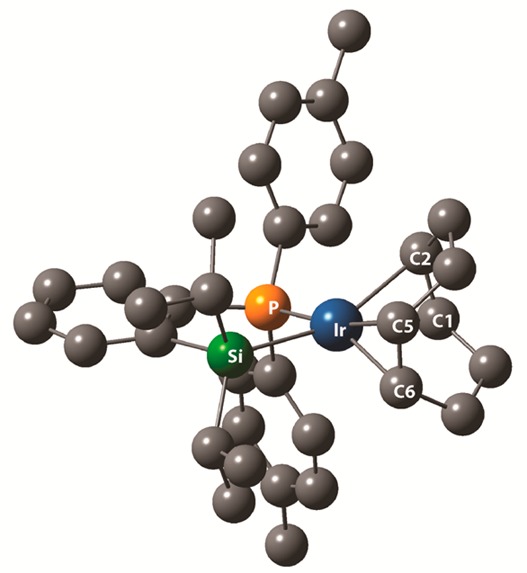
Crystal structure of compound 9 with H atoms omitted. Selected distances (Å) and angles (deg): Ir–Si (2.376(2)), Ir–P (2.260(2)), Ir–C1 (2.246(8)), Ir–C2 (2.217(8)), Ir–C5 (2.194(8)), Ir–C6 (2.161(8)), C1–C2 (1.386(12)), C5–C6 (1.357(13)), P–Ir–Si (83.53(6)), P–Ir–centroidC1–C2 (96.72), Si–Ir–centroidC5–C6 (95.39), centroidC1–C2–Ir–centroidC5–C6 (85.25).
There is a difference in the reaction rates when using pure 9 as precatalyst as opposed to in situ generated catalysts, with shorter reaction times being required for the isolated catalyst.
 |
1 |
Some substrates proved to be challenging for borylations carried out with ligand 5. For example, carbamates react sluggishly, and ketones give significant amounts of ketone reduction. Since Ir catalysts supported by N-donor ligands in most cases prove to be more reactive than their P-donor counterparts,16 we reasoned that an N-donor analog of 5 might exhibit better reactivity with carbamates and ketones. Thus, quinoline-based ligand 10 was prepared, and Ir catalysis with this ligand was evaluated relative to ligand 5 for compounds 11a and 11b.17
As shown in Table 3, ligand 10 outperforms its phosphine analog 5 for borylations of 11a and 11b. In the case of carbamate 11a, we see higher conversion, but slightly lower isolated yield to that reported by Sawamura for the related diethylcarbamate substrate. Perhaps more significantly, catalysis with 10 did not yield detectable borylation para to the carbamate, which is a minor byproduct in the silica-SMAP system. For phenyl ethyl ketone, ligand 5 gave the reduction to the alcohol upon workup as the major product. This unwanted reaction was almost completely eliminated by employing N-donor ligand 10. When compared to other homogeneous catalysts that effect ortho borylation of ketones,9 borylations employing 10 had shorter reaction times at considerably lower temperatures. Furthermore, borylations of t-butylbenzoate and benzamide 7a with ligand 10, using identical conditions to those used with ligand 5, gave 6m in 78% isolated yield after 3 h and 8a in 80% yield after 4 h, respectively. Significantly, ortho borylation of anisole was not found with ligand 10. These results show that modification of the silyl ligand framework can dramatically impact reactivity.
Table 3. Comparison of Silyl Ligands with Pendant P- and N-donors.

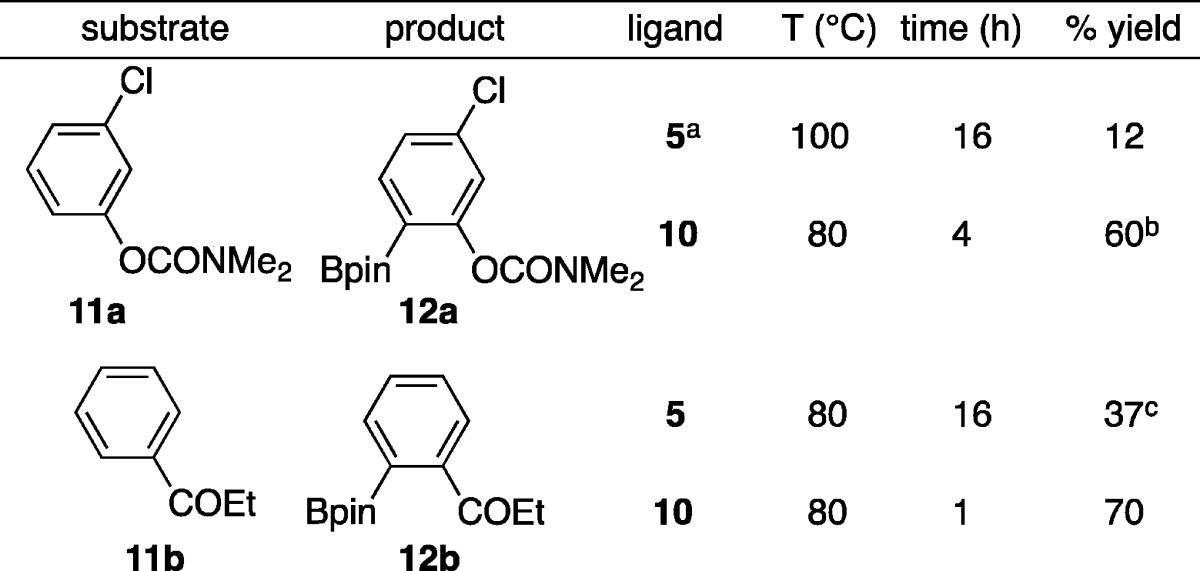
2.0 equiv B2pin2, 5 mol % 5, and 2.5 mol % [Ir(OMe)(cod)]2.
93% conversion of starting material was observed by GC.
Conversion based on GC. 56% conversion to 1-phenylpropan-1-ol was observed.
In summary, we have shown that Ir catalysts supported by silyl-tethered P- and N-donor ligands are effective for ortho borylation directed by a range of different functional groups. Because the ligands within the coordination sphere are bidentate and electron-rich, the activities are generally superior to homogeneous systems that utilize electron-poor ligands to favor unsaturated intermediates. We are exploring effects of varying the ligand framework on the reactivity in other directed C–H functionalizations.
Acknowledgments
We thank NIH (GM63188), NSF (GOALI-1012883), and ACS Green Chemistry Institute Pharmaceutical Roundtable for generous financial support and BoroPharm, Inc. for supplying B2pin2. We dedicate this paper to the memory of Professor Gregory Hillhouse.
Supporting Information Available
Full characterization, copies of all spectral data, experimental procedures, and X-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hurst T. E.; Macklin T. K.; Becker M.; Hartmann E.; Kügel W.; Parisienne-La Salle J. C.; Batsanov A. S.; Marder T. B.; Snieckus V. Chem.—Eur. J. 2010, 16, 8155. [DOI] [PubMed] [Google Scholar]
- Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890. [DOI] [PubMed] [Google Scholar]
- For a recent review of functional group directed borylation, see:Ros A.; Fernandez R.; Lassaletta J. M. Chem. Soc. Rev. 2014, 43, 3229. [DOI] [PubMed] [Google Scholar]
- Cho J.-Y.; Iverson C. N.; Smith M. R. III J. Am. Chem. Soc. 2000, 122, 12868. [Google Scholar]
- a Boebel T. A.; Hartwig J. F. J. Am. Chem. Soc. 2008, 130, 7534. [DOI] [PubMed] [Google Scholar]; b Robbins D. W.; Boebel T. A.; Hartwig J. F. J. Am. Chem. Soc. 2010, 132, 4068. [DOI] [PubMed] [Google Scholar]
- a Preshlock S. M.; Plattner D. L.; Maligres P. E.; Krska S. W.; Maleczka R. E. Jr.; Smith M. R. III Angew. Chem., Int. Ed. 2013, 52, 12915. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Roosen P. C.; Kallepalli V. A.; Chattopadhyay B.; Singleton D. A.; Maleczka R. E. Jr.; Smith M. R. III J. Am. Chem. Soc. 2012, 134, 11350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama T.; Isou H.; Kikuchi T.; Miyaura N. Chem. Commun. 2010, 159. [DOI] [PubMed] [Google Scholar]
- a Ros A.; Estepa B.; Lopez-Rodriguez R.; Alvarez E.; Fernandez R.; Lassaletta J. M. Angew. Chem., Int. Ed. 2011, 50, 11724. [DOI] [PubMed] [Google Scholar]; b López-Rodríguez R.; Ros A.; Fernández R.; Lassaletta J. M. J. Org. Chem. 2012, 77, 9915. [DOI] [PubMed] [Google Scholar]; c Ros A.; Lopez-Rodriguez R.; Estepa B.; Alvarez E.; Fernandez R.; Lassaletta J. M. J. Am. Chem. Soc. 2012, 134, 4573. [DOI] [PubMed] [Google Scholar]
- Itoh H.; Kikuchi T.; Ishiyama T.; Miyaura N. Chem. Lett. 2011, 40, 1007. [Google Scholar]
- a Roering A. J.; Hale L. V. A.; Squier P. A.; Ringgold M. A.; Wiederspan E. R.; Clark T. B. Org. Lett. 2012, 14, 3558. [DOI] [PubMed] [Google Scholar]; b Crawford K. M.; Ramseyer T. R.; Daley C. J. A.; Clark T. B. Angew. Chem., Int. Ed. 2014, 7589. [DOI] [PubMed] [Google Scholar]
- a Kawamorita S.; Ohmiya H.; Hara K.; Fukuoka A.; Sawamura M. J. Am. Chem. Soc. 2009, 131, 5058. [DOI] [PubMed] [Google Scholar]; b Kawamorita S.; Ohmiya H.; Sawamura M. J. Org. Chem. 2010, 75, 3855. [DOI] [PubMed] [Google Scholar]; c Yamazaki K.; Kawamorita S.; Ohmiya H.; Sawamura M. Org. Lett. 2010, 12, 3978. [DOI] [PubMed] [Google Scholar]; d Kawamorita S.; Murakami R.; Iwai T.; Sawamura M. J. Am. Chem. Soc. 2013, 135, 2947. [DOI] [PubMed] [Google Scholar]; e Kawamorita S.; Miyazaki T.; Ohmiya H.; Iwai T.; Sawamura M. J. Am. Chem. Soc. 2011, 133, 19310. [DOI] [PubMed] [Google Scholar]; f Kawamorita S.; Miyazaki T.; Iwai T.; Ohmiya H.; Sawamura M. J. Am. Chem. Soc. 2012, 134, 12924. [DOI] [PubMed] [Google Scholar]
- Recently, three-fold cross-linked polystyrene–triphenylphosphane hybrid systems have been developed. Their synthesis is simpler, and intial studies indicate that these function similar to silica-supported monophosphines:Iwai T.; Harada T.; Hara K.; Sawamura M. Angew. Chem., Int. Ed. 2013, 52, 12322. [DOI] [PubMed] [Google Scholar]
- Fang H.; Choe Y.-K.; Li Y.; Shimada S. Chem.—Asian J. 2011, 6, 2512. [DOI] [PubMed] [Google Scholar]
- Vanchura B. A.; Preshlock S. M.; Roosen P. C.; Kallepalli V. A.; Staples R. J.; Maleczka R. E. Jr.; Singleton D. A.; Smith M. R. III Chem. Commun. 2010, 46, 7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Wang L.; Chang S.-H.; Huang K.-L.; Chi Y.; Hung W.-Y.; Chen C.-M.; Lee G.-H.; Chou P.-T. Dalton Trans. 2013, 42, 7111. [DOI] [PubMed] [Google Scholar]
- Preshlock S. M.; Ghaffari B.; Maligres P. E.; Krska S. W.; Maleczka R. E. Jr.; Smith M. R. III J. Am. Chem. Soc. 2013, 135, 7572. [DOI] [PubMed] [Google Scholar]
- For an early example of a bis(quinoline)silyl ligand that has been used in dehydrogenative silylations of arenes, see:; a Stradiotto M.; Fujdala K. L.; Tilley T. D. Chem. Commun. 2001, 1200. [Google Scholar]; b Sangtrirutnugul P.; Tilley T. D. Organometallics 2007, 26, 5557. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.