

Abstract
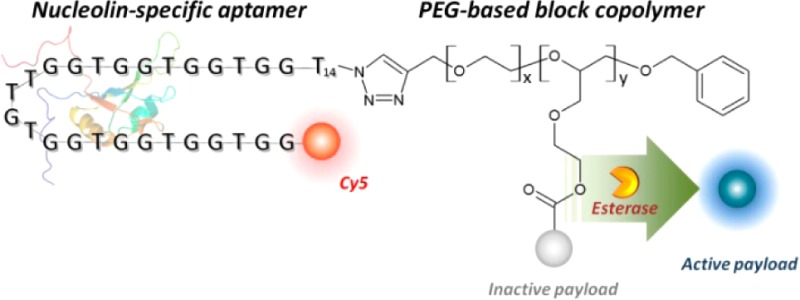
Viruses have evolved specialized mechanisms to efficiently transport nucleic acids and other biomolecules into specific host cells. They achieve this by performing a coordinated series of complex functions, resulting in delivery that is far more efficient than existing synthetic delivery mechanisms. Inspired by these natural systems, we describe a process for synthesizing chemically defined molecular constructs that likewise achieve targeted delivery through a series of coordinated functions. We employ an efficient “click chemistry” technique to synthesize aptamer-polymer hybrids (APHs), coupling cell-targeting aptamers to block copolymers that secure a therapeutic payload in an inactive state. Upon recognizing the targeted cell-surface marker, the APH enters the host cell via endocytosis, at which point the payload is triggered to be released into the cytoplasm. After visualizing this process with coumarin dye, we demonstrate targeted killing of tumor cells with doxorubicin. Importantly, this process can be generalized to yield APHs that specifically target different surface markers.
Introduction
Over many millions of years, viruses have evolved elegant and sophisticated mechanisms for selectively delivering biological payloads into host cells by performing a sequence of molecular functions in a coordinated and systematic manner.1,2 For example, adeno-associated viruses (AAV) selectively adsorb onto subsets of host cells that express specific surface markers.3,4 Then, the AAV is internalized by these host cells via endocytosis, after which it makes its escape from the endosome and releases its protein and DNA contents into the host cell. Indeed, its excellent delivery efficiency, coupled with the minimal pathogenicity of the virus, have made AAV a powerful biotechnological tool for delivering foreign DNA and other biomolecules into host organisms.5−7
The past decade has witnessed considerable effort to devise synthetic systems that mimic these natural molecular machines to achieve more effective drug delivery with fewer side effects.8,9 The systems that have made the greatest progress in the clinic to date are constructed by directly conjugating a targeting reagent (e.g., an antibody) to a drug.10,11 Two such antibody-drug conjugates have obtained approval from the U.S. Food and Drug Administration (FDA), and dozens more are now in clinical trials.12 Although most of the work to date has utilized monoclonal antibodies as the targeting reagent, nucleic acid aptamers are an alternative that offer additional advantages, including smaller size, chemically defined synthesis, and lower immunogenicity.13,14 Accordingly, several research efforts have demonstrated the potential of aptamers as a targeting moiety.15,16 For example, the Tan group has shown that aptamer-drug conjugates can facilitate drug uptake by specific target cells expressing an appropriate surface marker.17,18
Although these constructs that directly couple affinity reagent to drugs have shown promise thus far, we envision the potential for further gains in safety and efficacy through the development of “multifunctional” vehicles that—akin to a virus—achieve targeted cell entry and selective payload release in a stepwise fashion.19−21 More specifically, such a delivery mechanism would entail: (i) prolonged systemic circulation while carrying therapeutic agents in an inactive state, (ii) efficient tissue penetration to reach target cells, (iii) specific binding and internalization into target cells, and (iv) selective activation and release of therapeutic payload upon internalization. These features would be particularly valuable in the context of cancer therapeutics, where the drugs involved are typically highly toxic and the aim is to selectively eliminate subpopulations of tumor cells without harming healthy cells in the adjacent tissue.
Toward this end, the integration of aptamers with different classes of specialized biomaterials into a single, multifunctional molecular construct offers a compelling strategy for synthetic delivery vehicles. For example, the Langer and Farokhzad laboratories have demonstrated the potential utility of aptamers in this context, working extensively with drug-loaded polymeric nanoparticles coated with aptamers targeting tumor-specific membrane proteins and have shown that such constructs can enhance selective killing of tumor cells.22−24 We believe block copolymers constitute an especially promising class of polymers for the expansion of this strategy because of the diverse chemical functionalities that can be incorporated; furthermore, these molecules can be designed to incorporate large numbers of drug molecules in a chemically defined manner.25,26
In order to realize this potential, we have developed a mild yet highly effective synthesis strategy based on “click chemistry” that seamlessly integrates aptamers and block copolymers into a single, chemically defined molecular construct. Our methodology enables efficient and reproducible synthesis of multifunctional ‘aptamer-polymer hybrids (APHs)’ that combine the targeting aptamer with a drug-loaded multifunctional block copolymer, which secures its therapeutic payload in an inactive state (Figure 1A). Upon binding the target cell, the APH is internalized via endocytosis, at which point the covalent bonds linking the payload to the polymer scaffold are cleaved by the enzymatic milieu of the endosome. This selectively activates and releases the drug, which then diffuses into the cytoplasm of the target cell. Our synthesis scheme efficiently conjugates the two material components with a high yield without compromising the targeting efficiency of the aptamer or the programmed drug release functionality of the block copolymer (Figure 1B). As a model, we demonstrate APH constructs that specifically target tumor cells that overexpress the nucleolin surface marker. After confirming the coordinated molecular function of the APH based on the successful delivery of the fluorescent dye coumarin, we show that these APH constructs can selectively deliver the chemotherapeutic agent doxorubicin (DOX) into nucleolin-expressing tumor cells, enabling targeted killing of these cells. Furthermore, we show that our synthetic approach can be utilized with other aptamer sequences to achieve flexibility in binding to different surface markers, demonstrating the generality of our system for targeted drug delivery.
Figure 1.
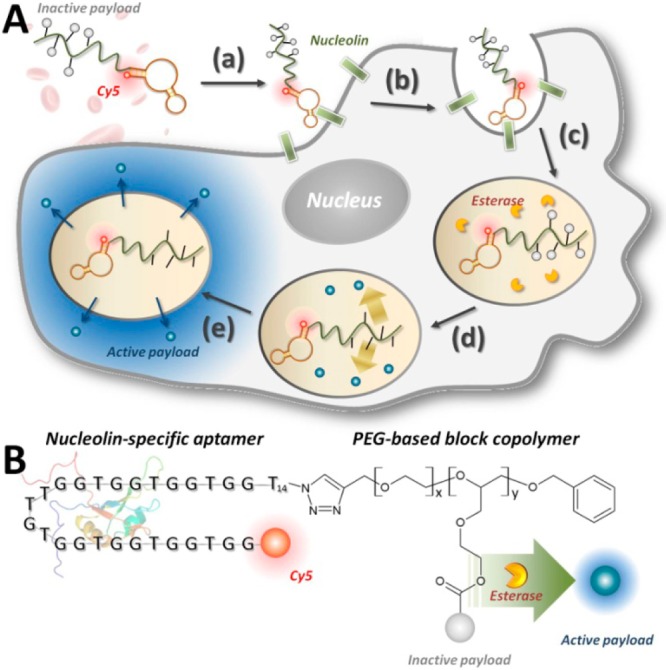
Multifunctional APH molecules achieve controlled, targeted drug delivery. (A) Mechanism of targeted drug delivery. (a) A PEGylated APH carrying multiple deactivated drug molecules binds a cancer target cell via aptamer-mediated recognition of nucleolin. (b) Binding triggers internalization via endocytosis. (c) Esterases within the endosome induce specific cleavage that (d) releases drug molecules from the APH scaffold, after which (e) the now-active small-molecule drugs can diffuse into the cytoplasm. (B) Components of the modular APH molecule. We conjugated a nucleolin-specific aptamer modified with Cy5 at its 5′ end to a PEG-based block copolymer via an efficient, ligand-accelerated CuAAC reaction. This polymer is loaded with multiple payload molecules, which are released within target cells via esterase-mediated cleavage.
Results and Discussion
Design of the APH
We designed our APH with a DNA aptamer27 that specifically binds to tumor cells that express nucleolin, a well-known tumor surface marker.28,29 The nucleolin aptamer sequence is provided in Table S1. Because nucleolin is known to undergo receptor cycling, it enables internalization of the APH into the tumor cells through endocytosis.30,31 The nucleolin aptamer is covalently conjugated to a block copolymer with repeat units of ethylene glycol and ethylene glycol vinyl glycidyl ether (EGVGE)32 using a “click-chemistry” scheme. We chose this polymer scaffold (11.3 kDa, see Scheme 1) because it offers excellent biocompatibility, prolonged circulation,33 and desirable size for efficient tissue penetration (radius ∼10 nm).34 The cell viabiliy assay also confirmed that this polymer scaffold is not cytotoxic to the cells (see Figure S1). Importantly, we designed the polymer backbone with multiple orthogonal reactive groups to accommodate multiple DOX molecules that are tethered through enzyme-cleavable linkers (see Scheme S1). These linkers are designed to be efficiently cleaved by esterases that are endogenous to the endosome of mammalian cells.35 Once cleaved, DOX efficiently escapes the endosome due to its small size and rapidly diffuses in the cytoplasm to reach the nucleus.
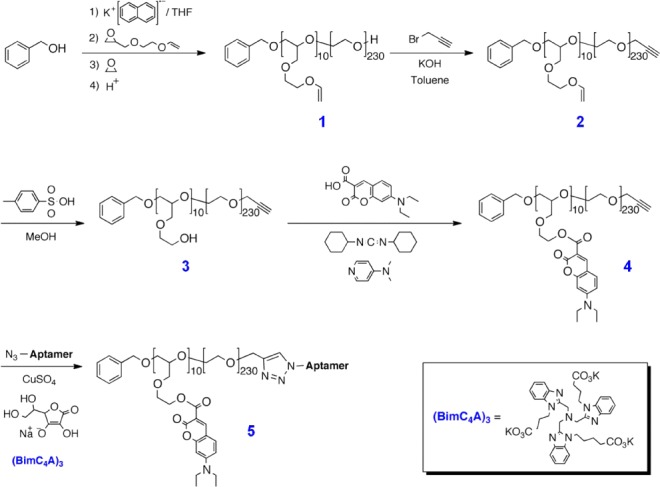
Scheme 1. Polymerization and Functionalization of Coumarin Derivative, 4, To Give Aptamer-Polymer Hybrid, 5.
Click Chemistry Scheme for Efficient Aptamer-Polymer Conjugation
APH synthesis requires efficient, site-specific conjugation of the DNA aptamer to the polymer scaffold without loss of function or binding affinity. To this end, we developed an improved synthetic strategy for coupling DNA aptamers to synthetic polymers based on the novel tricarboxylate ligand (BimC4A)3, which stabilizes Cu (I) during the copper-catalyzed azide–alkyne cycloaddition (CuAAC)36−38 (Scheme 1). In comparison to traditional CuAAC ligands, the use of (BimC4A)3 does not cause irreversible damage to the aptamers, leads to higher yield, and offers superior water solubility compared to tris(benzyltriazolylmethyl)amine (TBTA) and other ligands.39
The synthesis of our APH involves coupling of the nucleolin aptamer, which features a single azide at its 3′ terminus (see Table S1), with an ω-alkyne-functionalized polyether (Scheme 1 and Methods for details). After purification, dynamic light scattering of the APHs with concentrations ranging from 0.5 to 10 μM showed that the resulting APH consistently exhibited 10 ± 2 nm in radius (Figure 2A), and we found negligible evidence of micelle formation or aggregation at the measured concentrations (Figure 2A).
Figure 2.
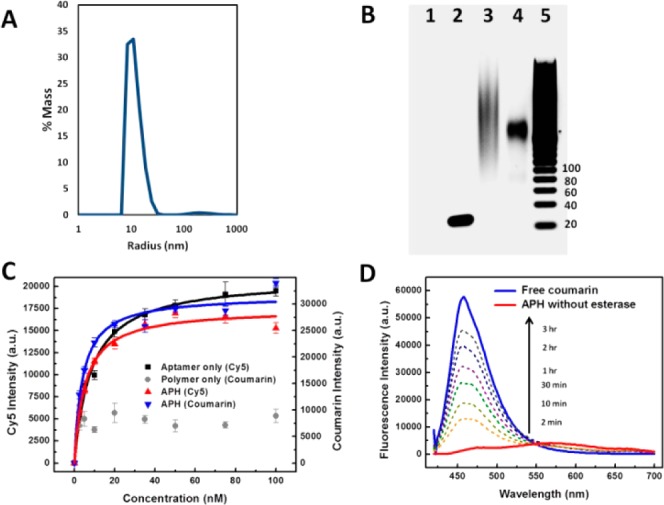
Characterizing functional integrity of the APH construct. (A) Dynamic light scattering measurements revealed a narrow radius distribution of ∼10 nm with negligible aggregation in phosphate-buffered saline buffer. (B) Gel electrophoresis shows that APH conjugation via (BimC4A)3 ligand-accelerated CuAAC greatly minimizes DNA damage. Lanes: 1, unconjugated polymer; 2, unconjugated aptamer (45 nt); 3, APH synthesized with (Bim)3; 4, APH synthesized with (BimC4A)3; 5, 20-bp ladder. (C) Affinity measurements from a bead-based nucleolin-binding assay show that aptamer target affinity is preserved within the APH construct. The unconjugated aptamer exhibits a Kd of 8.37 ± 0.75 nM (black), while APH molecules consisting of Cy5-labeled aptamer and coumarin-labeled polymer scaffold display similar Kds of 5.18 ± 0.72 and 4.01 ± 0.72 nM based on Cy5 (red) and coumarin (blue) intensities, respectively. In contrast, unconjugated polymer (gray) exhibits negligible nucleolin affinity. (D) Time-dependent fluorescence measurements confirm selective payload release. Untreated APHs emit a red-shifted, self-quenched signal (red), but esterase treatment shifts the peak fluorescence wavelength to that of free coumarin (blue), with a signal that increases over time as more coumarin is released (dashed lines).
We confirmed that CuAAC coupling causes minimal damage to the aptamer. This was clearly evident in the electrophoresis shown in Figure 2B. Specifically, APHs synthesized using conventional conjugation schemes with TBTA or the TBTA analogue (Bim)339 caused significant damage to the aptamer, which is evident in the heavy smearing of the band due to the fragmentation of DNA (Figure 2B, lane 3). In contrast, APH synthesis with (BimC4A)3 gave rise to a sharply defined single band, indicating minimal damage to the DNA aptamer (lane 4). As controls, the unconjugated aptamer (lane 2) formed a single band of the predicted size, whereas the unconjugated polymer scaffold exhibited no electrophoretic mobility due to its neutral charge and remained in the loading well (lane 1). Interestingly, when the polymer was loaded with coumarin dye molecules, it migrated toward the cathode due to the slightly positive charge resulting from protonation of coumarin (Figure S2).
Characterization of APH Function
We further confirmed the retention of aptamer function by comparing the equilibrium dissociation constant (Kd) of the aptamer in solution to that of the APH. A bead-based binding assay40 (see Methods) revealed a Kd of 8.37 ± 0.75 nM for Cy5-labeled nucleolin aptamer versus 5.18 ± 0.72 nM for the fully conjugated APH (Figure 2C), confirming that the conjugation scheme described above does not compromise the binding affinity of the aptamer. Importantly, the efficiency of quantitative polymerase chain reaction (qPCR) was comparable for aptamers conjugated to the polymer scaffold and unconjugated aptamers in solution (Figure S3). These data offer compelling evidence that our APH synthesis scheme using (BimC4A)3 does not affect aptamer functionality. Finally, we confirmed that the fully assembled APH retains the affinity of the aptamer by loading the polymer moiety with coumarin dye. This construct exhibited a Kd of 4.01 ± 0.72 nM (Figure 2C, blue) in our bead-based binding assay, essentially equivalent to that of the aptamer alone or the APH in the absence of a payload. As expected, the coumarin-loaded polymer scaffold showed negligible affinity for nucleolin in the absence of aptamer conjugation (Figure 2C, gray).
Our APH synthetic process can also be generalized to other aptamers as a means for targeting different cell-surface markers. To demonstrate this important feature, we used the same chemistry to conjugate DNA aptamers for thrombin41 and immunoglobulin E (IgE)42 to the same polymer scaffold. After verifying that our conjugation strategy preserves the integrity of the thrombin and IgE aptamers via gel electrophoresis (Figure S4) and qPCR (Figure S5), we measured the binding affinities of these APHs (Figures S6 and S7). In both cases, the difference in binding between the unconjugated aptamer and the APH was negligible, clearly demonstrating that the conjugation scheme does not affect the structure or function of the resulting hybrids. Although the underlying mechanism behind our ligand is not yet fully understood, we suspect that the negative charge of the (BimC4A)3 complex electrostatically repels the negatively charged phosphate backbone of DNA and thereby prevents Cu (I) from inflicting oxidative damage on the aptamer.
Finally, we verified that the therapeutic payload remains sequestered in an inactive state within the APH until it is cleaved by the enzymes in the endosome. To assess the efficiency of this cleavage reaction, we measured the release of coumarin by porcine liver esterase (PLE) as a function of time.35 Without PLE, we observe minimal fluorescence from the coumarin because it is tightly sequestered within the APH (Figure 2D), even after 3 h in either buffer or undiluted human serum (Figure S8). This latter result demonstrates that drug payloads should remain stably sequestered in the physiological environment prior to internalization by target cells.
However, in the presence of PLE, we observed a dramatic increase in fluorescence at 467 nm, the expected peak for free coumarin. Quantification of the data showed that 50% of the payload is released within 30 min at 15 U/mL PLE (Figure S9). As expected, the rate of coumarin release increased proportionally with the enzyme concentration (Figure S10). We confirmed that this process was enzyme dependent rather than chemically driven, as exposure to the acidic pH typically found in the endosome was insufficient to promote coumarin release (Figure S11). Taken together, these results confirm that our APH retains the binding affinity of the parent aptamer and requires the enzymatic activity of endosomal esterases to release their therapeutic payload.
Verification of APH Functionality and Mechanism
We used confocal fluorescence microscopy to verify targeted APH binding, internalization and payload release in live cells. As target cells, we used tumorigenic human breast epithelial cells (MCF-7) that overexpress nucleolin on their membrane, with nontumorigenic human breast epithelial cells (MCF-10A) that do not express nucleolin43 as negative control. In order to fluorescently track the payload and APH independently inside cells, we used coumarin as the model payload (blue fluorescence) and labeled the aptamer component of the APH with Cy5 (red fluorescence).
Our APHs exhibited highly selective binding to cells that overexpress nucleolin. With MCF-7 cells, we observed APH binding at the cell surface immediately after incubation, as indicated by strong red fluorescence along the cellular contour (Figures 3, top, and S12). In contrast, negligible binding was observed with MCF-10A cells (Figure 3, bottom), confirming that nucleolin expression is a prerequisite for specific binding. After 10 min, APH is internalized into endosomes, based on the localization of red fluorescence within the MCF-7 cells, and within 1 h, we observed evidence of cleavage of the ester linkages and endosomal escape of coumarin based on the permeation of blue fluorescence into the cytoplasm. After 4 h, efficient endosomal escape of coumarin was observed, with widespread diffusion throughout the cytoplasm. In contrast, the red fluorescence remained localized within the endosome at this late time-point, indicating that the polymer scaffold remained trapped, presumably due to its larger size. These data show that endosomal internalization efficiently triggers payload cleavage and release into the cellular interior. Critically, we observed a complete lack of blue fluorescence within MCF-10A cells even after 4 h, demonstrating that payload delivery does not occur without nucleolin-mediated APH binding and internalization (Figure 3, bottom).
Figure 3.
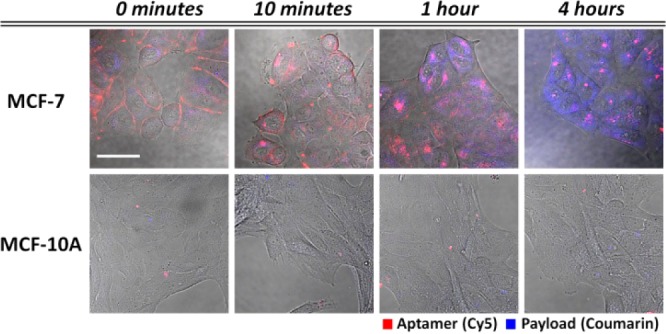
Live-cell imaging confirms APH-mediated, targeted coumarin delivery to nucleolin-expressing cells. We incubated APHs that couple a Cy5-labeled nucleolin aptamer (red) with a polymer scaffold loaded with coumarin (blue) with MCF-7 (top) and MCF-10A (bottom) cells, which express high and low levels of nucleolin, respectively. After 10 min, we washed the cells thoroughly; over the next 4 h, we observed nucleolin-mediated cell-surface binding, APH internalization and cytoplasmic payload release in MCF-7 but not MCF-10A cells. Scale bar = 40 μm.
The power of this modular coupling was clearly demonstrated through a series of control experiments that show how each component of the APH makes a critical contribution to its targeted delivery function. The deletion of the aptamer component rendered the polymer scaffold incapable of target recognition, and we observed no internalization by MCF-7 cells (Figure 4A). Similarly, a mixture of unconjugated aptamers and polymer scaffolds only showed localized red fluorescence within endosomes due to aptamer uptake but no blue fluorescence when incubated with MCF-7 cells (Figure 4B). Replacing the nucleolin aptamer in the APH with a Cy5-labeled IgE aptamer also prevented uptake by MCF-7 cells due to the lack of IgE receptors on their cell surface (Figure 4C).
Figure 4.
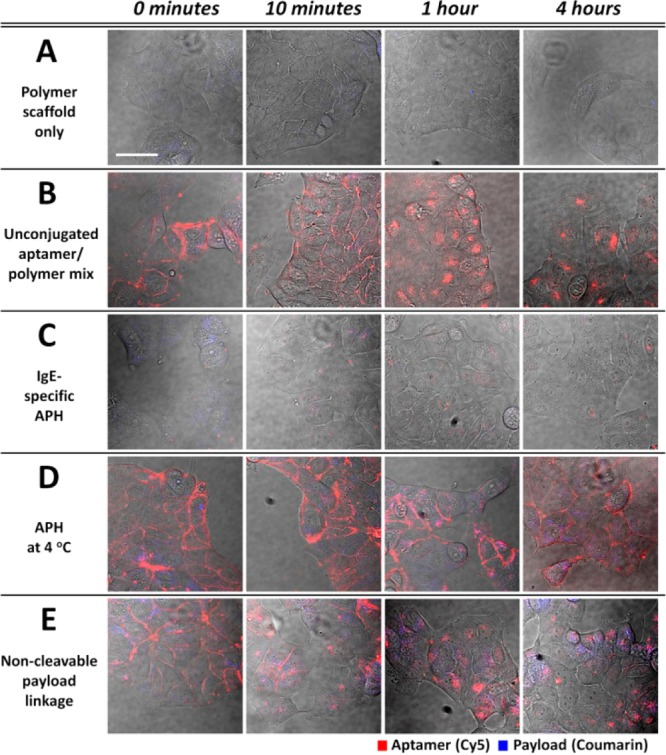
Control experiments demonstrate that each component of the APH contributes essentially to targeted delivery. (A) In the absence of a nucleolin-targeting aptamer, the polymer scaffold is no longer bound or internalized by MCF-7 cells. (B) MCF-7 cells incubated with an unconjugated mixture of nucleolin aptamers and polymer scaffolds internalized the aptamer but not the polymer. (C) APHs that target IgE are not bound or internalized by MCF-7 cells, which do not express this protein. (D) Nucleolin-targeting APHs bind MCF-7 cells but are no longer internalized at temperatures that inhibit endocytosis. (E) Coumarin release is greatly reduced when the payload is linked to the polymer scaffold via esterase-resistant bonds. Scale bar = 40 μm.
We confirmed that APH molecules are actively internalized via endocytosis rather than diffusion or another passive mechanism by incubating MCF-7 cells with nucleolin-targeting APHs at 4 °C, a temperature at which endocytotic pathways are inactive.44 Even after 4 h, red fluorescence remained entirely localized at the cellular surface, indicating that the APHs could successfully bind nucleolin but were no longer being internalized at this temperature (Figure 4D). Finally, we verified that payload release requires esterase-mediated cleavage in the endosome through the synthesis of an alternate APH wherein coumarin was coupled to the polymer scaffold through amide linkages (Scheme S2) that cannot be cleaved by esterases. In this case, only minimal blue fluorescence could be detected after 4 h at 37 °C (Figure 4E) with the magnitude of the signal being dramatically reduced in comparison to APHs with esterase-cleavable linkages (Figure 3, top row).
APHs Selectively Kill Target Cells
Finally, we constructed APH with the chemotherapeutic agent DOX as the payload and used it to selectively kill nucleolin-expressing target cells. To synthesize the APH, we conjugated the hydroxyl group of DOX to the polymer scaffold via the esterase-cleavable linker described above (Scheme S1). We found that DOX-loaded APH molecules efficiently killed MCF-7 cells at a concentration of 500 nM. We used a live/dead cell viability assay (see Methods) to measure the cytotoxicity of the DOX-loaded APHs (Figure 5) and determined that this treatment reduced cell viability by 64%. The potency of the APH was also readily apparent in microscopic images of treated cells, in which viability could be visually ascertained based on changes in cell morphology and adhesion characteristics (Figure S13A). However, the cytotoxicity of the APH is lower than equivalent, free DOX at a concentration of 5 μM, indicating that some ester linkages between the APH and the DOX would be still intact within the endosome.
Figure 5.

APH-mediated DOX delivery efficiently targets and kills cancer cells. MCF-7 cells incubated with DOX-loaded APHs for 4 days at 37 °C exhibited 36% viability, while the cells with free DOX yielded 18% viability relative to untreated controls. Similar treatment with either unconjugated aptamers or DOX-loaded polymers alone resulted in minimal cytotoxicity.
As demonstrated above with coumarin-loaded APHs, we found that each component of the APH plays an integral role in the targeted delivery of chemotherapy to nucleolin-expressing cancer cells. Equal concentrations of unconjugated nucleolin aptamers (Figure S13B) or DOX-loaded polymers (Figure S13C) alone had negligible effect on MCF-7 cell viability, even after 4 days of incubation, with levels of cell death that were essentially indistinguishable from untreated cells in culture media (Figure S13D). Based on our cell viability assay, cells treated with unconjugated aptamers or DOX-loaded polymers respectively exhibited ∼100% and 91% viability relative to untreated cells (Figure 5); the latter result confirms that the polymer keeps the drug safely sequestered in an inactive state, which is an important feature when delivering toxic therapeutics.45,46
Conclusion
In this work, we describe the design and synthesis of multifunctional drug-delivery vehicles that combine the advantages of aptamers and functional polymers to enable controlled, targeted therapy. The resulting APH constructs incorporate an aptamer component and a block copolymer component. The aptamer serves to selectively bind cell-surface markers specific to the target cell, such that the APH is subsequently internalized via an active endocytotic mechanism. In parallel, the polymer component incorporates multiple payload molecules, which are enzymatically cleaved from the scaffold and subsequently diffuse into the cytoplasm only after uptake of the APH in an endosomal compartment. As a result, drug-mediated cytotoxicity is restricted to target cells that express the surface marker of interest, and control experiments verified that there is minimal drug release in the absence of cellular binding and endosomal internalization.
Importantly, APH synthesis is achieved via a click chemistry scheme that offers reproducibility and high yield under mild conditions that ensure the structural and functional integrity of both the aptamer and polymer components. We showed that our synthesis strategy is modular and can be used with a variety of aptamers to target other cell surface markers. Finally, given that our scheme could be readily adopted for other polymer systems and functional nucleic acids such as DNAzymes,47 RNA riboswitches,48 and structure-switching aptamers,49,50 we believe our strategy of integrating aptamer technology with functional, biocompatible synthetic polymers into a single chemical platform may enable the development of other useful functional materials for biomedical applications.
Acknowledgments
This work was supported by the Institute for Collaborative Biotechnologies through the U.S. Army Research Office (W911NF-09-D-0001) (S.S.O., B.F.L., H.T.S., C.J.H.) and W911NF-10-2-0114 (H.T.S., S.S.O.), the Garland Intitiative (S.S.O., H.T.S.), the MRSEC Program of the National Science Foundation (NSF) under Award DMR-1121053 (F.A.L., M.J.R., C.J.H.) and the National Institutes of Health (NIH) as a Program of Excellence in Nanotechnology (HHSN268201000046C) (N.A.L., C.J.H.). Partial support from the NIH U54 DK093467 (H.T.S., S.S.O.), U01 HL099773-1 (H.T.S., S.S.O.), and the NRI-MCDB Microscopy Facility and the Spectral Laser Scanning Confocal supported by the Office of The Director, the NIH under Award S10OD010610 is gratefully acknowledged. We thank Prof. James A. Thomson, Prof. Patrick S. Daugherty, and Dr. Jack Riefert for their assistance in culturing cells.
Supporting Information Available
Synthetic procedures, experimental details, and other supporting data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Smith A. E.; Helenius A. Science 2004, 304, 237. [DOI] [PubMed] [Google Scholar]
- Marsh M.; Helenius A. Cell 2006, 124, 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W.; Zhang L.; Yan Z.; Engelhardt J. F. Gene Ther. 2005, 12, 873. [DOI] [PubMed] [Google Scholar]
- Buning H.; Perabo L.; Coutelle O.; Quadt-Humme S.; Hallek M. J. Gene Med. 2008, 10, 717. [DOI] [PubMed] [Google Scholar]
- Mingozzi F.; High K. A. Nat. Rev. Genet. 2011, 12, 341. [DOI] [PubMed] [Google Scholar]
- Manchester M.; Singh P. Adv. Drug Delivery Rev. 2006, 58, 1505. [DOI] [PubMed] [Google Scholar]
- Ma Y. J.; Nolte R. J. M.; Cornelissen J. J. L. M. Adv. Drug Deliver Rev. 2012, 64, 811. [DOI] [PubMed] [Google Scholar]
- Yoo J. W.; Irvine D. J.; Discher D. E.; Mitragotri S. Nat. Rev. Drug Discovery 2011, 10, 521. [DOI] [PubMed] [Google Scholar]
- Alvarez-Lorenzo C.; Concheiro A. Curr. Opin. Biotechnol. 2013, 24, 1167. [DOI] [PubMed] [Google Scholar]
- Scott A. M.; Wolchok J. D.; Old L. J. Nat. Rev. Cancer 2012, 12, 278. [DOI] [PubMed] [Google Scholar]
- Chari R. V. J.; Miller M. L.; Widdison W. C. Angew. Chem., Int. Ed. 2014, 53, 3796. [DOI] [PubMed] [Google Scholar]
- Mullard A. Nat. Rev. Drug Discovery 2013, 12, 329. [DOI] [PubMed] [Google Scholar]
- Xiao Z. Y.; Farokhzad O. C. ACS Nano 2012, 6, 3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe A. D.; Pai S.; Ellington A. Nat. Rev. Drug Discovery 2010, 9, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W. H.; Wang H.; Chen Y.; Zhang X. B.; Zhu H. Z.; Yang C. Y.; Yang R. H.; Liu C. Trends Biotechnol. 2011, 29, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Zhao Q. H.; Qiu L. Y. J. Controlled Release 2013, 171, 152. [DOI] [PubMed] [Google Scholar]
- Huang Y. F.; Shangguan D. H.; Liu H. P.; Phillips J. A.; Zhang X. L.; Chen Y.; Tan W. H. ChemBioChem 2009, 10, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. W.; Zhu G. Z.; Mei L.; Xie Y.; Ma H. B.; Ye M.; Qing F. L.; Tan W. H. J. Am. Chem. Soc. 2014, 136, 2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Cohen J. L.; Chu C. K.; Wich P. R.; Kierstead P. H.; Fréchet J. M. J. J. Am. Chem. Soc. 2012, 134, 15840–15848. [DOI] [PubMed] [Google Scholar]
- Cheng Z. L.; Al Zaki A.; Hui J. Z.; Muzykantov V. R.; Tsourkas A. Science 2012, 338, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan V. P.; Jain R. K. Nat. Mater. 2013, 12, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farokhzad O. C.; Jon S. Y.; Khademhosseini A.; Tran T. N. T.; LaVan D. A.; Langer R. Cancer Res. 2004, 64, 7668. [DOI] [PubMed] [Google Scholar]
- Farokhzad O. C.; Cheng J. J.; Teply B. A.; Sherifi I.; Jon S.; Kantoff P. W.; Richie J. P.; Langer R. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar S.; Kolishetti N.; Lippard S. J.; Farokhzad O. C. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsabahy M.; Wooley K. L. Chem. Soc. Rev. 2012, 41, 2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson Y. L.; Grinstaff M. W. Adv. Mater. 2012, 24, 3878. [DOI] [PubMed] [Google Scholar]
- Bates P. J.; Kahlon J. B.; Thomas S. D.; Trent J. O.; Miller D. M. J. Biol. Chem. 1999, 274, 26369. [DOI] [PubMed] [Google Scholar]
- Christian S.; Pilch J.; Akerman M. E.; Porkka K.; Laakkonen P.; Ruoslahti E. J. Cell Biol. 2003, 163, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otake Y.; Soundararajan S.; Sengupta T. K.; Kio E. A.; Smith J. C.; Pineda-Roman M.; Stuart R. K.; Spicer E. K.; Fernandes D. J. Blood 2007, 109, 3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates P. J.; Laber D. A.; Miller D. M.; Thomas S. D.; Trent J. O. Exp. Mol. Pathol. 2009, 86, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Reyes E. M.; Teng Y.; Bates P. J. Cancer Res. 2010, 70, 8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. F.; Wolffs M.; Delaney K. T.; Sprafke J. K.; Leibfarth F. A.; Hawker C. J.; Lynd N. A. Macromolecules 2012, 45, 3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooley K. L.; Hawker C. J.; Frechet J. M. J. Angew. Chem., Int. Ed. 1994, 33, 82–85. [Google Scholar]
- Suarez S.; Grover G. N.; Braden R. L.; Christman K. L.; Almutairi A. Biomacromolecules 2013, 14, 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir R. J.; Albertazzi L.; Willis J.; Khan A.; Kang T.; Hawker C. J. Angew. Chem., Int. Ed. 2011, 50, 3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong V.; Presolski S. I.; Ma C.; Finn M. G. Angew. Chem., Int. Ed. 2009, 48, 9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sagheer A. H.; Brown T. Chem. Soc. Rev. 2010, 39, 1388. [DOI] [PubMed] [Google Scholar]
- Lallana E.; Sousa-Herves A.; Fernandez-Trillo F.; Riguera R.; Fernandez-Megia E. Pharm. Res. 2012, 29, 1. [DOI] [PubMed] [Google Scholar]
- Rodionov V. O.; Presolski S. I.; Diaz D. D.; Fokin V. V.; Finn M. G. J. Am. Chem. Soc. 2007, 129, 12705. [DOI] [PubMed] [Google Scholar]
- Oh S. S.; Ahmad K. M.; Cho M.; Kim S.; Xiao Y.; Soh H. T. Anal. Chem. 2011, 83, 6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock L. C.; Griffin L. C.; Latham J. A.; Vermaas E. H.; Toole J. J. Nature 1992, 355, 564. [DOI] [PubMed] [Google Scholar]
- Wiegand T. W.; Williams P. B.; Dreskin S. C.; Jouvin M. H.; Kinet J. P.; Tasset D. J. Immunol. 1996, 157, 221. [PubMed] [Google Scholar]
- Soundararajan S.; Chen W.; Spicer E. K.; Courtenay-Luck N.; Fernandes D. J. Cancer Res. 2008, 68, 2358. [DOI] [PubMed] [Google Scholar]
- Punnonen E. L.; Ryhanen K.; Marjomaki V. S. Eur. J. Cell Biol. 1998, 75, 344. [DOI] [PubMed] [Google Scholar]
- Khandare J.; Minko T. Prog. Polym. Sci. 2006, 31, 359. [Google Scholar]
- Rautio J.; Kumpulainen H.; Heimbach T.; Oliyai R.; Oh D.; Jarvinen T.; Savolainen J. Nat. Rev. Drug Discovery 2008, 7, 255. [DOI] [PubMed] [Google Scholar]
- Willner I.; Shlyahovsky B.; Zayats M.; Willner B. Chem. Soc. Rev. 2008, 37, 1153. [DOI] [PubMed] [Google Scholar]
- Serganov A.; Patel D. J. Nat. Rev. Genet. 2007, 8, 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. W.; Cao Z. H.; Lu Y. Chem. Rev. 2009, 109, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. S.; Plakos K.; Xiao Y.; Eisenstein M.; Soh H. T. ACS Nano 2013, 7, 9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.