

Abstract
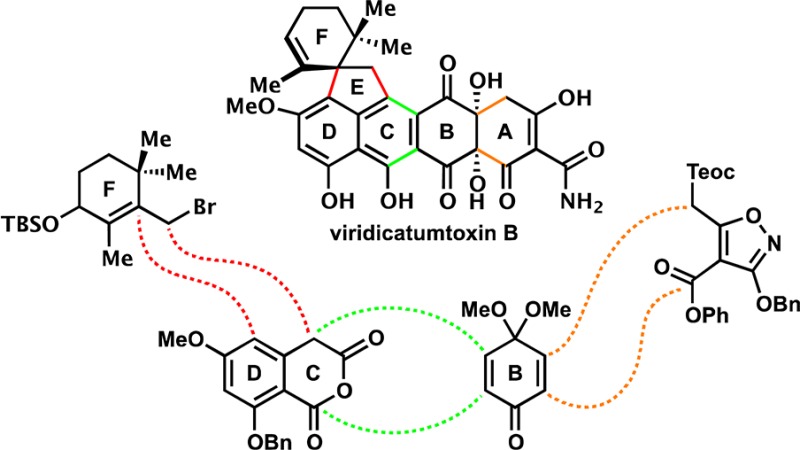
The details of the total synthesis of viridicatumtoxin B (1) are described. Initial synthetic strategies toward this intriguing tetracycline antibiotic resulted in the development of key alkylation and Lewis acid-mediated spirocyclization reactions to form the hindered EF spirojunction, as well as Michael–Dieckmann reactions to set the A and C rings. The use of an aromatic A-ring substrate, however, was found to be unsuitable for the introduction of the requisite hydroxyl groups at carbons 4a and 12a. Applying these previous tactics, we developed stepwise approaches to oxidize carbons 12a and 4a based on enol- and enolate-based oxidations, respectively, the latter of which was accomplished after systematic investigations that revealed critical reactivity patterns. The herein described synthetic strategy resulted in the total synthesis of viridicatumtoxin B (1), which, in turn, formed the basis for the revision of its originally assigned structure. The developed chemistry facilitated the synthesis of a series of viridicatumtoxin analogues, which were evaluated against Gram-positive and Gram-negative bacterial strains, including drug-resistant pathogens, revealing the first structure–activity relationships within this structural type.
Introduction
Within the class of tetracycline antibiotics, viridicatumtoxin B (1),1 viridicatumtoxin A (2),2 and spirohexaline (3)3 (Chart 1) are unique in that they include in their structures a geranyl-derived subunit in the form of a spirobicyclic system (ring system EF). In contrast to the majority of tetracyclines, these members of the group are also distinguished by their fungal, rather than bacterial, origins. The subject of this article is the pursuit of viridicatumtoxin B (1) by total synthesis, its full structural elucidation, and investigation of its antibacterial properties as well as those of selected synthetic analogues. The following brief historical overview places the present work and its aims in perspective within the field of tetracycline antibiotics.
Chart 1. Molecular Structures of Viridicatumtoxins 1–3.

The discovery of chlortetracycline (4, Chart 2a), the first tetracycline antibiotic, by B. M. Duggar of the American Cyanamid Corporation in the late 1940s ushered in a new subclass of antibacterial agents at the dawn of the golden era of antibiotics.4 The widespread success of tetracyclines in curing previously high-mortality-rate diseases bestowed on them the status of “wonder drug” shortly after their introduction into the clinic.5 Since the discovery of chlortetracycline and other first-generation tetracyclines [e.g., oxytetracycline (5) and tetracycline (6), Chart 2a], second-generation tetracyclines, including minocycline (7) and doxycycline (8) (Chart 2a), emerged with improved properties. More recently, third-generation tetracyclines such as tigecycline (9)6 and eravacycline (TP-434, 10)7 (Chart 2a) that overcome certain bacterial resistance mechanisms have been introduced.8
Chart 2. Molecular Structures of (a) Bacterial Tetracyclines and Designed Analogues and (b) Fungal Tetracyclines.

Most naturally occurring tetracyclines are produced by bacterial strains, although a few have been isolated from fungi. Thus, in addition to those shown in Chart 1 (1–3), hypomycetin (11),9 anthrotainin (TAN-1652, 12),10 TAN-1612 (13),11 and BMS-192548 (14)12 (Chart 2b) are fungal metabolites.
Due to their complex structures and important biological activities, tetracyclines have been the subject of numerous synthetic campaigns since the 1950s. Noteworthy achievements in tetracycline synthesis include those recorded by Woodward/Pfizer,13 Shemyakin,14 Muxfeldt,15 Barton,16 Wasserman/Scott,17 Stork,18 Tatsuta,19 and, more recently, Myers20 and Evans.21
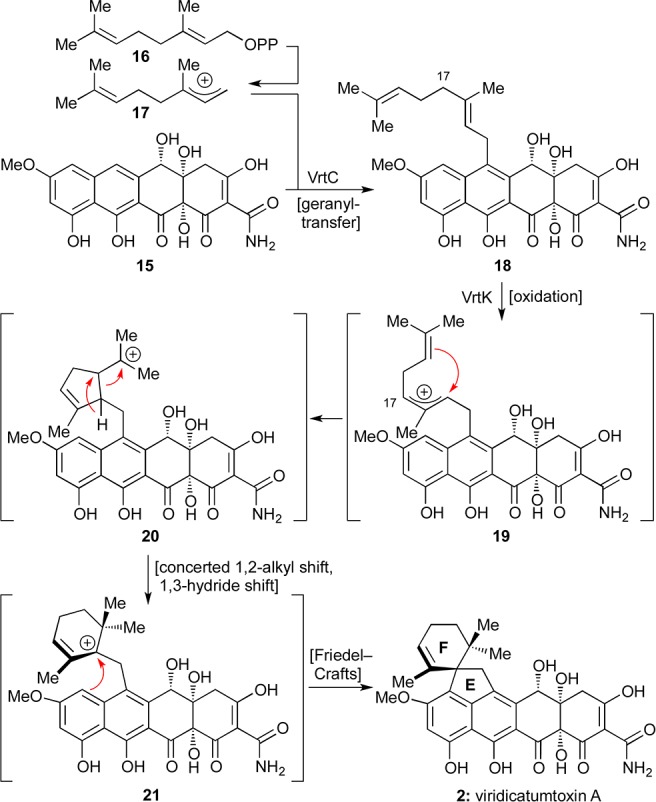
First isolated in 1973 from a Penicillium strain in South Africa, viridicatumtoxin A (2) yielded to X-ray crystallographic analysis in 1976.2,22 The biosynthesis of this antibiotic was studied by the groups of Vleggaar23 and, more recently, Tang and co-workers,24 the latter of whom proposed a complete biosynthetic pathway. Specifically, as shown in Scheme 1, it was suggested that the EF-spirosystem is formed from polyketide 15 and a unit of geranyl pyrophosphate (16) as facilitated by VrtC, a polyketide prenyltransferase.25 This reaction is followed by oxidative cyclization catalyzed by another Vrt enzyme (VrtK, a cytochrome P450-type enzyme)26 to afford viridicatumtoxin A (2) through transient intermediates 19–21 (on the basis of computational studies), as shown in Scheme 1.
Scheme 1. Proposed Biosynthesis of the Terpene-Derived Spirocyclic Region of Viridicatumtoxin A (2).
In 2008, Kim et al.1 reported the isolation of viridicatumtoxin B in small quantities along with viridicatumtoxin A (2) from Penicillium sp. FR11 and, on the basis of NMR spectroscopic analysis, assigned the hydroxy-epoxide structure 1′ (Chart 1) to the former. These investigators observed potent activities for viridicatumtoxins A and B against Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) (MIC = 0.25 and 0.5 μg/mL, respectively). Interestingly, a recent report suggested that the viridicatumtoxins exert their antibacterial properties through inhibition of UPP synthase, an important enzyme for bacterial peptidoglycan biosynthesis.3,27 This stands in contrast to the mode of action of other tetracyclines (e.g., 4–10, Chart 2a), which inhibit bacterial protein synthesis by binding to the 30S subunit of the ribosome.
In view of the scarcity of viridicatumtoxin B, its interesting but suspect structural assignment (i.e., 1′, Chart 1), and its important biological activity, we initiated a program directed toward its total synthesis. Herein, we describe details of our investigations that led to important new knowledge and insights with regard to this intriguing bioactive molecule.28
Results and Discussion
First-Generation Approach: Development of an Anthrone Alkylation and Lewis Acid-Mediated Spirocyclization
The intrigue surrounding the originally assigned structure of viridicatumtoxin B (1′) is derived from its uncommon epoxy-hemiacetal structural motif, its lipophilic spirocyclic domain, and its high oxygenation, particularly on ring B. Although the first and rather curious structural feature may have an explanation in this instance originating from the surrounding functionalities of the molecule, from the synthetic point of view, it was reasoned that it could be resolved by targeting either structure 1′ or 1 (Chart 1) and allowing their potential thermodynamic equilibration to provide an answer. The spirocycle provided a synthetic challenge that we decided to face following the proposed biosynthesis, which inspired a Friedel–Crafts-type approach (see Scheme 1). The remaining major challenge of a synthesis of viridicatumtoxin B was initially relegated to a rather speculative singlet oxygen [4 + 2] cycloaddition to an appropriately substituted aromatic ring (B). Finally, the construction of the enol–amide structural motif characteristic of most tetracyclines was to be derived from an isoxazole ring through hydrogenolysis, a well-tested and reliable tactic introduced by Stork and Hagedorn.18a
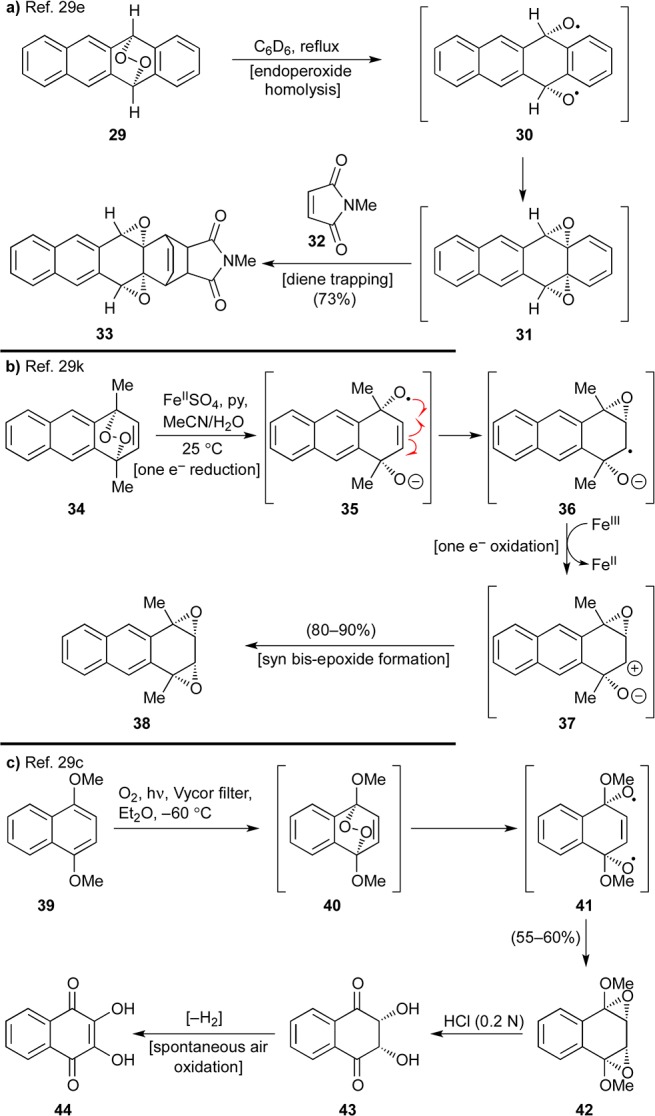
These considerations led to our first retrosynthetic analysis of viridicatumtoxin B as depicted in Scheme 2. Thus, it was reasoned that 1″ or 1 could be derived from bis-epoxide structure 22 (R = selectively cleavable protecting group), which could be traced to endoperoxide 23 through a radical-based rearrangement and thence to aromatic system 24 via selective oxygen addition to the electron-rich B-ring. This scenario was inspired by a number of previous relevant studies, highlights of which are shown in Scheme 3.29 Thus, endoperoxide 29 (generated from singlet oxygen and tetracene) was reported to undergo a thermally-induced rearrangement to bis-epoxide 31, presumably via diradical 30, and the latter was trapped with N-methylmaleimide (32) to afford adduct 33 (Scheme 3a).29e The regioselectivity of the latter reaction is of note in that it reflects different reactivities for the benzene and naphthalene systems. A similar type of fragmentation was induced within endoperoxide 34 by an iron(II) species, leading to bis-epoxide 38, as shown in Scheme 3b.29k This rearrangement is presumed to proceed through the sequential one-electron transfers involving transient species 35–37.30,31 Photochemically-induced homolytic rupture of endoperoxides is also possible as demonstrated in Scheme 3c.29c Specifically, endoperoxide 40, generated from naphthalene derivative 39, formed bis-epoxide 42, presumably via diradical 41. Further elaboration of 42 led first to syn diol 43 and thence dihydroxy quinone 44, demonstrating the accessibility and versatility of the endoperoxide moiety.32
Scheme 2. First-Generation Retrosynthetic Analysis through Endoperoxide 23.

Scheme 3. Precedent for Arene–Endoperoxide Rearrangements Based on (a) Thermal-, (b) Redox-, and (c) Photo-Induced O–O Bond Cleavage.
The synthesis of allylic chloride fragment 27 began with geranic acid (45) and followed a modified sequence based on a literature process,33 as shown in Scheme 4. Thus, acid-mediated cationic cyclization (H3PO4) of 45 followed by methylation (MeI, K2CO3) of the resulting acid produced methyl ester 46. Epoxidation of the latter with m-CPBA furnished a mixture of epoxide isomers which was treated with NaOMe to give the desired allylic alcohol methyl ester 47 (70% for four steps). Protection of the hydroxyl group of 47 (TBSCl, imid.) and subsequent reduction (DIBAL-H) then led to allylic alcohol 48 (91% for two steps). Attempted mesylation of 48 with MsCl/Et3N led to partial formation of allylic chloride 27, presumably via the initially formed mesylate 49. It was then found that treatment of 48 with MsCl/Et3N followed by addition of LiCl produced the allylic chloride 27 in satisfactory yield (48%).
Scheme 4. Synthesis of Allylic Chloride 27.
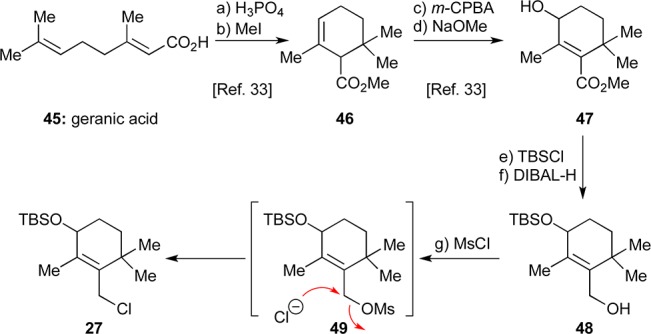
Reagents and conditions: (a) H3PO4 (0.2 equiv), toluene, reflux, 90 min; (b) MeI (3.9 equiv), K2CO3 (2.0 equiv), acetone, 25 °C, 15 h; (c) m-CPBA (1.2 equiv), CH2Cl2, 0 → 25 °C, 3 h; (d) NaOMe (1.5 equiv), MeOH, reflux, 17 h, 70% for four steps; (e) TBSCl (1.6 equiv), imidazole (2.0 equiv), CH2Cl2, 25 °C, 12 h; (f) DIBAL-H (2.7 equiv), CH2Cl2, −78 → 0 °C, 70 min, 91% for two steps; (g) MsCl (1.2 equiv), Et3N (1.5 equiv), DMAP (0.05 equiv), CH2Cl2, 25 °C, 14 h; then LiCl (1.0 equiv), 42 h, 48%. m-CPBA = meta-chloroperoxybenzoic acid, TBS = tert-butyldimethylsilyl, DIBAL-H = diisobutylaluminum hydride, Ms = methanesulfonyl, DMAP = 4-dimethylaminopyridine.
With allylic chloride 27 readily available, we next turned our attention to the construction of anthrone 28 (Scheme 5). Bromo-benzyl juglone 51 was prepared in 66% overall yield following a two-step literature procedure starting from juglone (50).34 Known Brassard diene 53 was prepared in one step starting from methyl 3-methoxybut-2-enoate (52) via formation of the corresponding enolate and subsequent trapping with TMSCl (99%).35 With these building blocks in hand, their fusion through a Diels–Alder reaction was performed by mixing the dienophile 51 with 3 equiv of diene 53 at −30 °C and allowing the reaction mixture to reach ambient temperature. The TMS ether of the initially formed Diels–Alder adduct 54 was cleaved with silica gel, which caused collapse of the ketal and spontaneous elimination of HBr (see 55, Scheme 5) followed by tautomerization, ultimately producing anthraquinone 56 in 90% overall yield. Similar types of cascade Diels–Alder/elimination sequences have been performed previously.36 Methylation of the free phenolic group of 56 (MeI, K2CO3) followed by regioselective deoxygenation (SnCl2, HCl/AcOH) and concomitant debenzylation then furnished anthrone 28 (86% yield for two steps).37
Scheme 5. Synthesis of Spirocycle 25 and Conformational Rationale for Its Stereoselective Formation.
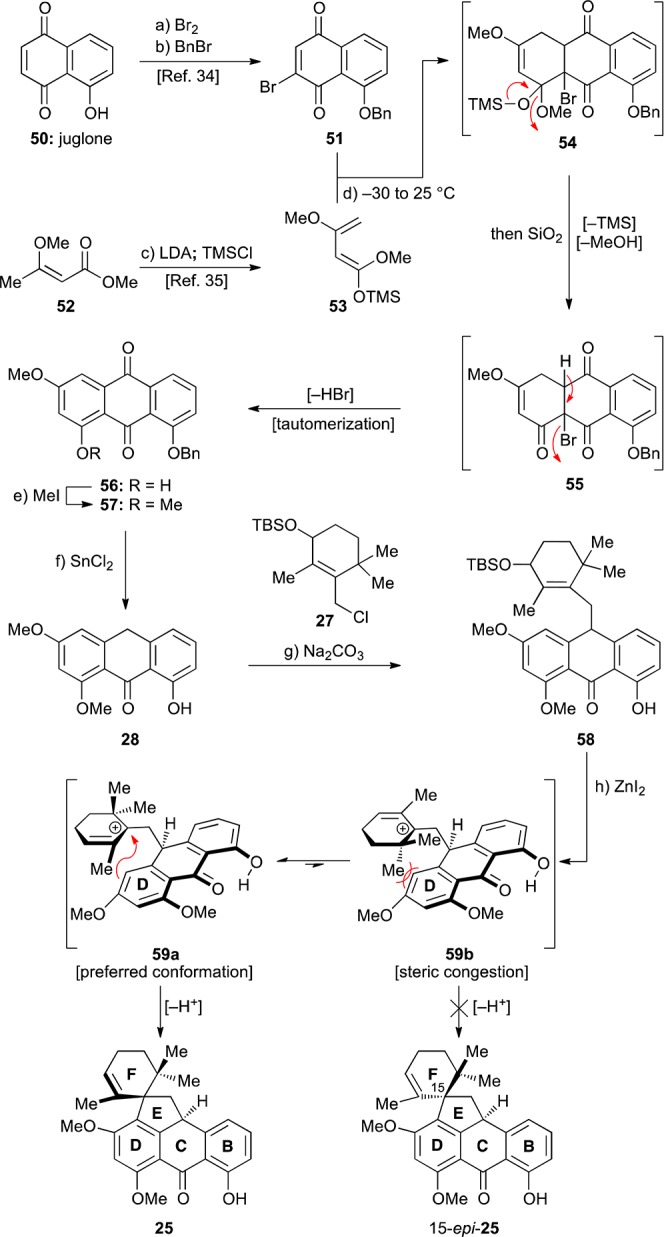
Reagents and conditions: (a) Br2 (1.03 equiv), AcOH, 25 °C, 30 min; then EtOH, reflux, 15 min; (b) BnBr (2.1 equiv), Ag2O (2.1 equiv), CH2Cl2, 25 °C, 18 h, 66% for two steps; (c) LDA (1.05 equiv), THF, −78 °C, 1 h; then TMSCl (1.2 equiv), −78 → 25 °C, 100 min, 99%; (d) 51 (1.0 equiv), 53 (3.0 equiv), CH2Cl2, −30 → 25 °C, 70 min; then SiO2 (excess), 1 h, 90%; (e) MeI (5.0 equiv), K2CO3 (5.0 equiv), DMF, 65 °C, 14 h; (f) SnCl2 (7.0 equiv), AcOH:HCl 10:1, 50 °C, 86% for two steps; (g) 27 (1.0 equiv), KI (0.1 equiv), Na2CO3 (2.5 equiv), acetone, 50 °C, 18 h, 51%, ca. 1:1 dr; (h) ZnI2 (0.78 equiv), CH2Cl2, reflux, 25 h, 34%. Bn = benzyl, THF = tetrahydrofuran, LDA = lithium diisopropylamide, TMS = trimethylsilyl.
Previous reports have detailed the successful alkylation of the methylene position of anthrones with simple alkyl halides.38 These studies also provided precedent for alkylation at the methylene position in preference to the free phenolic position(s), the latter being deactivated by hydrogen bonding with the adjacent carbonyl group. Nevertheless, the alkylation of an anthrone with a more complex electrophile was unprecedented and had not been utilized previously in the context of tetracycline synthesis. It was, therefore, pleasing to find that union of anthrone 28 with chloride 27 was successful when performed at 50 °C, employing Na2CO3 as the base in the presence of catalytic quantities of KI, providing the desired product 58 in 51% yield (ca. 1:1 dr). It is worth noting that the success of this reaction depended on degassing the solution and conducting the reaction in the absence of light39 to prevent oxidative radical dimerization of the anthrone substrate.40
Initial attempts to form the EF spirosystem were based on precedents that typically employed unprotected allylic alcohols41 or allylic acetates42 as the cationic precursors. However, most attempts to remove the TBS group of intermediate 58 were complicated with side-products, including elimination or allylic substitution products (e.g., allylic methyl ether with HCl/MeOH). These observations led us to hypothesize that it might be possible to directly ionize the TBS ether of 58, thereby eliminating the need for the deprotection step. After a brief screen of acidic conditions, it was found that ZnI2 indeed promoted the desired transformation in 34% yield. As shown in Scheme 5, portionwise treatment of allylic TBS ether 58 in refluxing CH2Cl2 with ZnI2 produced spirocyclic compound 25 as a single detectable diastereomer. This reaction presumably proceeds via ionization of the TBS ether to generate allylic cation 59a/b. Then, intramolecular Friedel–Crafts-type reaction with the more electron-rich arene (ring D) followed by loss of a proton produces the observed product 25. We did not isolate any product resulting from Friedel–Crafts reaction on the B-ring, although some elimination products were observed. While the yield of this transformation was, for the time, rather modest, it was sufficient to allow us to probe the following steps. The stereochemical configuration of 25 was assigned by comparison with subsequent intermediates whose structures were unambiguously deduced from X-ray crystallographic studies (see below). The observed diastereoselectivity of the spirocyclization (58 → 25) can be rationalized by invoking the transition state resulting from conformer 59a as having the lowest energy barrier, as shown in Scheme 5. The transition state resulting from alternative conformer 59b, required to afford the other (15-epi-25, undesired) diastereomer, suffers from unfavorable interactions between one of the methyl groups and the arene, as indicated in structure 59b. These interactions are apparently not present in the transition state leading to the observed product (59a → 25).
As shown in Scheme 6, we next needed to perform a phenolic oxidation to render the B-ring of the growing molecule electrophilic, and thereby susceptible, to nucleophilic attack from a negatively charged A-ring isoxazole (i.e., anion of 26, see Scheme 2). The prototypical sequence of events for achieving this objective would involve PIDA [PhI(OAc)2]- or PIFA [PhI(TFA)2]-mediated oxidation of the phenolic moiety of 25 in MeOH to give intermediate 60, which may undergo a second oxidation to yield the desired quinone monoketal 61.43 In reality, however, 25 proved intransigent to these projected transformations, leaving quinone monoketal 61 and its relatives, 60 and 64, elusive under a variety of conditions tested. These included hypervalent iodine-, molecular oxygen-, and metal-based oxidative conditions (see Supporting Information for details). Instead, benzylic oxidation leading to quinomethide 62 was observed in several cases, as well as decomposition under different conditions. In a few cases, undesired products involving oxidation of the F-ring were also observed.
Scheme 6. Attempted Phenolic Oxidation of 25 and 63.

Reagents and conditions: (a) PhI(OAc)2 (3.0 equiv), MeOH, 25 °C, 43 h, 37% for 62 + 26% recovered 25; (b) K2CO3 (2.4 equiv), MeOH, 25 °C, 10 min, 54%; (c) PhI(OAc)2 (2.5 equiv), CH2Cl2:H2O 7:1, 0 → 25 °C, 55 min, 4% for 64, 13% for 62, 45% recovered 63. PIDA = phenyliodonium diacetate.
Eventually, it was found that spirocycle 25 could be tautomerized to its desmotropic form 63 under basic conditions (i.e., K2CO3, Scheme 6).44 When the latter compound was treated with PIDA in CH2Cl2/H2O the desired quinone 64 could be isolated in only 4% yield, along with substantial quantities of benzylic oxidation product 62 (13%) and recovered starting material (45%). A number of different oxidants (i.e., Fremy’s salt, O2/salcomine catalyst, PIFA) were examined in efforts to improve the efficiency of the transformation of 63 to quinone 64, but unfortunately to no avail.
Unable to access quinone 64 in sufficient quantities, it was decided to focus our attention on an approach that would directly bring in the A-ring from an earlier stage, which would circumvent the problematic B-ring phenolic oxidation barriers we faced. Despite its failure to break through these barriers, however, the initial approach provided us with useful information and methods for the construction of the anthrone and allylic halide substrates and established the required spirocyclization reaction.
Second-Generation Approach: Attempts To Install the C4a and C12a Hydroxyl Groups via Dearomatization Pathways
The second-generation retrosynthesis of substrate 24, required for our endoperoxide rearrangement approach (see Scheme 2), is shown in Scheme 7. Thus, to avoid the oxidation problems discussed above, the subtarget molecule 24 was dissected through the C-ring, which retrosynthetically defined three fragments: allylic chloride 27, known homophthalic anhydride 66,45 and AB-enone 65. The latter could be further disassembled into known quinone monoketal 67(46) and Stork–Hagedorn isoxazole 26.18a
Scheme 7. Second-Generation Retrosynthetic Analysis of Viridicatumtoxin B (1 or its isomer 1″) through Endoperoxide Precursor 24.

The synthesis of requisite AB-enone 65 commenced from the known Stork–Hagedorn isoxazole 26, which was prepared using a modification of the literature procedure (see Scheme 8). Thus, the process for the synthesis of 26 involved acylation of dimethyl malonate (68) (MgCl2, Et3N, AcCl, 96%),47 methylation of the resulting enol (69) with Me2SO4 (54%), cyclization of 70 with H2NOH to afford compound 71 (48%), and benzylation of the resulting hydroxy-isoxazole (BnBr, Ag2O, 67%). The best conditions we found for the benzylation reaction were those employing Ag2O which provided the O-Bn in preference to the N-Bn product in ratios of up to 11:1. Alternative conditions gave unacceptably low selectivity ratios (Cs2CO3/BnBr: O:N ca. 1.25:1; TfOH/BnTCA: O:N ca. 1:7.5) in agreement with Kornblum’s rule for ambident nucleophiles.48 A procedure using phenyldiazomethane as the electrophile as used by Stork and co-workers18a was considered undesirable due to safety concerns. With isoxazole 26 readily available, we next attempted its conjugate addition to quinone monoketal 67, as shown in Scheme 8. Thus, deprotonation of the methyl group of 26 with LiHMDS, followed by addition of quinone monoketal 67 to the resulting anion (26′), however, led to 1,2-addition product 72, rather than the desired 1,4-product.
Scheme 8. Synthesis and 1,2-Addition of Stork–Hagedorn Isoxazole 26 to Quinone Monoketal 67.

Reagents and conditions: (a) MgCl2 (1.0 equiv), Et3N (2.0 equiv), AcCl (1.0 equiv), MeCN, 0 → 25 °C, 23 h, 96%; (b) Me2SO4 (1.3 equiv), K2CO3 (1.3 equiv), DMF, 0 → 25 °C, 17 h, 54%; (c) H2NOH·HCl (1.4 equiv), NaOMe (3.1 equiv), MeOH, 0 → 25 °C, 24 h, 48%; (d) BnBr (1.2 equiv), Ag2O (1.5 equiv), DMF, 25 °C, 18 h, 67%; (e) LiHMDS (1.4 equiv), THF, −78 °C, 40 min; then 67 (1.0 equiv), −78 → −30 °C, 45 min, 62%. Ac = acetyl, DMF = dimethylformamide, HMDS = hexamethyldisilazide.
Faced with this predicament, we decided to install an electron-withdrawing group at the isoxazole methyl group, which we hoped would make the resulting nucleophile softer due to its vinylogous diketone nature.18a Toward that end, and as shown in Scheme 9, treatment of isoxazole 26 with LiHMDS followed by quenching with methyl chloroformate provided 73 (96% yield). Michael addition of the latter to quinone monoketal 67 was then achieved under basic conditions (NaOMe) to give adduct 74 in 71% yield as an inconsequential ca. 3:1 mixture of diastereomers. Dieckmann condensation within 74 induced by NaH then led to tricycle 75 as a single diastereomer (3J4,4a = 11.2 Hz), presumably through epimerization at C4 during the reaction. Demethylation and subsequent decarboxylation of 75 was then carried out with Me3SnOH49 at elevated temperatures, providing 76 in 39% yield for the two steps. Of note here is the observation that the use of LiOH proved unsuccessful in promoting this transformation. Subsequent oxidation of the latter substrate (i.e., 76) by way of selenide formation/oxidation/syn-elimination furnished compound 79 (54% for the two steps), through intermediate 77 and transient species 78. Finally, benzylation of the free phenolic moiety within 79 (BnBr, K2CO3, 94%) afforded the desired AB-ring system fragment 65 (mp = 103 °C, Et2O). The structure of this key intermediate was unambiguously confirmed by X-ray crystallographic analysis (see ORTEP representation, Figure 1).
Scheme 9. First Synthesis of AB-Quinone Monoketal 65.

Reagents and conditions: (a) LiHMDS (2.2 equiv), THF −78 °C, 30 min; then MeOCOCl (1.0 equiv), −78 °C, 45 min, 96%; (b) 67 (1.0 equiv), NaOMe (1.0 equiv), MeOH, 25 °C, 18 h, 71%, ca. 3:1 dr; (c) NaH (3.9 equiv), toluene, 0 → 90 °C, 2.5 h; (d) Me3SnOH (9.0 equiv), ClCH2CH2Cl, 80 °C, 21 h, 39% for two steps; (e) PhSeCl (1.9 equiv), py (2.1 equiv), CH2Cl2, 0 °C, 1.5 h; (f) H2O2 (excess), CH2Cl2, 0 °C, 54% for two steps; (g) BnBr (3.0 equiv), K2CO3 (4.0 equiv), acetone, 25 → 60 °C, 6.5 h, 94%. py = pyridine.
Figure 1.
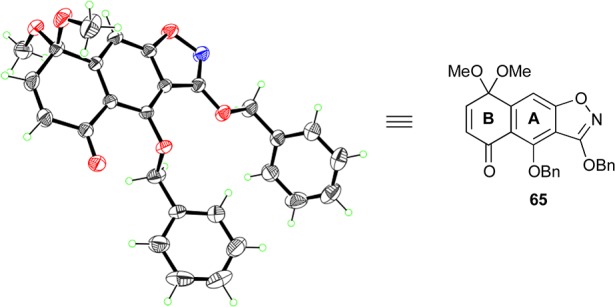
ORTEP representation of AB-quinone monoketal 65. Thermal ellipsoids at 30% probability. Gray = carbon, red = oxygen, blue = nitrogen, green = hydrogen.
While the above route provided sufficient quantities of fragment 65 for preliminary studies, scaling up the synthetic sequence, especially the ester saponification with Me3SnOH, proved inconvenient. To circumvent this problem, we elected to change the C4-ester group to a more labile one. Alternative moieties considered were the ethoxyethyl ester, originally employed by Stork,18a and the cyclic anhydride 80 (see Scheme 10), which was envisioned to undergo a one-pot Michael–Dieckmann/decarboxylation cascade sequence. Eventually, however, we selected a Teoc ester due to its anticipated orthogonality to the rest of the functionalities within the molecule. Thus, and as shown in Scheme 10, installation of the Teoc ester at C4 of isoxazole 26 was accomplished using a similar protocol as for the methyl ester (LiHMDS; then TeocCl, 88%) to afford derivative 81. Michael reaction of the latter with 67 (t-BuOK) then gave adduct 82 (71% yield, ca. 2.4:1 dr, inconsequential). At this point, it was gratifying to observe that the Teoc group could be removed in a straightforward fashion using TBAF at 25 °C to furnish decarboxylated product 83 (85% yield). Dieckmann cyclization within 83, again induced by NaH, provided intermediate 76 (80% yield) which intersected the previously described route to 65 (see Scheme 9).
Scheme 10. Improved Synthesis of AB-Quinone Monoketal 65 through the Use of a Teoc Ester.

Reagents and conditions: (a) LiHMDS (2.2 equiv), THF, −78 °C, 30 min; then TeocCl (1.1 equiv), −78 °C, 1 h, 88%; (b) 67 (1.0 equiv), t-BuOK (0.2 equiv), toluene, 25 °C, 4 h, 71%, ca. 2.4:1 dr; (c) TBAF (1.2 equiv), THF, 25 °C, 45 min, 85%; (d) NaH (4.0 equiv), toluene, 0 → 110 °C, 4 h, 80%. TBAF = tetra-n-butylammonium fluoride.
With AB-enone 65 in hand, our attention turned toward the synthesis of the desired ABCD-ring fragment 88 (Scheme 12) through union of the former with a suitable CD-ring precursor. Due to the observed 1,2-addition of isoxazole anion 26′ to quinone monoketal 67 (see Scheme 8), we anticipated the need for a soft CD-ring nucleophile. Toward this end, we selected the known homophthalic anhydride 66 [readily prepared in four steps from 4-chlororesorcinol (84) through intermediate 85 as shown in Scheme 11].45 Anhydride 66 has traditionally been used (after deprotonation with a strong base, typically NaH or LDA) in Tamura-type Diels–Alder reactions with quinones.50 However, we found that substrate 66 could be united with AB-enone 65 using DBU as the base at 60 °C to afford pentacyclic system 88 (82% yield, see Scheme 12). A 1H NMR-tube experiment to monitor the reaction between 65 and 66 suggested that it proceeds via a distinct stepwise mechanism (intermediates 86 and 87 were tentatively observed; see selected peaks, Figure 2), as opposed to a concerted [4 + 2] cycloaddition followed by decarboxylation. Additionally, this experiment indicated that the reaction required approximately 20 h for completion. Elimination of methanol from cyclization product 88, induced by exposure to TFA in CH2Cl2, then led to elaborated anthrone 89 in quantitative yield.
Scheme 12. Synthesis of Intermediate 88 through a Michael–Dieckmann/Decarboxylation Cascade Sequence and Spirocyclization of Hexacycle 90 to Heptacycle 24.

Reagents and conditions: (a) 65 (1.0 equiv), DBU (3.0 equiv); then 66 (1.3 equiv), MeCN, 25 → 60 °C, 18 h, 82%; (b) TFA (4.9 equiv), CH2Cl2, 25 °C, 20 min, quant; (c) Et3N (2.0 equiv), MsCl (1.7 equiv), CH2Cl2, −50 °C, 1 h; then LiBr (3.5 equiv), THF, −50 → −20 °C, 1 h, quant; (d) 91 (1.5 equiv), Na2CO3 (9.1 equiv), DMF, 25 °C, 45 min, 78%, ca. 6:1 dr; (e) BF3·OEt2 (0.6 equiv), CH2Cl2, 0 °C, 1 h, 55%. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TFA = trifluoroacetic acid.
Scheme 11. Preparation of Cyclic Anhydride Fragment 66.
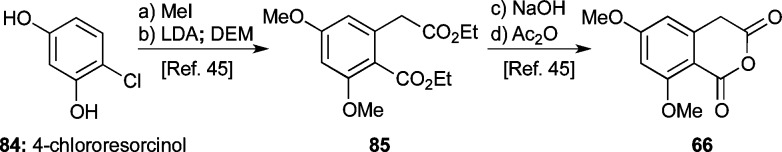
Reagents and conditions: (a) MeI (4.0 equiv), K2CO3 (8.0 equiv), acetone, reflux, 15 h, 91%; (b) NaH (6.0 equiv), diethyl malonate (4.0 equiv), THF, 0 °C, 2.5 h; then LDA (1.0 equiv), THF, 0 °C, 3.5 h, 65%; (c) MeOH:2 M NaOH 1:1, reflux, 12 h, 97%; (d) Ac2O (4.2 equiv), toluene, reflux, 2 h, 99%. DEM = diethyl malonate.
Figure 2.

Monitoring the coupling of cyclic anhydride 66 and AB-quinone monoketal 65 by 1H NMR spectroscopy (MeCN-d3, 500 MHz): (a) 1H NMR spectra of starting materials 65 and 66; (b) 1H NMR spectra during reaction (0, 5.5, and 20 h); (c) 1H NMR spectrum of product 88 after purification.
Attempts to alkylate anthrone 89, however, with chloride 27 (Scheme 12) under the conditions employed for the alkylation of the simpler anthrone 28 (Scheme 5) were thwarted by the insolubility of 89 in acetone and the low reactivity of 27, necessitating a search for a new protocol to achieve the desired union of the two partners. Thus, with further experimentation, it was found that upon using the corresponding allylic bromide 91 (prepared from allylic alcohol 48 by treatment with MsCl, Et3N, and LiBr in quantitative yield, Scheme 12) and changing the solvent from acetone to DMF, the required alkylation proceeded smoothly at ambient temperature and within 1 h to provide the desired coupling product 90 in 78% yield.
With intermediate 90 available, and in an effort to prepare subtarget 24 (Scheme 12), we then proceeded to apply our original spirocyclization conditions (i.e., ZnI2, CH2Cl2, reflux, see Scheme 5), but unfortunately encountered side-products, presumed to arise from elimination,51 demethylation,52 and/or benzylic oxidation reactions. We reasoned that such side reactions could be suppressed by using a nonmobile counterion on the activating agent, a hypothesis that prompted us to test BF3·OEt2, a catalyst that held promise to also induce the pending spirocyclization at lower temperatures. Indeed, exposure of 90 to substoichiometric amounts of this Lewis acid at 0 °C led to the formation of the desired heptacycle 24 in 55% yield with minimum amounts of side-products. The configuration of 24 was confirmed as shown in Scheme 12 by NOESY experiments.
Scheme 13 depicts our attempts to realize the singlet oxygen cascade sequence from 93 (derived from 24 via benzoate derivative 92) to 97 through postulated intermediates 94–96. Thus, heptacyclic phenol 24 was converted to its benzoate 92 (BzCl, Et3N, 79% yield) and thence to hexacyclic system 93 through hydrogenolysis (Pd black, H2, quantitative yield) of the N–O bond and the two benzyl ether moieties. Our reasoning for the protection of the phenolic group of 24 was based on the assumption that this change would promote the aromatization of ring C within 92 by removing the possibility for a H-bond to the C-ring carbonyl group. This assumption, however, proved erroneous, as both 92 and 93 were found to exist exclusively in their C-ring keto forms as opposed to their enol (phenolic) forms. In retrospect, this outcome is not surprising in light of previous reports, which indicated that conjugation of a naphthalene ring (as in 92 and 93) with a ketone provides thermodynamic stability greater than that of its phenolic counterpart (as in the phenolic tautomers of 92 and 93).53
Scheme 13. Preparation of Endoperoxide Precursor 93 and Expected Pathway to Oxygenated Species 97.

Reagents and conditions: (a) BzCl (15 equiv), Et3N (25 equiv), DMAP (0.06 equiv), CH2Cl2, 25 °C, 45 min, 79%; (b) Pd black (5.0 equiv), 1,4-dioxane:MeOH 1:1, H2, 25 °C, 20 min, quant; (c) O2, TPP (cat.), CH2Cl2, sun lamp, −78 °C. Bz = benzoyl, TPP = tetraphenylporphyrin.
Anticipating the singlet oxygen-initiated cascade sequence of reactions shown in Scheme 13 (93 → 94 → 95 → 96 → 97) based on precedented chemistry29 (see Scheme 3), we proceeded to react substrate 93 with singlet oxygen. This transformation, however, could not be realized. Thus, bubbling of O2 through a solution of substrate 93 in CH2Cl2 in the presence of catalytic quantities of TPP and with sun lamp irradiation at −78 °C rapidly produced a product that proved, upon warming, too labile for characterization. Prolonged irradiation at −78 °C produced no detectable amounts of bis-epoxide 97. We then attempted to tautomerize the C-ring of 93 to its corresponding phenolic form under basic conditions with the hope that this maneuver (see above, Scheme 6) might lead to a favorable result in the singlet oxygen reaction. However, the previous conditions used for the corresponding keto–phenol tautomerization (25 → 63, K2CO3, MeOH, Scheme 6) failed to yield the desired product. Other efforts to achieve the desired outcome included attempts to form and rearrange the endoperoxide using different sensitizers on substrates with and without the isoxazole ring, the latter scenario being inspired by Myers’s hypothesis that the isoxazole moiety may act as a self-sensitizer.20a These experiments failed to produce any of the proposed intermediates in Scheme 13 (i.e., 94–97).
On the basis of these findings, we were forced to explore alternative phenolic oxidation protocols in an effort to install oxidation at C12a (Scheme 14). Attempted phenolic oxidations of substrate 24, or its corresponding hydrogenolyzed counterpart 98, were unsuccessful. A number of other conditions, including hypervalent iodine reagents, Fremy’s salt, Pb(OAc)4, and various metals with molecular oxygen, were also unsuccessful. Typically either no reaction was observed or the substrate decomposed, especially for the more electron-rich substrate 98. Furthermore, phenol 101 was synthesized as a possible substrate for C12a hydroxylation through a sequence involving PIFA oxidation to the quinone, benzoyl ester formation (BzCl, 65% for two steps), and regioselective debenzylation (MgBr2·OEt2, 75%). Phenolic oxidation of this substrate (i.e., 101), however, also failed to produce the desired C12a oxidation product 102. The challenges faced here with respect to installation of the C12a hydroxyl group are reminiscent of previous synthetic efforts toward tetracyclines, including those of Woodward,13 Muxfeldt,15 Stork,18 and, in particular, Barton.54
Scheme 14. Phenolic Oxidation Attempts To Hydroxylate at C12a Heptacycle 24 and Derivatives 98 and 101.

Reagents and conditions: (a) Pd black (5.0 equiv), H2, 1,4-dioxane:MeOH, 1:1, 25 °C, 30 min, quant.; (b) PIFA (1.1 equiv), DMF:H2O 9:1, 40 °C, 2 h; (c) BzCl (4.4 equiv), Et3N (10 equiv), DMAP (0.06 equiv), CH2Cl2, 25 °C, 5 min, 65% for two steps; (d) MgBr2·OEt2 (1.0 equiv), Et2O:benzene 1:7, 25 °C, 2 h, 75%. PIFA = phenyliodonium bis(trifluoroacetate).
The failure to install the obligatory hydroxyl groups at C4a and C12a through dearomatization pathways refocused our attention on alternative tactics, the choice of which was based on the intelligence gathered thus far. This information pointed away from an aromatic A-ring as a precursor to the desired oxygenation.
Third-Generation Approach: Stepwise Strategies To Install the C4a and C12a Hydroxyl Groups
Our first attempts to install the coveted hydroxyl groups onto the substrate via enol or enolate chemistry focused on the rather simple AB-ring fragment 76 (Scheme 15). As shown in this scheme, treatment of 76 with DMDO gave a mixture of C12a-hydroxylated compounds 103 and 12a-epi-103 (ca. 3:5 ratio, 79% yield combined, chromatographically separated, inconsequential) whose configurations were established through analysis of their NOESY correlations and (2,3)J coupling constants (see structure 12a-epi-103). We envisioned that the extrusion of methanol from 103 might result in the formation of methyl enol ether 105, whose selective epoxidation would furnish 106. Such a strategy would be conceptually similar to that of Myers, which features a fully functionalized AB-ring enone system. However, the elimination of methanol (103 → 104 → 105, Scheme 15) turned out to be surprisingly problematic. A variety of Lewis- or Brønsted-acidic conditions failed to promote this transformation, leading instead to various decomposition side-products, including lactone ring expansion products involving the C12a hydroxyl group. We reasoned that these failures may be due to mismatched electronics which did not allow for the formation of the oxonium moiety adjacent to the enone system. We then speculated that the presence of an aromatic ring adjacent to the oxonium system (see structure 104, Scheme 15) might facilitate the formation of such a species.
Scheme 15. Early Stage Installation of the C12a Hydroxyl Group and Failed Attempts To Install Oxygenation at C4a.
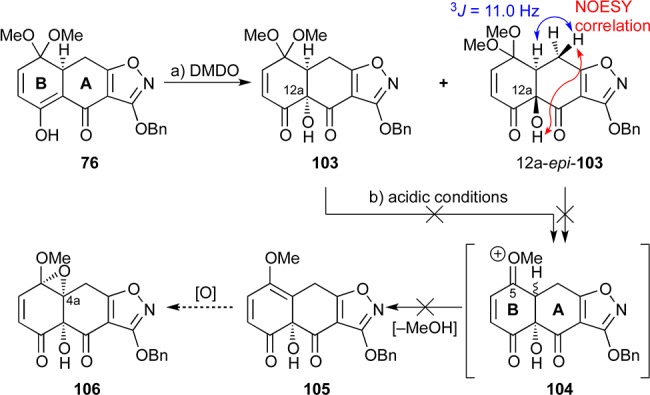
Reagents and conditions: (a) DMDO (1.7 equiv), acetone:H2O 9:1, 25 °C, 1.5 h, 29% for 103, 50% for 12a-epi-103; (b) conditions included several Lewis and Brønsted acids. DMDO = dimethyldioxirane.
With these postulates in mind, a new strategy was devised based on the retrosynthetic analysis shown in Scheme 16, which most notably circumvents an aromatic A-ring and postpones the C4a/C12a oxygenations to the later stages of the projected synthesis. Thus, viridicatumtoxin B structure 1″ was disconnected through the A-ring with a Michael–Dieckmann reaction similar to that which had successfully been deployed in the second-generation strategy. This analysis unveiled isoxazole fragment 81 and BCDEF pentacycle 61 as potential key building blocks. The latter was envisioned to be derived from fragments 91, 66, and 67 through a pathway similar to that of the second-generation approach involving homophthalic anhydride/quinone monoketal cyclization, anthrone alkylation, and Lewis acid-mediated spirocyclization (see Scheme 12).
Scheme 16. Third-Generation Retrosynthetic Analysis of Viridicatumtoxin B Structure 1″.

As shown in Scheme 17, a similar set of reaction conditions as those used to prepare 24 (see Scheme 12) were employed to synthesize spirocyclic compound 60. Thus, DBU-promoted Michael–Dieckmann/decarboxylation cascade (107, 50% yield) and CSA-catalyzed aromatization (quant.) gave anthrone 108. Anthrone alkylation with the allylic bromide 91 then gave the alkylated compound 109 (75%, ca. 1:1 dr, inconsequential). The steric congestion around the newly generated C–C bond of the latter compound manifested itself in some unusual NMR spectroscopic features. Thus, all three methyl groups of the F-ring in 109 showed very broad signals, which suggested hindered rotation around this region of the molecule at 298 K, thereby forcing the methyl groups to experience a number of different magnetic environments. Supporting this notion was the fact that when the 1H NMR spectrum of 109 was acquired at 338 K in DMSO-d6, the methyl signals sharpened considerably. Spirocyclization of 109 proceeded in the presence of BF3·OEt2 as before, producing the desired pentacyclic compound 60 in 82% yield. An X-ray crystallographic analysis of 60 [mp = 114–116 °C (EtOAc:CHCl3 1:1), see ORTEP, Figure 3] unambiguously proved its spectroscopically derived structural assignment.55
Scheme 17. Synthesis of BCDEF Pentacycle 60.

Reagents and conditions: (a) 66 (1.0 equiv), 67 (3.0 equiv), DBU (3.0 equiv), MeCN, 60 °C, 15 h, 50%; (b) CSA (0.02 equiv), CH2Cl2, 25 °C, 30 min, quant.; (c) 91 (1.2 equiv), Na2CO3 (10 equiv), DMF, 25 °C, 1 h, 75%, ca. 1:1 dr; (d) BF3·OEt2 (0.2 equiv), CH2Cl2, 0 °C, 20 min, 82%. CSA = (±)-camphor-10-sulfonic acid.
Figure 3.
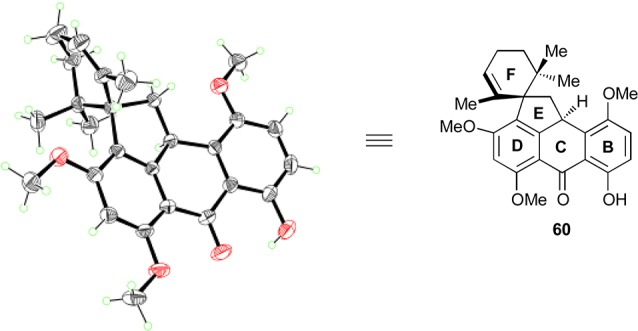
ORTEP representation of spirocycle 60. Thermal ellipsoids at 30% probability. Gray = carbon, red = oxygen, green = hydrogen.
With key intermediate 60 now accessible, we turned next to its phenolic oxidation. Although initially concerned, due to the previous recalcitrance of substrate 25 (see Scheme 6) to undergo phenolic oxidation, these trepidations proved to be unfounded.
Thus, in an initial experiment, treatment of spirocycle 60 with PIDA (3.0 equiv) led to the formation of desired quinomethide–quinone monoketal 111 (54% yield) and side-product 112 (22%, Scheme 18). The stereochemical configuration of the latter species was initially assigned on the basis of a NOESY experiment and was later unambiguously confirmed through X-ray crystallographic analysis [mp = 201–203 °C (EtOAc:CH2Cl2 1:1), see ORTEP representation, Figure 4]. The formation of both 111 and 112 in the presence of excess PIDA in MeOH at 25 °C is apparently a consequence of the competing pathways shown in Scheme 18. Thus, initial phenolic oxidation of 60 affords 61, which suffers elimination of MeOH induced by the in situ-generated AcOH leading to phenol p-quinomethide 110 (isolated and characterized), whose phenolic oxidation furnishes 111. Intermediate 61, however, may also undergo phenolic oxidation, furnishing 112. The facility by which substrate 60 undergoes phenolic oxidation as compared to its simpler counterpart 25 mentioned above (see Scheme 6) is attributed to the extra methoxy group residing on ring B of the former. Attempts to convert side-product 112 to the desired compound 111 under acidic, basic, or thermal conditions failed.
Scheme 18. Optimization of Phenolic Oxidation of Substrate 60 to p-Quinomethide 111.

Reagents and conditions: (a) PhI(OAc)2 (3.0 equiv), MeOH, 25 °C, 2.8 h, 54% for 111 + 22% for 112; (b) PhI(OAc)2 (1.2 equiv), MeOH:CH2Cl2 1:1, 0 → 25 °C, 1 h; (c) CSA (0.07 equiv), CH2Cl2, 0 °C, 30 min; (d) PhI(OAc)2 (1.2 equiv), MeOH, 25 °C, 1.5 h, 78% overall for b, c, and d; (e) NaCNBH3 (1.2 equiv), MeOH, 0 → 25 °C, 45 min, 29%, unoptimized.
Figure 4.
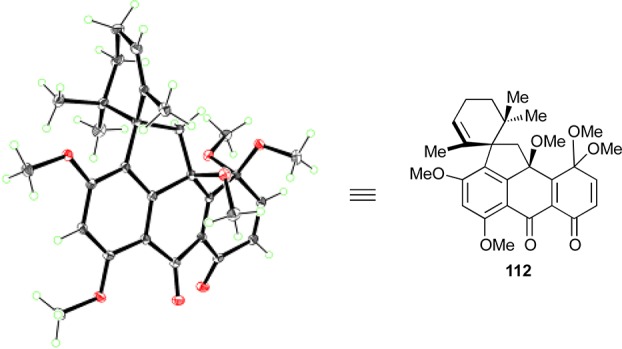
ORTEP representation of side-product 112. Thermal ellipsoids at 30% probability. Gray = carbon, red = oxygen, green = hydrogen.
Efforts to optimize the conversion of 60 to 111 led to the preferred three-step sequence (via 61 and 110) shown in Scheme 18. Thus, employment of limited quantities of PIDA (1.2 equiv), MeOH:CH2Cl2 (1:1) as solvent, and lower temperature (0 → 25 °C), generated intermediate 61 in high yield. The crude product in CH2Cl2 was then treated with catalytic quantities of CSA at 0 °C to provide p-quinomethide 110, which was subjected to further phenolic oxidation with PIDA in MeOH to afford 111 in 78% overall yield for the three steps. It should be mentioned here that substrate 61 proved intransigent to the desired pending reactions (i.e., Michael–Dieckmann cascade), leading instead to MeOH-elimination product 110. Access to substrate 111 opened the way forward, despite the need for reduction of the p-quinomethide to the desired phenolic structural motif at a later stage. The feasibility of such a transformation was briefly explored at this stage, leading to the finding that it could be achieved with NaCNBH3 (Scheme 18, 111 → 61, 29% yield, unoptimized).
The next task was the union of pentacyclic enone 111 with isoxazole 81 through the Michael–Dieckmann sequence of reactions as indicated in Scheme 19. On the basis of our success with this sequence on the simpler systems discussed above (i.e., Schemes 9 and 10), we were rather optimistic for favorable results in this instance as well. However, initial experimentation with methyl ester 81 as the isoxazole equivalent proved otherwise. Thus, although the first step (Michael reaction, Scheme 19) of the anticipated fusion of 111 with 81 proceeded well, leading to Michael adduct 113 (t-BuOK cat., 74% yield, mixture of four diastereoisomers), conditions for the subsequent step (Dieckmann reaction) proved elusive despite considerable experimentation (a variety of basic conditions typically led to recovery of unreacted 113 or decomposition). Removal of the Teoc group from 113 led to 115 and its C15-epimer (15-epi-115, ca. 2:1 dr)56 (TBAF, 60% yield), which, however, also failed to undergo the coveted Dieckmann cyclization (to 116, Scheme 19). Reduction of the quinomethide moiety of 115/15-epi-115 furnished 117/15-epi-117 (NaCNBH3, 82% yield, ca. 2:1 dr). The dibenzyl ether (119/15-epi-119) of the latter was also prepared (NaH, BnBr, TBAI cat., 85% yield, ca. 2:1 dr). Neither 117/15-epi-117 nor 119/15-epi-119 entered the desired Dieckmann reaction to afford the expected heptacyclic systems 118/15-epi-118 or 120/15-epi-120, respectively. Similar problems were reported by Stork and Kahne.57
Scheme 19. Failed Attempts To Close the A-Ring through Dieckmann Condensations.

Reagents and conditions: (a) 81 (1.1 equiv), t-BuOK (0.2 equiv), toluene, 25 °C, 45 min, 74%, mixture of four diastereomers; (b) TBAF (1.2 equiv), THF, 25 °C, 10 min, 60%, ca. 2:1 dr; (c) NaCNBH3 (5.0 equiv), THF, 0 °C, 2 min, 82%, ca. 2:1 dr; (d) BnBr (10 equiv), NaH (10 equiv), TBAI (0.15 equiv), toluene, 25 → 100 °C, 3.75 h, 85%, ca. 2:1 dr. TBAI = tetra-n-butylammonium iodide.
Faced with these difficulties and inspired by the work of White58 and Myers,20d we opted to employ the phenyl ester counterpart of isoxazole 81 (see 123, Scheme 20a). Isoxazole phenyl ester 123 was prepared from methyl ester 26 via 121 and 122 through saponification (NaOH, 99% yield), esterification with PhOH under Mitsunobu conditions (PPh3, DIAD, 78% yield),59 and Teoc attachment (LiHMDS, TeocCl, 86% yield) as shown in Scheme 20a. It is of note that several standard ester-forming reactions (e.g., mixed anhydride, acid chloride, DCC) failed to bring about the desired esterification between 121 and PhOH. Gratifyingly, quinone monoketal 111 reacted smoothly with phenyl ester isoxazole 123 in the presence of a slight excess of t-BuOK to give directly the desired heptacyclic compound 114 as a ca. 1:1 mixture with its C-15 diastereoisomer (15-epi-114) in 94% yield (Scheme 20b).60 The stereochemical relationship of H4 and H4a within 114 was evident from the observed coupling constant 3J4,4a = 9.9 Hz, indicative of the diaxial disposition of these protons. The impressive rapidity (<15 min at 25 °C) of the Michael–Dieckmann cascade to afford the heptacyclic product (111 → 114 + 15-epi-114) is a testament to the activating power of the phenyl ester moiety in such circumstances.
Scheme 20. Synthesis of Phenyl Ester 123 (a) and Its Successful Michael–Dieckmann Cyclization with Enone 111 (b).

Reagents and conditions: (a) NaOH (1.9 equiv), H2O:EtOH 3:10, 25 °C, 3 h, 99%; (b) PPh3 (1.05 equiv), PhOH (1.05 equiv), DIAD (1.05 equiv), THF, reflux, 3 h, 78%; (c) LiHMDS (2.2 equiv), THF, −78 °C, 30 min; then TeocCl (2.2 equiv), −78 °C, 2 h, 86%; (d) 123 (1.1 equiv), t-BuOK (1.2 equiv), toluene, 25 °C, 15 min, 94%, ca. 1:1 dr. DIAD = diisopropyl azodicarboxylate.
We initially explored the C12a hydroxylation of substrate 114, with the Teoc group still intact, as shown in Scheme 21. Thus, hydroxylation of substrate 114 at C12a with DMDO (conditions a) at −78 °C produced the desired compound 124 (together with its C15 epimer 15-epi-124, ca. 2:1 dr) but only as a minor product (9% combined yield). The major product in this reaction was seven-membered lactone 125 formed together with its C15-epimer (15-epi-125, 21% combined yield, ca. 2:1 dr) through rearrangement/ring expansion of presumed transient intermediate 126 + 15-epi-126 (see Scheme 21). C12a hydroxylation of substrate 114 + 15-epi-114 was also achieved with m-CPBA, this time leading exclusively to the desired hydroxy compound 124 + 15-epi-124 (ca. 2:1 dr) in 71% combined yield based on 24% recovered starting material. However, attempts to remove the Teoc group from the latter mixture with HF·Et3N led to product 127 + 15-epi-127 from which the Teoc had been removed and the B-ring had been expanded to a seven-membered lactone moiety, presumably through rearrangement of fleeting intermediate 128 + 15-epi-128 (or its Teoc counterpart which suffers Teoc removal concomitantly or subsequently), as depicted in Scheme 21. Exposure of 124 (+ 15-epi-124) to TBAF led to decomposition and partial recovery of starting material.
Scheme 21. C12a Hydroxylation of Substrate 114.
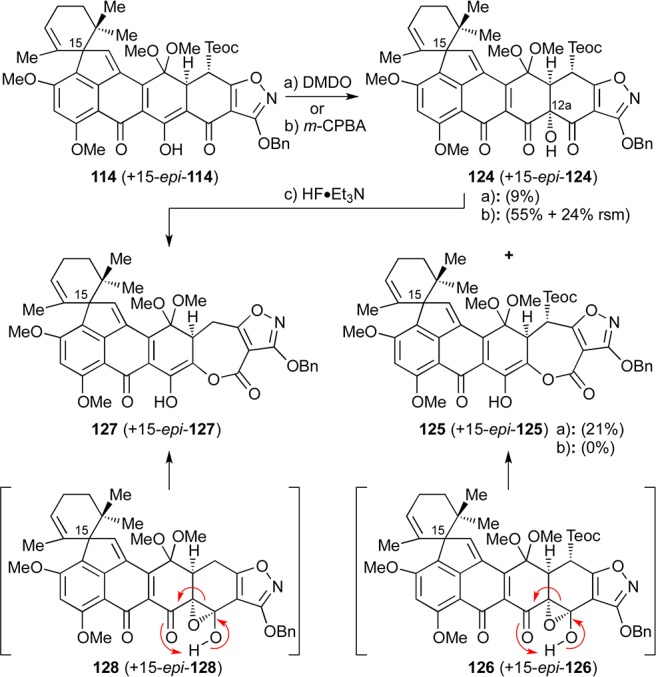
Reagents and conditions: (a) DMDO (2.8 equiv), acetone, −78 °C, 3.5 h, 21% for 125 (ca. 2:1 dr), 9% for 124 (ca. 2:1 dr); (b) m-CPBA (2.7 equiv), CH2Cl2, −78 °C, 1.5 h, 55% for 124 (71% brsm, ca. 2:1 dr) + 24% recovered 114; (c) HF·Et3N (100 equiv), DMSO, 60 °C, 2 h, 43%, ca. 2:1 dr. DMSO = dimethyl sulfoxide.
The difficulties of removing the Teoc group in the presence of the C12a hydroxyl group led us to explore the alternative sequence in which the Teoc moiety was removed prior to the hydroxylation step. To this end, and as shown in Scheme 22, 114 (+ 15-epi–114) was treated with excess TBAF in THF, furnishing small amounts of the desired product 116 (+ 15-epi-116, 16% combined yield) and unreacted starting material (33%). Presumably, this failure was caused by deprotonation of the diketone moiety adjacent to the isoxazole (see 129) that rendered the desired loss of TMSF, ethylene, and carbon dioxide less facile (see 129 → 130). Faced with this predicament, we undertook a thorough investigation of conditions to achieve the desired Teoc group removal from 114 (+ 15-epi-114), including ZnCl2, ZnF2, MgBr2, NH4F, and SiF4,61,62 but unfortunately to no avail. Disappointingly, the use of HF·Et3N led to the desired product 116 (+ 15-epi-116) in 28% combined yield, albeit with substantial amounts of side-product 131 (30% yield), presumably formed through an air-oxidation process.63 Indeed, it was not until we combined Fürstner’s TBAF–NH4F conditions64 with solvent degassing (to prevent air oxidation of the A-ring) that we reached a solution to this problem, obtaining a pleasing 98% yield of the coveted deprotected product 116 (+15-epi-116) (Scheme 22).
Scheme 22. Teoc Removal from 114 and C12a Hydroxylation of 116.
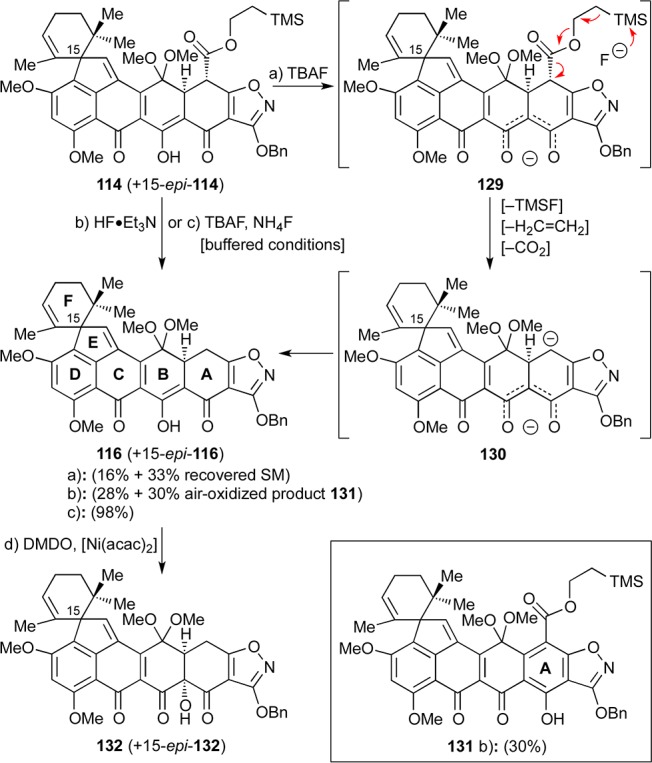
Reagents and conditions: (a) TBAF (5.3 equiv), THF, 25 °C, 1 h, 16% for 116 (ca. 2:1 dr) + 33% recovered 114; (b) HF·Et3N (excess), DMSO, 25 → 60 °C, 3 h, 28% for 116 (ca. 2:1 dr) + 30% for 131; (c) TBAF (10 equiv), NH4F (20 equiv), THF, 25 °C, 5 min, 98%, ca. 2:1 dr; (d) DMDO (7.6 equiv), [Ni(acac)2] (0.2 equiv), acetone, −78 → −65 °C, 6.5 h, 61%, ca. 2:1 dr. acac = acetylacetonate.
Having solved the Teoc removal problem, we turned our attention to the C12a hydroxylation task using 116/15-epi-116 as a substrate. Our initial use of DMDO in acetone at low temperature (i.e., −78 °C) proved irreproducible, gave low conversion to product due to substrate insolubility, and suffered from TLC monitoring problems. To improve the reaction, we resorted to the use of Ni(acac)2, an additive known to facilitate DMDO oxidations through coordination.65 Indeed, portionwise addition of DMDO to a stirred mixture of 116/15-epi-116 and catalytic amounts of Ni(acac)2 in acetone at −78 to −65 °C led to 61% combined yield of C12a hydroxylated product 132/15-epi-132 (ca. 2:1 dr) as shown in Scheme 22. Both diastereomers were of the syn stereochemical configuration with regard to the C12a hydroxyl and C4a hydrogen residues, presumably as a consequence of the concavity of the AB ring system. It should be noted at this point that at temperatures above −40 °C, epoxidation of the F-ring olefinic site began to occur. Other oxidizing agents such as m-CPBA, Davis oxaziridine,66 and magnesium monoperoxyphthalate (MMPP) proved unsuccessful in providing the desired C12a-hydroxylated compound (i.e., 132 +15-epi-132).
With the C12a hydroxylation problem solved, the challenge of installing the C4a hydroxyl group became the next task, a mission that was to prove even more intransigent than the one before it. Our initial attempts to achieve this goal are shown in Scheme 23. Thus, reduction of the quinomethide moiety of 132/15-epi-132 with NaCNBH3 led to the now chromatographically separable derivatives 133 and 15-epi-133. Gratifyingly, one of the two isomers (more polar) crystallized readily from EtOAc [mp = 213–215 °C (decomp)] and yielded to X-ray crystallographic analysis, establishing its stereochemical configuration as 15-epi-133 (see ORTEP representation, Figure 5), and as a consequence, that of its less polar epimer as the desired diastereomer 133.55 Several attempts to prepare methylenol ether 134 as a potential precursor of the desired 4a-hydroxylated compound 136 through selective epoxidation (to afford 135) and thence hydrolysis, or through 137 via hydrolysis, enolization, and oxidation, failed. Efforts to generate 134 from 133 employing a variety of conditions, including Lewis acids or protic acids, or under anhydrous conditions (see Supporting Information), however, did not give the desired product but led instead to ketone 137. Similarly, various conditions to carry out the desired enol ether formation at the quinomethide stage (compound 132/15-epi-132) also failed. In addition, a number of transketalization reactions with dimethyl ketal 133 directed at forming an oxa-seleno mixed ketal67 (intended to be used as a precursor for a selenoxide/syn elimination sequence to produce the desired methylenol ether 134) ended without success. Attempts to achieve C4a hydroxylation of ketone 137 through enolization followed by oxidation also failed, typically resulting in aromatization to afford 138 (Scheme 23) through β-elimination/tautomerization (see Supporting Information for more details).
Scheme 23. Initial Strategy for C4a Hydroxylation.

Reagents and conditions: (a) NaCNBH3 (10 equiv), THF, −78 °C, 1.5 h, 51% for 133 + 19% for 15-epi-133, chromatographically separated; (b) various Lewis and Brønsted acids: see the Supporting Information; (c) THF:2 M aq. HCl 10:1, 25 °C, 3.5 h, 98%; (d) various conditions: see the Supporting Information.
Figure 5.
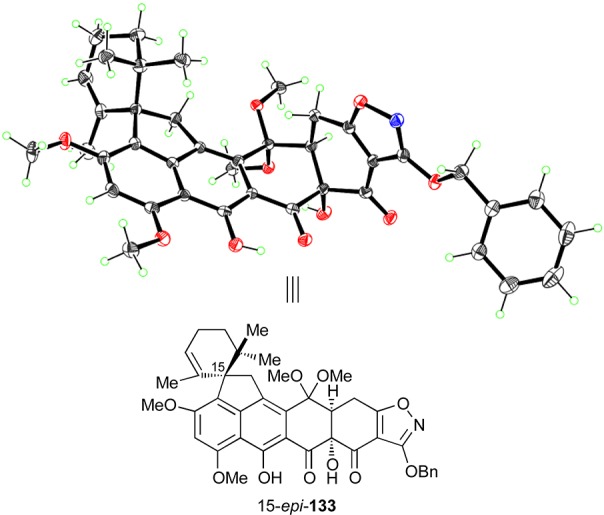
X-ray-derived ORTEP representation of compound 15-epi-133. Thermal ellipsoids at 30% probability. Gray = carbon, blue = nitrogen, red = oxygen, green = hydrogen.
We then explored the possibility of achieving C4a hydroxylation using dimethyl ketal 133 (single diastereomer) to generate olefinic compound 141, whose directed dihydroxylation68 was expected to yield 142 (see Scheme 24). To this end, exposure of 133 to NaCNBH3 in AcOH produced chromatographically separable methyl ethers 140 (40%) and 5-epi-140 (32%), presumably through reactive oxonium species 139.69 The configuration of 140 (and by deduction, of 5-epi-140) was assigned on the basis of 2D NMR (NOESY) spectroscopic analysis and the observed 3J4a,5 = 8.7 Hz coupling constant (see structure 140, bottom of Scheme 24). Numerous attempts (e.g., Lewis and protic acids) to generate olefin 141 from 140 or 5-epi-140 through elimination of MeOH, however, failed. In several cases, the bis-elimination/tautomerization product 143 was isolated instead. Attempted solvolysis of isomer 5-epi-140 by heating in trifluoroethanol at 70 °C as a means to reach olefin 141 led, surprisingly, to trifluoroethyl ester 144 (70% yield, Scheme 24), presumably through a retro-Dieckmann ring opening reaction.70
Scheme 24. Attempts To Generate C4a,5-Olefin 141 as a Substrate for Dihydroxylation.
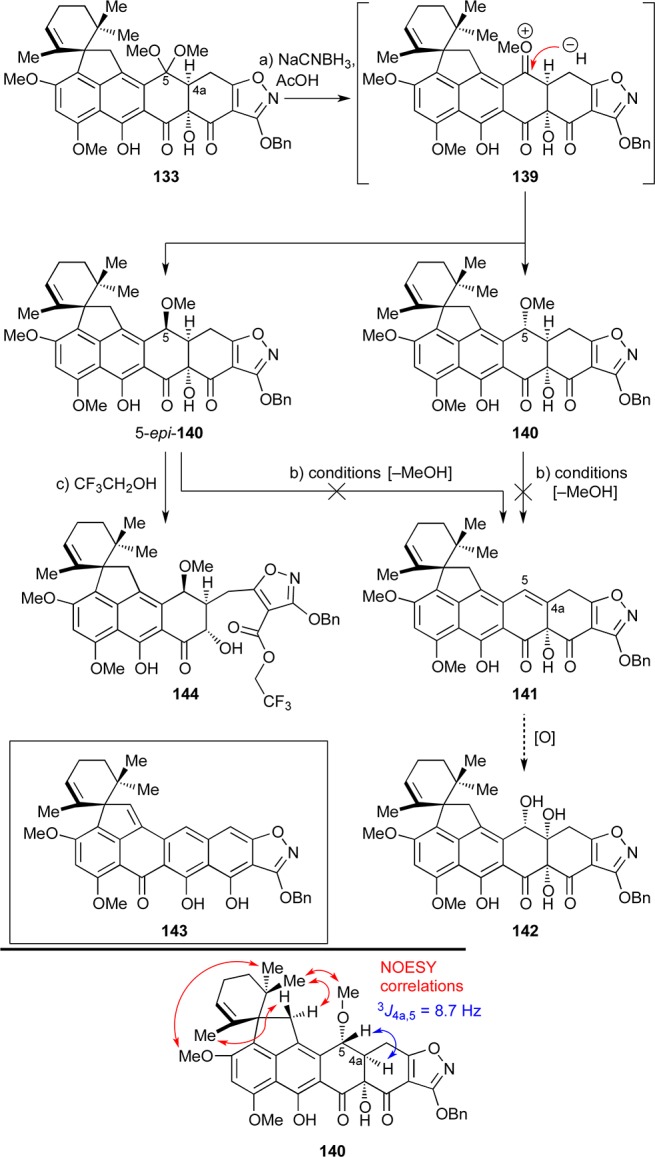
Reagents and conditions: (a) NaCNBH3 (4.3 equiv), AcOH, 25 °C, 40 min, 40% for 140, 32% for 5-epi-140, chromatographically separated; (b) various Lewis and Brønsted acids; (c) CF3CH2OH, 70 °C, 2 h, 70%.
Unable to reach the Δ4a,5-compound 141 (Scheme 24), we turned to the Δ4,4a-compound 145 as an alternative substrate for the desired C4a hydroxylation. It was envisioned that 145 could be reached from 133 through an oxidative process and then converted to the desired hydroxylated product 147 via selective formation and regioselective reductive opening of epoxide 146, as outlined in Scheme 25. As shown in Scheme 26, execution of this plan began with formation of presumed trianion 148 (KHMDS, −78 °C) followed by quenching with PhSeCl (−78 → −40 °C) to afford phenyl selenide 149 as a single isomer (46% yield + 26% recovered starting material). The configuration of the phenylseleno group within 149 was tentatively assigned on steric grounds (i.e., concavity of the cis-fused AB-ring system). Treatment of selenide 149 with DMDO at −78 °C produced a complex mixture from which seven-membered lactone 152 could be isolated (46% yield), presumably formed through the indicated pathways involving selenoxide 150, olefin 145, and either acylketene 151 (pathway A)71,72 or hydroxy-epoxide 153 (pathway B) (see Scheme 26). Other oxidizing agents (e.g., H2O2, m-CPBA) led to similar results. In an attempt to shut down the hypothesized hydroxy-epoxide pathway, we prepared TMS ether 154 from 149 (TMSOTf, Et3N, 62% yield, Scheme 26) and employed it as a substrate in the DMDO oxidation, only to observe the same ring expansion product 152, albeit in only 7% yield (with concomitant loss of the TMS residue). This outcome seems to tentatively support pathway A, rather than B, for the formation of 152 from 154 (and 149), although due to the lability of the TMS group the possibility of both pathways operating concurrently cannot be excluded.
Scheme 25. Proposed Elaboration of 133 to C4a-Hydroxylated Compound 147 through C4,4a-Olefin 145 and Epoxide 146.

Scheme 26. Attempted Preparation of Olefin 145.

Reagents and conditions: (a) KHMDS (3.3 equiv), THF, −78 °C, 1 h; then PhSeCl (3.5 equiv), −78 → −40 °C, 1 h, 46% (62% brsm) + 26% recovered 133; (b) from 149: DMDO (1.0 equiv), THF, −78 °C, 20 min, 46%; from 154: DMDO (1.5 equiv), THF, −78 °C; then 25 °C, 14 h, 7%; (c) TMSOTf (30 equiv), Et3N (40 equiv), CH2Cl2, 0 °C, 5 min, 62%. OTf = trifluoromethanesulfonate.
A number of other attempts to prepare Δ4,4a-substrates such as 157 (Scheme 27) and 145 (Scheme 28) were made. Thus, TFA-induced hydrolysis of ketal 149 gave triketone 155 (52% yield) which was expected to facilitate the formation of 157 through selenoxide formation (156) and syn-elimination (see Scheme 27). Unfortunately, however, oxidation of selenide 155 (H2O2) did not lead to the expected olefin 157, but rather to the expanded keto-lactone 158 (35% yield), presumably through selenoxide 156 (see Scheme 27). Similarly, hydroxylation of 133 with Davis oxaziridine through its trianion (KHMDS, −78 °C, 52% yield) followed by ketal hydrolysis (TFA, 63% yield) furnished secondary alcohol 160, whose attempted dehydration (e.g., through the corresponding mesylate) also failed (see Scheme 28). Iodide 159 (Scheme 28) was also prepared from 133 (KHMDS; NIS, 37%) as a potential precursor to olefinic compound 145, through oxidation and syn-elimination of the corresponding iodoso compound.73 Attempts to effect the latter transformation, however, by m-CPBA oxidation failed. Moreover, attempted Mukaiyama dehydration74 from the trianion derived from 133 also did not give the desired olefin 145 (Scheme 28).
Scheme 27. Attempted Preparation of Enone 157.

Reagents and conditions: (a) TFA:CH2Cl2 (1:2), 0 °C, 40 min, 52%; (b) H2O2 (4.5 equiv), CH2Cl2, 0 °C, 10 min, 35%.
Scheme 28. Other Attempts To Synthesize Olefinic Compounds 141 and 153.

Reagents and conditions: (a) KHMDS (3.8 equiv), THF, −78 °C, 1 h; then Davis oxaziridine (3.5 equiv), −78 → −40 °C, 1 h, 52%; (b) TFA:CH2Cl2 (1:3), 0 → 25 °C, 45 min, 63%; (c) KHMDS (3.6 equiv), THF, −78 °C, 80 min; then NIS, −78 → −35 °C, 1 h, 37%. NIS = N-iodo-succinimide.
Faced with these obstacles toward Δ4,4a-substrates 145 and 157, we decided to return to Δ4a,5-substrate 141 which we had previously considered but later abandoned (see Scheme 24). Inspired by a report from the Tatsuta group,75 we reasoned that a 1,3-directed reduction of the C5-carbonyl moiety at the 137 stage could give us access to the C5-hydroxy compound 162, whose dehydration would lead to the targeted substrate 141 that could serve as a precursor to the desired trihydroxylated intermediate 142 (Scheme 29). However, when 137 was treated with NaBH(OAc)3, we instead isolated the 1,2-directed reduction product 164 (35% yield, Scheme 29), along with other unidentifiable products. After further unsuccessful attempts to secure the targeted C5-hydroxy derivative of our heptacyclic compound, we opted to pursue the newly emerged 1,2-directed reduction product 164 as a possible substrate for advancement to viridicatumtoxin B. The rationale behind this choice included the expectation that this intermediate and its derivatives could be resistant to elimination and aromatization side reactions involving rings A and B as previously observed (see Schemes 23 and 24). Brief experimentation led to an improved protocol for the selective reduction of triketone 137 [NaBH(OAc)3, EtOAc:acetone] that delivered alcohol 164 in 53% yield (Scheme 30). The newly generated hydroxy group within the latter compound was protected as a TBS ether through the use of excess TBSOTf (most likely needed due to steric difficulties caused by the concavity of the AB-ring system) to afford compound 165 (64% yield). Conversion of the latter compound to its trianion (166) with KHMDS followed by quenching with Davis oxaziridine led to the coveted C4 hydroxylated product 167 in 29% yield (47% based on 39% recovered starting material). Alternative bases (LiHMDS, NaHMDS, or LDA) or equivalents of base did not improve the outcome of this reaction. The configuration of the newly generated C4a hydroxyl moiety within 167 was deduced from NOESY correlations as shown in Scheme 30 (bottom).
Scheme 29. Attempted Preparation of C5-Hydroxy Compound 162 through 1,3-Directed Reduction and Observation of 1,2-Directed Reduction Product 164.
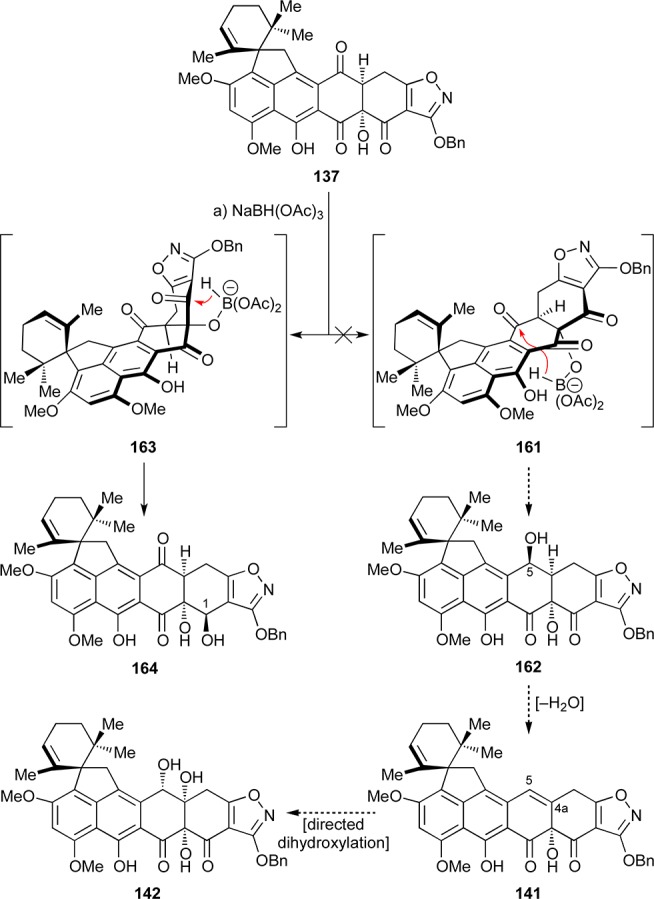
Reagents and conditions: (a) NaBH(OAc)3 (1.3 equiv), EtOH:THF 1:1, 0 → 25 °C, 3.25 h, 35%.
Scheme 30. Successful C4a Hydroxylation of 164 through Oxidation of Its Trianion.
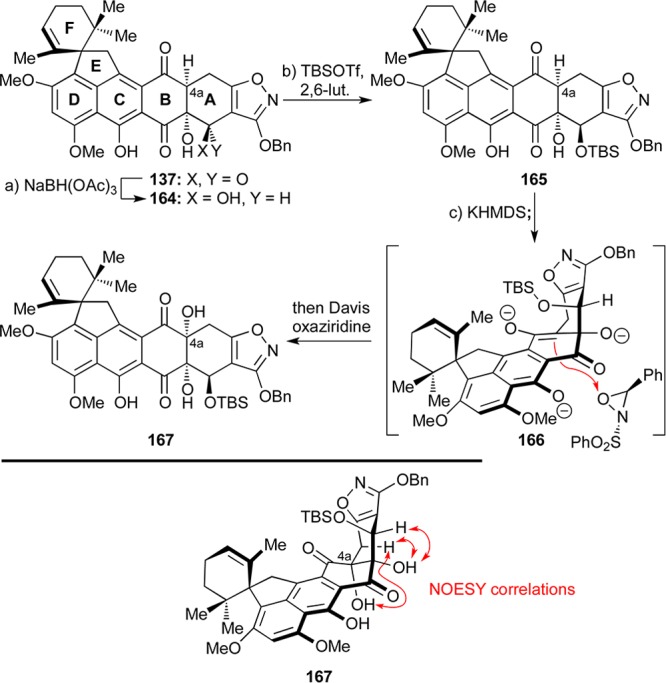
Reagents and conditions: (a) NaBH(OAc)3 (1.2 equiv), EtOAc:acetone 1:1, 40 °C, 3.5 h, 53%; (b) TBSOTf (35 equiv), 2,6-lut. (53 equiv), ClCH2CH2Cl, 5 → 25 °C, 1.8 h, 64%; (c) KHMDS (3.5 equiv), THF, −78 °C, 65 min; then Davis oxaziridine (4.0 equiv), −78 °C, 40 min, 29% for 167 (47% brsm) + 39% recovered 165. lut. = lutidine.
In an effort to improve the C4a hydroxylation reaction, we targeted bis-TBS ether 168 (Scheme 31) as a substrate, counting on its dianion to undergo the desired transformation more efficiently. Bis-silylation of 164 with excess TBSOTf required the use of Hünig’s base and led to 168 in 57% yield. Interestingly, however, treatment of 168 with KHMDS at −78 °C led rapidly to aromatized product 172 (43% yield), which, in addition to elimination of the OTBS group from its A-ring, had also suffered removal of the TBS group from the phenolic oxygen. These observations suggest that the presumed initially formed dianion 169 is too reactive entering the proposed cascade sequence shown in Scheme 31. Thus, attack of the tertiary alkoxide within 169 on the adjacent carbonyl moiety generates a new alkoxide anion that abstracts the TBS group from the adjacent phenolic position to afford epoxide species 170. Prompted by the adjacent enolate anion, the epoxide within the latter species undergoes facile β-elimination/opening with concomitant loss of the B-ring OTBS group to afford diketone dianion 171. The generation of the newly established anion at C4 within 171 is facilitated by the isoxazole ring on one side and the quinone moiety on the other. Species 171 then suffers OTBS elimination/ring A aromatization, leading to observed product 172 upon protonation.
Scheme 31. Synthesis and Unsuccessful Attempt To C4a Hydroxylate Bis-TBS Ether 168.
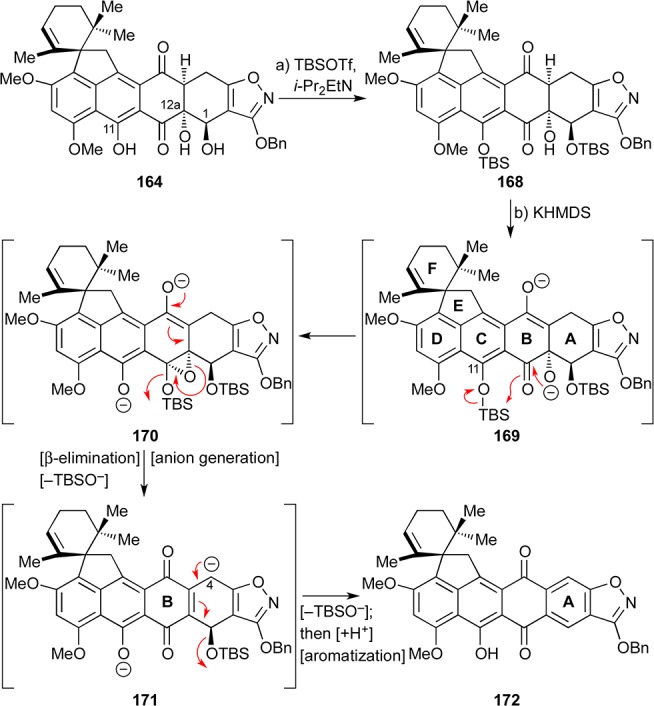
Reagents and conditions: (a) TBSOTf (20 equiv), i-Pr2EtN (30 equiv), CH2Cl2, 0 → 25 °C, 33 min, 57%; (b) KHMDS (2.5 equiv), THF, −78 °C, 45 min, 43%.
Taken together, these extensive investigations on the C4a hydroxylation defined the required structural features for the substrate as (a) protected hydroxyl group at C1 (ring A), (b) free hydroxyl group at C12a (rings A/B), and (c) free phenolic moiety at C11 (ring C). Straying away from any one of these key structural motifs led to either no conversion or rearranged products of undesired structures.
At this point, we decided to move forward and test the remaining steps of the devised synthetic strategy toward viridicatumtoxin B that required reinstallment of the carbonyl group at C1, selective removal of the methyl group from the C10 methyl ether, and rupture of the isoxazole moiety to provide the required enol amide functionality at the terminus of the molecule. To this end and as shown in Scheme 32, silyl ether 167 was desilylated (HF·py, 66% yield) to afford C1 hydroxy compound 173 which was assumed to exist in equilibrium with its hemiacetal form 173′ based on its broad 1H and 13C NMR spectroscopic features. Oxidation of this mixture with DMP resulted in the formation of ketone 136 (32% yield, unoptimized) through reaction of 173, which apparently drives the equilibrium in the right direction for funneling both isomers (173 and 173′) toward the desired product (136). The obligatory removal of the methyl group from the C10 phenolic group, however, proved difficult despite the inspiring precedent for such deprotections in the tetracycline series.19,20b Thus, attempts to achieve this goal with BCl3, BBr3, AlCl3/EtSNa, and 9-I-BBN76 failed, leading to either decomposition and/or cleavage of the benzyl ether as indicated by LCMS detection of hydroxy-isoxazole 174 (but not 175).
Scheme 32. Synthesis of Triketone 136.

Reagents and conditions: (a) HF·py (excess), MeCN, 0 → 55 °C, 25 h, 66%; (b) DMP (5.2 equiv), ClCH2CH2Cl, 0 → 40 °C, 4 h, 32%; (c) BCl3, BBr3, AlCl3/EtSNa, or 9-I-BBN. DMP = Dess–Martin periodinane, BBN = borabicyclo[3.3.1]nonane.
In contrast to this disappointing result, the rupture of the isoxazole ring planned as the ultimate step of the synthesis proved feasible with substrate 136 under hydrogenation conditions in the presence of Pd black, furnishing 10-O-methylviridicatumtoxin B (176, 57% yield) as shown in Scheme 33. Interestingly, the spectroscopic data of this compound confirmed its hydroxy-keto form 176 rather than the originally assigned1 (to viridicatumtoxin B) hydroxy-epoxide structure (i.e., 1′, Chart 1).
Scheme 33. Total Synthesis of 10-O-Methylviridicatumtoxin B.

Reagents and conditions: (a) H2, Pd black (5.3 equiv), MeOH:1,4-dioxane 1:1, 25 °C, 8 min, 57%.
The path-pointing results and intelligence discussed above served as the basis for the final strategy and drive toward viridicatumtoxin B.
Total Synthesis and Structural Revision of Viridicatumtoxin B: Final Drive
Scheme 34 presents the final retrosynthetic analysis and successful strategy that led to the total synthesis of viridicatumtoxin B. Thus, cyclic anhydride 177, quinone monoketal 67, allylic bromide 91, and isoxazole 123 were confidently defined as the required building blocks for the intended route (based on the studies described above). The high modularity of the strategy (i.e., four building blocks of approximately equal sizes) and the accessibility of the starting materials appeared attractive for further development and designed-analogue construction. The adoption of the benzyl ether protecting groups at both the anhydride (177) and isoxazole (123) fragments offered the advantage of their concurrent removal as the last step in the synthesis. The overall plan called for the union of the anhydride (i.e., 177) and quinone monoketal (i.e., 67) fragments through (1) a Michael–Dieckmann/decarboxylation cascade to cast the C ring, (2) an SN2 alkylation to attach the F ring, (3) a Lewis acid-catalyzed spirocyclization to cast the E ring, and (4) a Michael–Dieckmann sequence to fuse the A-ring as outlined in Scheme 34.
Scheme 34. Final Retrosynthetic Analysis of Viridicatumtoxin B (1).

Access to building blocks 67, 91, and 123 has already been discussed above, leaving only the preparation of the benzyl-protected anhydride fragment 177 to be described here. Its synthesis and union with the other fragments (67, 91, and 123) are summarized in Scheme 35. Thus, regioselective demethylation of the known diethyl ester 85 with BBr3 followed by benzylation (BnBr, Ag2O) led to benzyl ether 178 in 66% overall yield. Saponification of the ester moieties of the latter (aq NaOH) and heating of the resulting dicarboxylic acid (179) with Ac2O as previously reported45 furnished desired anhydride 177 in 90% overall yield.
Scheme 35. Synthesis of the Carbon Framework 187 of Viridicatumtoxin B with a C10-OBn Protecting Group.

Reagents and conditions: (a) BBr3 (1.35 equiv), CH2Cl2, −78 → 25 °C, 30 min; (b) BnBr (1.1 equiv), Ag2O (1.9 equiv), DMF, 25 °C, 15 h, 66% for two steps; (c) NaOH (27 equiv), H2O:EtOH 5:7, reflux, 15 h; (d) Ac2O (1.1 equiv), toluene, reflux, 1 h, 90% for two steps; (e) 67 (3.0 equiv), NaH (3.0 equiv), THF, 0 °C, 45 min; then 25 °C, 1 h; (f) DBU (5.0 equiv), toluene, 65 °C, 4.5 h, 54% for two steps; (g) CSA (0.02 equiv), CH2Cl2, 25 °C, 30 min, 99%; (h) 91 (1.1 equiv), Na2CO3 (10 equiv), DMF, 25 °C, 1 h, 77%, ca. 1:1 dr; (i) BF3·OEt2 (0.10 equiv), CH2Cl2, 0 °C, 20 min, 73%; (j) PIDA (1.2 equiv), MeOH:CH2Cl2 1:1, 0 → 25 °C, 1 h; (k) CSA (0.07 equiv), CH2Cl2, 0 °C, 5 min, 85% for two steps; (l) PIDA (1.2 equiv), MeOH:CH2Cl2 10:1, 25 °C, 1.5 h, 90%; (m) 123 (1.1 equiv), t-BuOK (1.2 equiv), toluene, 25 °C, 15 min, 91%, ca. 2:1 dr; (n) TBAF (10 equiv), NH4F (20 equiv), degassed THF, 25 °C, 5 min, 86%, ca. 2:1 dr.
Anhydride 177 was reacted with quinone monoketal 67 through the action of NaH (Michael–Dieckmann sequence) followed by treatment of the coupling product with DBU (decarboxylation) to afford tricycle 180 in 54% overall yield. Elimination of MeOH from the latter induced by CSA gave the corresponding anthrone (99% yield), which was alkylated with allylic bromide fragment 91 in the presence of Na2CO3 to furnish intermediate 181 (77% yield). Exposure of 181 to catalytic amounts of BF3·OEt2 afforded spirocycle 182 in 73% yield. The latter was then treated with PIDA in MeOH:CH2Cl2 to give the initially formed quinone monoketal 183 and thence with CSA to afford the phenol p-quinomethide 184 (85% yield overall). Phenolic oxidation of the latter with PIDA in MeOH:CH2Cl2 then delivered 185 in 90% yield, as observed previously with the corresponding C10-methyl series of substrates (see Scheme 18). Coupling of 185 with isoxazole phenyl ester fragment 123 was effected with t-BuOK (Michael reaction/Dieckmann condensation) furnishing heptacyclic compound 186 together with its unseparable C15 epimer (15-epi-186, ca. 2:1 dr, 91% combined yield). Removal of the now-superfluous Teoc group was accomplished through application of the developed desilylation/decarboxylation procedure (TBAF/NH4F) to furnish advanced intermediate 187 together with its unseparable isomer 15-epi-187 (ca. 2:1 dr) in 86% combined yield.
The next step in the synthesis entailed hydroxylation of the molecule at C12a (see Scheme 36), a process that was plagued with increased insolubility issues of the benzyl-protected substrate 187/15-epi-187 as compared to the methyl-protected version employed in the earlier generation route. This necessitated the use of DMDO in the presence of Ni(acac)2 in CH2Cl2 (instead of acetone); the efficiency of the reaction remained similar as the previous result after recycling of recovered starting material, yielding C12a-hydroxylated compound 188 together with its 15-epimer (15-epi-188, 35% yield, 50% yield after one recycle, ca. 2:1 dr, Scheme 36). Employing a solution of DMDO in CH2Cl277 did not result in a noticeable improvement of conversion or yield, which might be attributed to the decreased stability of DMDO in that solvent. Exposure of the quinomethide mixture (188 + 15-epi-188) to NaCNBH3 gave chromatographically separable reduced products 189 (39% yield) and 15-epi-189 (19% yield) whose configurations were assigned by comparisons of their NMR spectroscopic data to those of their corresponding C10-OMe counterparts (as discussed above, the structures of the latter were unambiguously assigned through X-ray crystallographic analysis, see Scheme 23 and Figure 5). Isomer 189 was then advanced to TBS ether 192 as shown in Scheme 36. Thus, treatment of 189 with HCl in aqueous THF led quantitatively to triketone 190, whose reduction with NaBH(OAc)3 furnished C1-hydroxy compound 191 in 47% yield. The latter was then silylated with TBSOTf in the presence of 2,6-lutidine to afford 192 (61% yield).
Scheme 36. Synthesis of Enolate-Oxidation Precursor 192.

Reagents and conditions: (a) [Ni(acac)2] (0.2 equiv), DMDO (5.1 equiv), CH2Cl2, −78 → −60 °C, 6.5 h, 36% (ca. 2:1 dr, 60% brsm), 50% after one recycle; (b) NaCNBH3 (10 equiv), THF, −78 → −60 °C, 90 min, 39% for 189, 19% for 15-epi-189, chromatographically separated; (c) 2 N aq. HCl:THF 1:10, 25 °C, 5 h, quant.; (d) NaBH(OAc)3 (1.2 equiv), EtOAc:acetone 1:1, 40 °C, 105 min, 47%; (e) TBSOTf (40 equiv), 2,6-lut. (60 equiv), CH2Cl2, 0 → 25 °C, 1 h, 61%.
The final stretch of the synthesis of viridicatumtoxin B is depicted in Scheme 37. The challenging C4a hydroxylation of precursor 192 proceeded in the presence of KHMDS (added first at −78 °C to form the presumed trianion of 192) and Davis oxaziridine (added thence at −78 °C) to afford hydroxylated product 193 (20% yield + 45% recovered starting material). Desilylation of 193 with HF·py furnished polyhydroxy compound 194, whose equilibrium with its lactol form (194′) was evident from the 1H NMR spectra (CDCl3, 500 MHz) at ambient temperature (broad signals) and −40 °C (two sets of sharp signals, ca. 2:1 ratio). (This was the same phenomenon we encountered with their 10-methoxy counterparts 173/173′, Scheme 32, as discussed above). Oxidation of the so-obtained mixture (194 + 194′) with DMP then gave triketone 195 in 66% yield, with the lactol form 194′ being funneled into the oxidation pathway by the equilibrium. Pleasantly, hydrogenation of the latter compound (Pd black, H2) produced synthetic viridicatumtoxin B (1) in 98% yield through cleavage of the two benzyl ethers, rupture of the isoxazole N–O bond, and tautomerization of the resulting hydroxy-imine to the desired primary amide.
Scheme 37. Completion of the Total Synthesis of Viridicatumtoxin B (1).

Reagents and conditions: (a) KHMDS (3.4 equiv), THF, −78 °C, 1 h; then Davis ox. (3.9 equiv), −78 °C, 1.7 h, 20% of 193 + 45% recovered 193; (b) HF·py (excess), MeCN, 0 → 50 °C, 25 h, 61%; (c) DMP (3.0 equiv), ClCH2CH2Cl, 0 → 50 °C, 7.5 h, 66%; (d) H2, Pd black (4.9 equiv), 1,4-dioxane:MeOH 1:1, 25 °C, 8 min, 98%.
The physical properties of synthetic viridicatumtoxin B were consistent with those of the authentic natural product and our proposed structure (1). The 1H NMR data of synthetic viridicatumtoxin B (1) were in good agreement with those reported in the literature.1 However, we observed a 13C NMR signal at δ = 194.1 ppm for C5, whereas the reported chemical shift for the proposed C5 epoxy-hemiacetal in the originally assigned structure was δ = 116.4 ppm. Therefore, a detailed re-examination of the authentic carbon NMR spectrum (obtained from Professor W. G. Kim) of natural viridicatumtoxin B was conducted. Indeed, the signal near δ = 194 ppm was observable in the authentic spectrum of the natural product. We assume that, due in part to the minute amounts of this complex natural product isolated, an erroneous interpretation of the HMBC spectrum seemingly supported the original structural assignment (see the Supporting Information for more details and copies of the authentic spectra). We therefore revised the originally proposed structure of viridicatumtoxin B (1′)1 to that shown in Scheme 37 (i.e., 1). Furthermore, our synthetic material crystallized from CH2Cl2/EtOH in suitable form for X-ray crystallographic analysis [mp = 245–247 °C (decomp)] and proved unambiguously its structure as shown in 1 (see ORTEP representation, Figure 6).55
Figure 6.
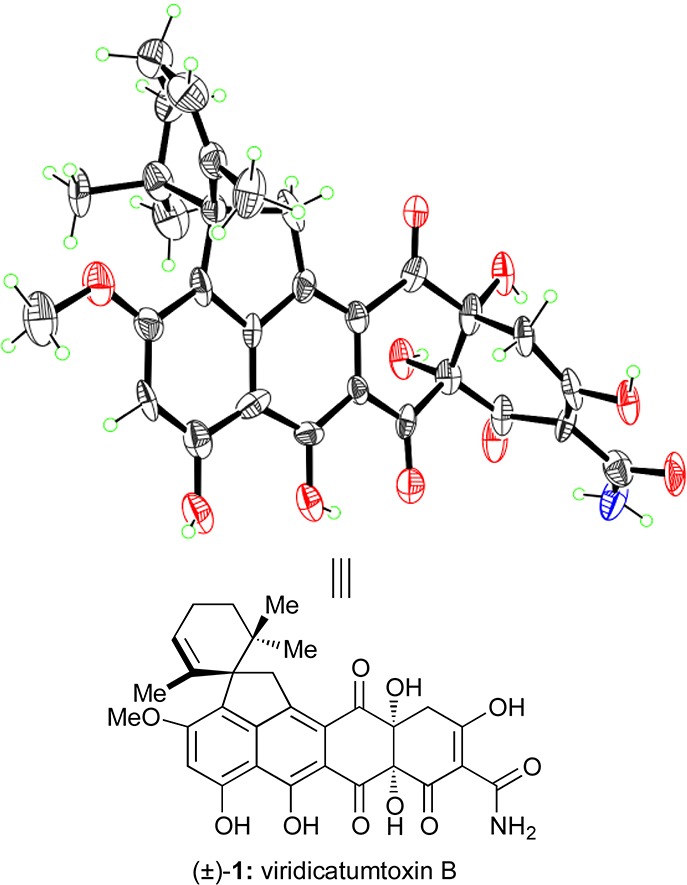
X-ray-derived ORTEP representation of synthetic viridicatumtoxin B (1). Thermal ellipsoids at 30% probability. Gray = carbon, blue = nitrogen, red = oxygen, green = hydrogen.
Biological Evaluation of Synthetic (±)-Viridicatumtoxin B and Analogues
Employing the developed synthetic technologies in this research program, we were able to access not only (±)-viridicatumtoxin B [(±)-1] but also a number of analogues that are simpler and easier to synthesize for biological evaluation (see Chart 3 for structures). Specifically, analogues (±)-V2, (±)-V3, (±)-V4, (±)-V5, and (±)-V6, all lacking the C4a hydroxyl group so cumbersome to install, were synthesized (see Supporting Information for their synthesis) and, together with (±)-1, were tested against a number of bacterial strains and compared to natural viridicatumtoxin B [(+)-1, reported values1], natural viridicatumtoxin A [(+)-2, obtained from Professor Yi Tang], minocycline (Minocin, 7), and tigecycline (Tygacil, 9) (see Chart 3 for structures).
Chart 3. Molecular Structures of Natural Viridicatumtoxin A (2), Synthetic Viridicatumtoxin B (1), Synthesized Viridicatumtoxin Analogues (V2–V6), and Tetracycline Drugs Minocycline (7) and Tigecycline (9).

As shown in Table 1, all of the viridicatumtoxins and analogues tested exhibited antibacterial efficacy against Gram-positive bacteria [(E. faecalis S613, E. faecium 501, and methicillin-resistant Staphylococcus aureus 371 (MRSA 371)] but were largely inactive against Gram-negative bacteria (i.e., A. baumannii AB210). Thus, synthetic viridicatumtoxin B [(±)-1] exhibited comparable antibacterial properties against these strains (E. faecalis S613, E. faecium 501, and MRSA 371: MIC = 1, 0.5, and 4 μg/mL, respectively) to those reported for natural viridicatumtoxin B [(+)-1] against similar strains (E. faecalis KCTC5191, E. faecium KCTC3122, MRSA CCARM3167: MIC = 2, 0.5, and 0.5 μg/mL, respectively), despite the racemic nature of the former. The potencies of synthetic (±)-1 were also comparable to those reported1 for natural viridicatumtoxin A [(+)-2] against similar strains (see Table 1).
Table 1. Minimum Inhibitory Concentration (MIC) Data of Compounds against Gram-Positive and Gram-Negative Bacteria and Comparison with Selected Literature Data.
| Gram-(+) |
Gram-(−) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| this studya |
ref (1) |
this studya | ref (1) |
||||||
| entry | E. faecalis S613 | E. faecium 501 | MRSA 371 | E. faecalis KCTC5191b | E. faecium KCTC3122b | MRSA CCARM3167b | A. baumannii AB210 | A. calcoaceticus KCTC2357b | E. coli CCARM1356b |
| (−)-7 | 4 | 4 | 2 | – | – | – | 4 | – | – |
| (−)-9 | 0.5 | 0.5 | 1 | – | – | – | 0.5 | – | – |
| (±)-1 | 1 | 0.5 | 4 | 2c | 0.5c | 0.5c | 64 | 1c | >64c |
| (+)-278 | 1 | 1 | 4 | 4 | 1 | 0.25 | 64 | 2 | >64 |
| (±)-V2 | 0.5 | 0.5 | 2 | – | – | – | 64 | – | – |
| (±)-V3 | 4 | 2 | 8 | – | – | – | 64 | – | – |
| (±)-V4 | 4 | 4 | 4 | – | – | – | 64 | – | – |
| (±)-V5 | 1 | 1 | 8 | – | – | – | 64 | – | – |
| (±)-V6 | 0.5 | 0.5 | 2 | – | – | – | 64 | – | – |
Viridicatumtoxin analogue (±)-V2, lacking the C4a hydroxyl group, displayed high potency against the same strains (E. faecalis S613: MIC = 0.5 μg/mL; E. faecium 501: MIC = 0.5 μg/mL; MRSA 371: MIC = 2 μg/mL) leading to the conclusion that this functionality is not necessary for antibacterial activity in this subclass of tetracyclines. Of note is the loss of considerable potency in going from the natural to the opposite C15 configuration [analogues (±)-V3 and (±)-V4] as shown in Table 1. Interestingly, methyl ethers (±)-V5 and (±)-V6, also lacking the C4a hydroxyl moiety, demonstrated potent antibacterial properties against E. faecalis S613 [(±)-V5: MIC = 1 μg/mL; (±)-V6: MIC = 0.5 μg/mL], E. faecium 501 [(±)-V5: MIC = 1 μg/mL; (±)-V6: MIC = 0.5 μg/mL], and MRSA 371 [(±)-V5: MIC = 8 μg/mL; (±)-V6: MIC = 2 μg/mL]. These results further support the conclusion that the C4a hydroxyl group of the viridicatumtoxin analogues is not necessary for biological activity (see Table 1). Finally, despite the previously reported activity of viridicatumtoxins against several Gram-negative bacterial strains,1 our tested compounds were inactive against A. baumannii AB210, consistent with previous reports suggesting that the C4-dimethylamino residue is important for imparting the broad-spectrum activity observed for both minocycline (7) and tigecycline (9).79 Incorporation of such a moiety into the viridicatumtoxin scaffold could expand their antibacterial profile as well as improve their pharmacological properties.79
In preliminary experiments to probe the mode-of-action of viridicatumtoxin analogues, time-kill assays were performed to measure the killing of E. faecalis S613 by viridicatumtoxin A (2) and (±)-V6 alongside tigecycline (9). The motivation for this study was previous reports that tetracycline analogues with an aromatic C-ring [e.g., viridicatumtoxin A (2) and (±)-V6] act via a bactericidal mechanism as opposed to a bacteriostatic one (i.e., inhibition of the bacterial ribosome).80 Our time-kill assays clearly indicated that both viridicatumtoxin A (2) and (±)-V6 act bacteriostatically and not bactericidally (see Supporting Information for additional data).81 Although (±)-V6 did not meet the criterion for bactericidal activity, the ability of (±)-V6 to kill E. faecalis S613 was similar to the clinically used and bacteriostatic antibiotic tigecycline (9). If viridicatumtoxins [i.e., viridicatumtoxin A (2) and (±)-V6] are indeed inhibitors of the bacterial ribosome, as opposed to inhibitors of UPP synthase as suggested by Tomoda and co-workers,3 then based on the structure of tigecycline (9) bound to the Thermus thermophilus ribosome, the C4–C7 positions of viridicatumtoxins are likely ideal sites for further modifications, as those positions do not directly interact with the ribosome.82
Conclusions
In summary, we described herein the evolution of synthetic strategies toward viridicatumtoxin B [(±)-1] that finally led to the development of a viable route for the total synthesis of this intriguing natural product. During the course of our studies, we discovered various chemical transformations and acquired synthetic knowledge expected to prove valuable for the synthesis of other fungal tetracyclines and related compounds. In particular, we delineated tactics and strategic requirements for the installation of the hydroxyl groups at C4a and C12a. Other important findings include the anthrone alkylation/spirocyclization reactions to create the congested EF-spirosystem as well as the Michael–Dieckmann cascade reactions that led to the construction of rings A and C. Most notably, our synthetic efforts led to structural revision and configurational assignment of viridicatumtoxin B. Furthermore, application of the developed synthetic technologies allowed the preparation of a number of simpler analogues of the natural product endowed with antibacterial properties comparable with those of viridicatumtoxins A and B, despite their racemic nature. The described chemistry paves the way for further studies, including an asymmetric synthesis of viridicatumtoxins and the design, synthesis, and biological evaluation of other analogues for potential applications as antibacterial agents.83
Acknowledgments
We thank Drs. D. H. Huang and L. Pasternack for NMR spectroscopic, Drs. G. Siuzdak and C. Pennington for mass spectrometric, and Drs. R. K. Chadha and C. Moore for X-ray crystallographic assistance. We thank Prof. W.-G. Kim for graciously providing scanned NMR spectra of natural viridicatumtoxin B. We additionally thank Prof. Yi Tang (UCLA) for generously providing a sample of natural viridicatumtoxin A. K.C.N. acknowledges financial support from the National Institutes of Health (USA) (grant AI055475), the Skaggs Institute of Research, the Cancer Prevention & Research Institute of Texas (CPRIT), and The Welch Foundation. Fellowships to C.R.H.H. (NSF Graduate Research Fellowship), C.N. (Feodor Lynen Research Fellowship, Alexander von Humboldt Foundation), H.A.I. (Marie-Curie International Outgoing Fellowship, European Commission), and A.E. (Fundación Alfonso Martín Escudero) are gratefully appreciated. Y.S. acknowledges financial support from the National Institutes of Health (USA) (grant AI080714).
Supporting Information Available
Experimental procedures and characterization data for key compounds (pdf and cif files). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Department of Chemistry, Rice University, 6100 Main St., Houston, Texas 77005.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author Contributions
⊥ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zheng C. J.; Yu H. E.; Kim E. H.; Kim W. G. J. Antibiot. 2008, 61, 633–637. [DOI] [PubMed] [Google Scholar]
- Hutchison R. D.; Steyn P. S.; van Rensberg S. J. Toxicol. Appl. Pharmacol. 1973, 24, 507–509. [DOI] [PubMed] [Google Scholar]
- Inokoshi J.; Nakamura Y.; Hongbin Z.; Uchida R.; Nonaka K.; Masuma R.; Tomoda H. J. Antibiot. 2013, 66, 37–41. [DOI] [PubMed] [Google Scholar]
- Duggar B. M. Ann. N.Y. Acad. Sci. 1948, 51, 177–181. [DOI] [PubMed] [Google Scholar]
- Dougherty T. J.; Pucci M. J.. Antibiotic Discovery and Development; Springer: New York, 2012. [Google Scholar]
- Tally F. T.; Ellestad G. A.; Testa R. T. J. Antimicrob. Chemother. 1995, 35, 449–452. [DOI] [PubMed] [Google Scholar]
- Sutcliffe J. A.; O’Brien W.; Fyfe C.; Grossman T. H. Antimicrob. Agents Chemother. 2013, 57, 5548–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra I.; Roberts M. Microbiol. Mol. Biol. R. 2001, 65, 232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breinholt J.; Jensen G. W.; Kjaer A.; Olsen C. E.; Rosendahl C. N. Acta Chem. Scand. 1997, 51, 855–860. [Google Scholar]
- Wong S. M.; Kullnig R.; Dedinas J.; Appell K. C.; Kydd G. C.; Gillum A. M.; Cooper R.; Moore R. J. Antibiot. 1993, 46, 214–221. [DOI] [PubMed] [Google Scholar]
- Ishimaru T.; Tsuboya S.; Saijo T. (Takeda Chem. Ind. Ltd.). JP Patent 06,40995A, March 17, 1993.
- a Kodukula K.; Arcuri M.; Cutrone J. Q.; Hugill R. M.; Lowe S. E.; Pirnik D. M.; Shu Y. Z.; Fernandes P. B.; Seethala R. J. Antibiot. 1995, 48, 1055–1059. [DOI] [PubMed] [Google Scholar]; b Shu Y. Z.; Cutrone J. F. Q.; Klohr S. E.; Huang S. J. Antibiot. 1995, 48, 1060–1065. [DOI] [PubMed] [Google Scholar]
- a Conover L. H.; Korst J. J.; Butler K.; Woodward R. B.; Johnston J. D. J. Am. Chem. Soc. 1962, 84, 3222–3224. [Google Scholar]; b Korst J. J.; Johnston J. D.; Butler K.; Bianco E. J.; Conover L. H.; Woodward R. B. J. Am. Chem. Soc. 1968, 90, 439–457. [Google Scholar]
- Gurevich A. I.; Karapetyan M. G.; Kolosov M. N.; Korobko V. G.; Onoprienko V. V.; Popravko S. A.; Shemyakin M. M. Tetrahedron Lett. 1967, 131–134. [DOI] [PubMed] [Google Scholar]
- a Muxfeldt H.; Hardtman G.; Kathawal F.; Vedejs E.; Mooberry J. B. J. Am. Chem. Soc. 1968, 90, 6534–6536. [DOI] [PubMed] [Google Scholar]; b Muxfeldt H.; Haas G.; Hardtmann G.; Kathawala F.; Mooberry J. B.; Vedejs E. J. Am. Chem. Soc. 1979, 101, 689–701. [DOI] [PubMed] [Google Scholar]
- For a review, see:; a Barton D. H. R. Pure Appl. Chem. 1971, 25, 5–23. [DOI] [PubMed] [Google Scholar]; Barton et al. published a series of “back to back” papers on their work, starting with:; b Barton D. H. R.; Magnus P. D. J. Chem. Soc. C 1971, 2164–2166. [DOI] [PubMed] [Google Scholar]
- Wasserman H. H.; Lu T. J.; Scott A. I. J. Am. Chem. Soc. 1986, 108, 4237–4238. [Google Scholar]
- a Stork G.; Hagedorn A. A. J. Am. Chem. Soc. 1978, 100, 3609–3611. [Google Scholar]; b Stork G.; Yee Y. K. Can. J. Chem. 1984, 62, 2627–2628. [Google Scholar]; c Stork G.; LaClair J. J.; Spargo P.; Nargund R. P.; Totah N. J. Am. Chem. Soc. 1996, 118, 5304–5305. [Google Scholar]
- Tatsuta K.; Yoshimoto T.; Gunji H.; Okado Y.; Takahashi M. Chem. Lett. 2000, 646–647. [Google Scholar]
- a Charest M. G.; Siegel D. R.; Myers A. G. J. Am. Chem. Soc. 2005, 127, 8292–8293. [DOI] [PubMed] [Google Scholar]; b Charest M. G.; Lerner C. D.; Brubaker J. D.; Siegel D. R.; Myers A. G. Science 2005, 308, 395–398. [DOI] [PubMed] [Google Scholar]; c Brubaker J. D.; Myers A. G. Org. Lett. 2007, 9, 3523–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sun C. X.; Wang Q.; Brubaker J. D.; Wright P. M.; Lerner C. D.; Noson K.; Charest M. G.; Siegel D. R.; Wang Y. M.; Myers A. G. J. Am. Chem. Soc. 2008, 130, 17913–17927. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kummer D. A.; Li D. R.; Dion A.; Myers A. G. Chem. Sci. 2011, 2, 1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wright P. M.; Myers A. G. Tetrahedron 2011, 67, 9853–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wzorek J. S.; Knopfel T. F.; Sapountzis I.; Evans D. A. Org. Lett. 2012, 14, 5840–5843. [DOI] [PubMed] [Google Scholar]
- a Kabuto C.; Silverton J. V.; Akiyama T.; Sankawa U.; Hutchison R. D.; Steyn P. S.; Vleggaar R. J. Chem. Soc., Chem. Commun. 1976, 728–729. [Google Scholar]; b Silverton J. V.; Kabuto C.; Akiyama T. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1982, 38, 3032–3037. [Google Scholar]
- a De Jesus A. E.; Hull W. E.; Steyn P. S.; Van Heerden F. R.; Vleggaar R. J. Chem. Soc., Chem. Commun. 1982, 902–904. [Google Scholar]; b Horak R. M.; Maharaj V. J.; Marais S. F.; Van Heerden F. R.; Vleggaar R. J. Chem. Soc., Chem. Commun. 1988, 1562–1564. [Google Scholar]
- Chooi Y. H.; Cacho R.; Tang Y. Chem. Biol. 2010, 17, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chooi Y. H.; Wang P.; Fang J. X.; Li Y.; Wu K.; Wang P.; Tang Y. J. Am. Chem. Soc. 2012, 134, 9428–9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chooi Y. H.; Hong Y. J.; Cacho R. A.; Tantillo D. J.; Tang Y. J. Am. Chem. Soc. 2013, 135, 16805–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama N.; Inokoshi J.; Tomoda H. Molecules 2013, 18, 204–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Nilewski C.; Hale C. R. H.; Ioannidou H. A.; ElMarrouni A.; Koch L. G. Angew. Chem., Int. Ed. 2013, 52, 8736–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dufraisse C.; Rigaudy J.; Basselier J.-J.; Cuong N. K. C. R. Acad. Sci. 1965, 260, 5031–5036. [Google Scholar]; b Rigaudy J. Pure Appl. Chem. 1968, 16, 169–186. [Google Scholar]; c Rigaudy J.; Deletang C.; Basselier J.-J. C. R. Acad. Sci. Paris 1969, 268, 344–347. [Google Scholar]; d Rigaudy J.; Sparfel D. Bull. Chem. Soc. Fr. 1972, 3441–3446. [Google Scholar]; e Rigaudy J.; Sparfel D. Tetrahedron 1978, 34, 113–121. [Google Scholar]; f Sparfel D.; Gobert F.; Rigaudy J. Tetrahedron 1980, 36, 2225–2235. [Google Scholar]; g Santamaria J.; Rigaudy J. Tetrahedron 1980, 36, 2453–2457. [Google Scholar]; h Rigaudy J.; Scribe P.; Brelière C. Tetrahedron 1981, 37, 2585–2593. [Google Scholar]; i Rigaudy J.; Caspar A.; Lachgar M.; Maurette D.; Chassagnard C. Bull. Chem. Soc. Fr. 1992, 129, 16–24. [Google Scholar]; j Rigaudy J.; Lachgar M.; Saad M. M. A. Bull. Chem. Soc. Fr. 1994, 131, 177–187. [Google Scholar]; k Rigaudy J.; Lachgar M.; Caspar A.; Chassagnard C. Bull. Chem. Soc. Fr. 1996, 133, 481–490. [Google Scholar]; l Rigaudy J.; Lachgar M. Tetrahedron Lett. 1997, 38, 2267–2270. [Google Scholar]
- For cobalt-mediated endoperoxide rearrangements, see:Boyd J. D.; Foote C. S.; Imagawa D. K. J. Am. Chem. Soc. 1980, 102, 3641–3642. [Google Scholar]
- Other iron species are capable of inducing these rearrangements, see:Kamata M.; Satoh C.; Kim H.-S.; Wataya Y. Tetrahedron Lett. 2002, 43, 8313–8317. [Google Scholar]
- For reviews on endoperoxide rearrangements, see:; a Waldemar A.; Balci M. Tetrahedron 1980, 36, 833–858. [Google Scholar]; b Balci M. Chem. Rev. 1981, 81, 90–108. [Google Scholar]
- Kadás I.; Árvai G.; Horváth G. Org. Prep. Proced. 1998, 30, 79–85. [Google Scholar]
- Tietze L. F.; Singidi R. R.; Gericke K. M. Chem.—Eur. J. 2007, 13, 9939–9947. [DOI] [PubMed] [Google Scholar]
- Savard J.; Brassard P. Tetrahedron 1984, 40, 3455–3464. [Google Scholar]
- For selected examples, see ref (34) andGrunwell J. R.; Karipides A.; Wigal C. T.; Heinzman S. W.; Parlow J.; Surso J. A.; Clayton L.; Fleitz F. J.; Daffner M.; Stevens J. E. J. Org. Chem. 1991, 56, 91–95. [Google Scholar]
- Cameron D. W.; Raverty W. D. Aust. J. Chem. 1976, 29, 1523–1533. [Google Scholar]
- a Schaltegger A.; Steiger W. Arch. Pharm. 1986, 319, 575–576. [Google Scholar]; b Müller K.; Gürster D.; Piwek S.; Wiegrebe W. J. Med. Chem. 1993, 36, 4099–4107. [DOI] [PubMed] [Google Scholar]
- Thoma K.; Holzmann C. Eur. J. Pharm. Biopharm. 1998, 46, 201–208. [DOI] [PubMed] [Google Scholar]
- a Stoll A.; Becker B.; Helfenstein A. Helv. Chem. Acta. 1950, 33, 313–316. [Google Scholar]; b Geiger K. Chem. Ber. 1974, 107, 2976–2984. [Google Scholar]; See also:; c Müller K.; Eibler E.; Mayer K. K.; Wiegrebe W.; Klug G. Arch. Pharm. 1986, 319, 2–9. [Google Scholar]; d Yamaguchi M.; Hasebe K.; Uchida M.; Higashi H.; Minami T. Bull. Chem. Soc. Jpn. 1989, 62, 2745–2747. [Google Scholar]; e Kuhnert N.; Molod H. Y. Tetrahedron Lett. 2005, 46, 7571–7573. [Google Scholar]
- a Davis B. R.; Hinds M. G.; Johnson S. J. Aust. J. Chem. 1985, 38, 1815–1825. [Google Scholar]; b Namba K.; Yamamoto H.; Sasaki I.; Mori K.; Imagawa H.; Mugio N. Org. Lett. 2008, 10, 1767–1770. [DOI] [PubMed] [Google Scholar]; c Yadav J. S.; Thirupathaiah B.; Al Khazim AlGhamdi A. Eur. J. Org. Chem. 2012, 2072–2076. [Google Scholar]; d Wang J.; Zhang L.; Jing Y.; Huang W.; Zhou X. Tetrahedron Lett. 2009, 50, 4978–4982. [Google Scholar]; See also:; e Pittman C. U. Jr.; Miller W. G. J. Am. Chem. Soc. 1973, 95, 2947–2956. [Google Scholar]; f Miller W. G.; Pittman C. U. Jr. J. Org. Chem. 1974, 39, 1955–1956. [Google Scholar]; g Gassman P. G.; Ray J. A.; Wenthold P. G.; Mickelson J. W. J. Org. Chem. 1991, 56, 5143–5146. [Google Scholar]
- a Ma S.; Zhang J. Tetrahedron 2003, 59, 6273–6282. [Google Scholar]; b Werle S.; Fey T.; Neudörfl J. M.; Schmalz H.-G. Org. Lett. 2007, 9, 3555–3558. [DOI] [PubMed] [Google Scholar]; c Liu H.; Wang Y.-H.; Zhu L.-L.; Li X.-X.; Zhou W.; Chen Z.; Hu W.-X. Tetrahedron Lett. 2011, 52, 2990–2993. [Google Scholar]
- Moriarty R. M.; Prakash O. Org. React. 2004, 327–415. [Google Scholar]
- a Meyer K. H. Justus Liebigs Ann. Chem. 1911, 379, 37–78. [Google Scholar]; b Thomson R. H. Q. Rev. Chem. Soc. 1956, 10, 27–43. [Google Scholar]; c Kündig E. P.; Enríquez García A.; Lomberget T.; Bernardinelli G. Angew. Chem., Int. Ed. 2006, 45, 98–101. [DOI] [PubMed] [Google Scholar]; d Reviriego F.; Alkorta I.; Elguero J. J. Mol. Struct. 2008, 891, 325–328. [Google Scholar]
- Bauta W. E.; Lovett D. P.; Cantrell W. R. Jr.; Burke B. D. J. Org. Chem. 2003, 68, 5967–5973. [DOI] [PubMed] [Google Scholar]
- Pelter A.; Elgendy S. Tetrahedron Lett. 1988, 29, 677–680. [Google Scholar]
- Rathke M. W.; Cowan P. J. J. Org. Chem. 1985, 50, 2622–2624. [Google Scholar]
- Kornblum N.; Smiley R. A.; Blackwood R. K.; Iffland D. C. J. Am. Chem. Soc. 1955, 77, 6269–6280. [Google Scholar]
- Nicolaou K. C.; Estrada A. A.; Zak M.; Lee S. H.; Safina B. S. Angew. Chem., Int. Ed. 2005, 44, 1378–1382. [DOI] [PubMed] [Google Scholar]
- a Tamura Y.; Wada A.; Kita Y. Tetrahedron Lett. 1981, 22, 4283–4286. [Google Scholar]; For a review on cyclic anhydrides in cycloaddition reactions, see:; b González-López M.; Shaw J. T. Chem. Rev. 2009, 109, 164–189. [DOI] [PubMed] [Google Scholar]
- a Ohta Y.; Hirose Y. Chem. Lett. 1972, 263–266. [Google Scholar]; For a discussion on the role of the counterion in cationic cyclizations, see:; b Pronin S. V.; Shenvi R. A. Nat. Chem. 2012, 4, 915–920. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S.; Nedachi M.; Yokoyama H.; Hirai Y. Tetrahedron Lett. 1999, 40, 7363–7365. [Google Scholar]
- a Raczyńska E. D.; Kosińska W. Chem. Rev. 2005, 105, 3561–3612. [DOI] [PubMed] [Google Scholar]; b Cyrański M. K. Chem. Rev. 2005, 105, 3773–3811. [DOI] [PubMed] [Google Scholar]; c Korth H.-G.; Mulder P. J. Org. Chem. 2013, 78, 7674–7682. [DOI] [PubMed] [Google Scholar]
- Barton D. H. R.; Ley S. V.; Magnus P. D.; Rosenfeld M. N. J. Chem. Soc., Perkin Trans. 1 1977, 567–572. [PubMed] [Google Scholar]
- Crystallographic data for compounds 60 (CCDC 941202), 15-epi-133 (CCDC 941203), and 1 (945867) have previously been reported by us (see ref (28)). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- For the sake of clarity, and because fragment 111 is racemic, we elected to define the newly generated stereocenters at C4 and C4a as those shown in Scheme 20, which results in the configuration at C15 appearing as a mixture (hence 15-epi). Both diastereomers shown are formed along with their enantiomeric counterparts.
- Kahne D. E. Ph.D. Thesis, Columbia University, 1986. [Google Scholar]
- a White J. D.; Nolen E. G. Jr.; Miller C. H. J. Org. Chem. 1986, 51, 1150–1152. [Google Scholar]; b White J. D.; Demnitz F. W. J.; Xu Q.; Martin W. H. C. Org. Lett. 2008, 10, 2833–2836. [DOI] [PubMed] [Google Scholar]
- Fitzjarrald V. P.; Pongdee R. Tetrahedron Lett. 2007, 48, 3553–3557. [Google Scholar]
- Alternative batches of material displayed varying diastereomeric ratios ranging from 1:1 to 3:1, favoring the desired C15 spirocycle configuration, albeit in reduced yield, indicating some levels of diastereomeric enhancement during chromatography.
- Gioeli C.; Balgobin N.; Josephson S.; Chattopadhyaya J. B. Tetrahedron Lett. 1981, 22, 969–972. [Google Scholar]
- a Corey E. J.; Yang Y. K. Tetrahedron Lett. 1992, 33, 2289–2290. [Google Scholar]; b Nicolaou K. C.; Reddy K. R.; Skokotas G.; Sato F.; Xiao X. Y.; Hwang C. K. J. Am. Chem. Soc. 1993, 115, 3558–3575. [Google Scholar]
- Denmark S. E.; Kobayashi T.; Regens C. S. Tetrahedron 2010, 66, 4745–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürstner A.; Weintritt H. J. Am. Chem. Soc. 1998, 120, 2817–2825. [Google Scholar]
- Adam W.; Smerz A. K. Tetrahedron 1996, 52, 5799–5804. [Google Scholar]
- Davis F. A.; Stringer O. D. J. Org. Chem. 1982, 47, 1774–1775. [Google Scholar]
- a Ikota N.; Ganem B. J. Org. Chem. 1978, 43, 1607–1608. [Google Scholar]; b Clarembeau M.; Cravador A.; Dumont W.; Hevesi L.; Krief A.; Lucchetti J.; Van Ende D. Tetrahedron 1985, 41, 4793–4812. [Google Scholar]; c Krief A.; Hobe M.; Badaoui E.; Bousbaa J.; Dumont W.; Nazih A. Synlett 1993, 707–709. [Google Scholar]; d Santi C.; Santoro S.; Testaferri L.; Tiecco M. Synlett 2008, 1471–1474. [Google Scholar]
- a Donohoe T. J.; Blades K.; Moore P. R.; Waring M. J.; Winter J. J. G.; Helliwell M.; Newcombe N. J.; Stemp G. J. Org. Chem. 2002, 67, 7946–7956. [DOI] [PubMed] [Google Scholar]; b Donohoe T. J.; Bataille C. J. R. Org. React. 2012, 1–48. [Google Scholar]
- Phillips S. T.; Shair M. D. J. Am. Chem. Soc. 2007, 129, 6589–6598. [DOI] [PubMed] [Google Scholar]
- Schadt F. L.; Bentley T. W.; Schleyer P. v. R. J. Am. Chem. Soc. 1976, 98, 7667–7674. [Google Scholar]
- Experimental evidence has been reported that supports the existence of a transient, ring-opened intermediate. See:; a Hlavka J. J.; Bitha P.; Boothe J. H. J. Am. Chem. Soc. 1968, 90, 1034–1037. [DOI] [PubMed] [Google Scholar]; b Scott A. I.; Yamaguchi E.; Chung S.-K. Tetrahedron Lett. 1975, 16, 1369–1372. [Google Scholar]; For related rearrangements, see:; c Stork G. Chem. Ind. 1955, 915–916. [Google Scholar]; d Barton D. H. R.; Scott A. I. J. Chem. Soc. 1958, 1767–1772. [Google Scholar]
- Miller B. Acc. Chem. Res. 1975, 8, 254–256. [Google Scholar]
- Horiuchi C. A.; Ji S. J.; Matsushita M.; Chai W. Synthesis 2004, 202–204. [Google Scholar]
- Mukaiyama T.; Matsuo J.; Kitagawa H. Chem. Lett. 2000, 29, 1250–1251. [Google Scholar]
- Tatsuta K.; Fukuda T.; Ishimori T.; Yachi R.; Yoshida S.; Hashimoto H.; Hosokawa S. Tetrahedron Lett. 2012, 53, 422–425. [Google Scholar]
- a Köster R.; Seidel G. In Organometallic Syntheses; King R. B.; Eisch J. J., Eds.; Elsevier: Amsterdam, 1998; Vol. 4, pp 440–442. [Google Scholar]; b Fürstner A.; Seidel G. J. Org. Chem. 1997, 62, 2332–2336. [DOI] [PubMed] [Google Scholar]
- Gibert M.; Ferrer M.; Sánchez-Baeza F.; Messeguer A. Tetrahedron 1997, 53, 8643–8650. [Google Scholar]
- Professor Yi Tang (UCLA) is acknowledged for providing natural viridicatumtoxin A [(+)-2] obtained from P. aethiopicum. In this study, that material was used for antibacterial testing. For the comparison data, compound (+)-2 was isolated by Kim and co-workers from Penicillium sp. FR11 (see ref (1)).
- Nelson M.; Hillen W.; Greenwald R. A.. Tetracyclines in Biology, Chemistry, and Medicine; Birkhauser Verlag: Basel, 2001; p 26. [Google Scholar]
- a Oliva B.; Gordon G.; McNicholas P.; Ellestad G.; Chopra I. Antimicrob. Agents Chemother. 1992, 36, 913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chopra I. Antimicrob. Agents Chemother. 1994, 38, 637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Clinical and Laboratory Standards Institute (CLSI) defines a bactericidal compound as one capable of inducing a ≥ 3 log10 drop in colony-forming units (CFU)/mL.
- Jenner L.; Starosta A. L.; Terry D. S.; Mikolajka A.; Filonava L.; Yusupov M.; Blanchard S. C.; Wilson D. N.; Yusupova G. Proc. Natl. Acad. Sci., U. S. A. 2013, 110, 3812–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review on the role of chemical synthesis in antibacterial drug discovery, see:Wright P. M.; Seiple I. B.; Myers A. G. Angew. Chem., Int. Ed. 2014, 10.1002/anie.201310843. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.