

Abstract
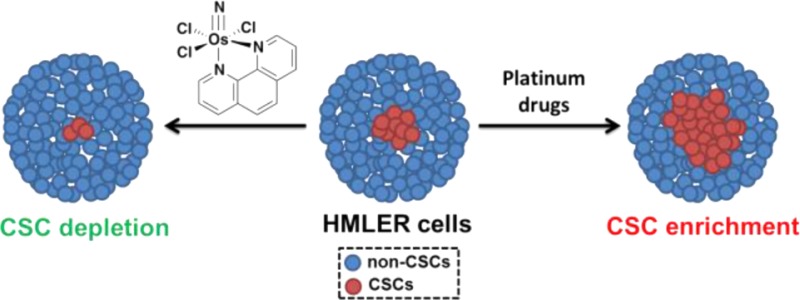
The effect of a newly developed osmium(VI) nitrido complex, 1, on breast cancer stem cells (CSCs) is reported. The complex displays selective toxicity for HMLER breast cancer cells enriched with CD44-positive, CSC-like cells over the same cells having reduced CSC character. Remarkably, 1 also reduces the proportion of CSCs within a heterogeneous breast cancer cell population and irreversibly inhibits the formation of free-floating mammospheres to an extent similar to that of salinomycin, a natural product that targets CSCs. Detailed mechanistic studies reveal that in breast cancer cells 1 induces DNA damage and endoplasmic reticulum stress, the latter being responsible for the CSC selectivity. The anti-CSC properties of 1 provide a strong impetus for the development of new metal-based compounds to target CSCs and to treat chemotherapy-resistant and relapsed tumors.
Cancer relapse is strongly linked to the existence of cancer stem cells (CSCs), a small sub-population of tumor cells that have the ability to self-renew, differentiate, and form secondary or tertiary tumors.1 Conventional chemotherapy and radiotherapy are ineffective against CSCs.6 Although current therapies effectively reduce tumor mass by destroying the bulk of cancer cells, they cannot remove CSCs, which persist and generate new tumors, often of a far more aggressive nature. To improve clinical outcomes, treatments must have the ability to kill the entirety of cancer cells, including CSCs. Compounds capable of selectively killing CSCs and disrupting the microenvironments supporting these cells are currently the subject of intense research.10 Several potential CSC therapeutic targets have been identified, such as cell surface markers14 and various deregulated signaling pathways,21 but there is still no clinically approved drug that specifically targets CSCs. A recent high-throughput screen of ∼16,000 compounds, including commercial libraries and collections of natural extracts, found only four members—salinomycin, abamectin, etoposide, and nigericin—to exhibit prominent CSC specificity.25 Thus, there is an urgent need to discover new, selective compounds to add to this limited arsenal of anti-CSC agents.
The quest for new CSC-targeting compounds has been severely hampered by the inability to obtain and sustain CSC-rich cell cultures.26 A recent study has shown, however, that enriched populations of CSCs can be achieved by modifying HMLER breast cancer cells through short hairpin RNA (shRNA)-mediated inhibition of the CDH1 gene, which encodes E-cadherin.27 Another study by the same group demonstrated that CSC-enriched cultures could be generated by treating HMLER cells with non-CSC-specific compounds, such as paclitaxel and staurosporine.25 The latter approach relies on the ability of the non-CSC-specific agents to kill bulk cancer cells, leaving stem-like cells untouched. Here we sought to use these tools to investigate the CSC-targeting ability of metal-based compounds, including the newly developed osmium(VI) nitrido series, 1–3 (Figure 1). We recently reported their synthesis and anti-proliferative properties.28 Encouragingly, 1–3 display selective toxicity for cancer cells over healthy cells and no cross-resistance with cisplatin, a clinically administered anticancer drug. In the current study we examine the anti-CSC activity of 1–3 as well as some well-established Pt(II)- and Pt(IV)-based antineoplastic agents, also illustrated in Figure 1. The Os and Pt compounds were prepared using previously reported methods.28,29 Prior to carrying out cellular studies, the stability of 1, taken as a representative member of the osmium(VI) nitrido series, in MEGM cell culture media was established by UV–vis spectroscopy (Figure S1).
Figure 1.
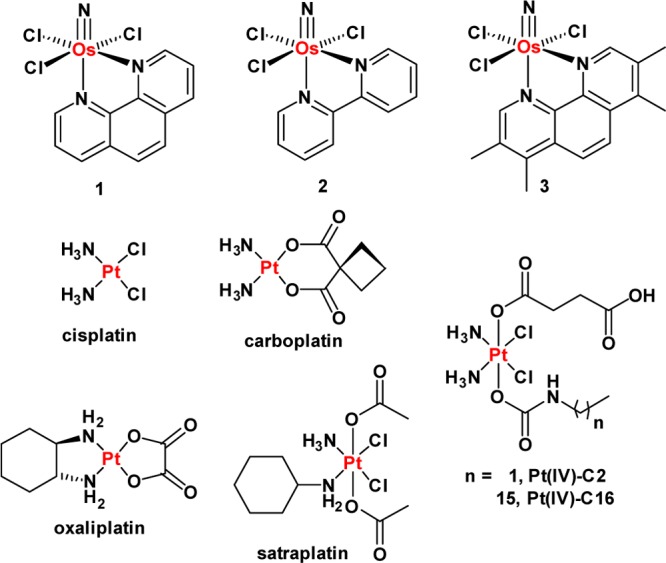
Chemical structures of the platinum(II and IV) and the osmium(VI) nitrido complexes (1–3) under investigation.
Experimentally transformed HMLER breast cancer cells33 were used to assess the CSC specificity of the metal complexes. Under standard cell culture conditions, HMLER cells contain an inherent CSC population of 5–8%. HMLER CSCs overexpress CD44,14 a cell surface glycoprotein involved in cell signaling, adhesion, and migration,34 and thus are defined by a CD44high marker profile. Following reported protocols, CSC-enriched HMLER cells were generated by paclitaxel treatment (10 nM for 4 days; Figure S2).25 This strategy enabled access to CSC-enriched (>30%), CD44high HMLER cells (hereafter referred to as HMLERtax cells).
The anti-proliferative properties of 1–3 against HMLER and HMLERtax cells were assessed using the MTT assay. Identical studies were also performed with compounds known to have CSC-selective potency, such as salinomycin and abamectin. FDA-approved Pt(II) anticancer drugs cisplatin, carboplatin, and oxaliplatin,35 and Pt(IV) pro-drugs such as satraplatin37 and the recently developed fatty-acid mimics Pt(IV)-C2 and Pt(IV)-C16,32 were also investigated. The IC50 values (concentration required to induce 50% inhibition) were derived from dose–response curves (Figures 2A and S3–S12) and are summarized in Table 1. Osmium complexes 1–3 displayed micromolar toxicity toward both cell lines. Moreover, the 1,10-phenanthroline-bearing complex 1 exhibited selective toxicity for HMLERtax cells over HMLER cells (2.3-fold). Salinomycin and abamectin also killed HMLERtax cells preferentially over HMLER cells (8.3- and 2.1-fold, respectively). None of the Pt-based agents showed CSC selectivity. In fact, oxaliplatin, satraplatin, and Pt(IV)-C16 exhibited 2–4.5-fold greater toxicity for HMLER cells over HMLERtax cells (Figures S9, S10, and S12). This result is consistent with the tendency of Pt compounds to induce CSC enrichment rather than CSC depletion (vide infra). Overall, the anti-proliferative data suggest that 1 can selectively reduce the viability of CSC-enriched HMLERtax cells over CSC-depleted HMLER cells, in the same order of magnitude as salinomycin and abamectin, two of the most selective CSC-targeting compounds identified to date. Although salinomycin displays better selectivity for CSCs than 1, 1 exhibits a larger toxicity differential (the concentration difference between the IC50 values for HMLER and HMLERtax cells). These properties are highly desirable for selecting CSC drug candidates in preclinical studies.
Figure 2.
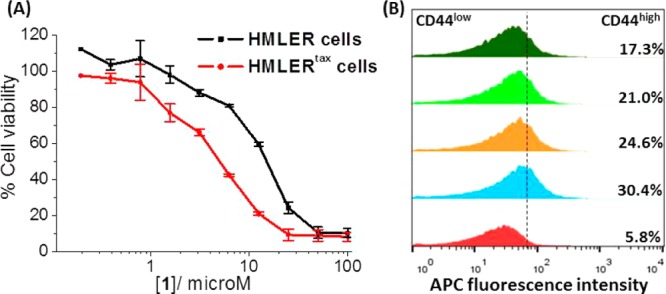
(A) Average dose–response curves for the treatment of HMLER and HMLERtax cells with 1 (n = 30 for each point). (B) Representative histograms displaying the red fluorescence emitted by anti-CD44 APC antibody-stained HMLER cells (red), HMLERtax cells (blue), and HMLERtax cells treated with 1 (5 μM, orange; 10 μM, light green; 20 μM, dark green) for 4 days followed by 4 days recovery in compound-free MEGM media.
Table 1. IC50 Values of Tested Compounds against HMLER and HMLERtax Cells.
| compound | HMLER IC50 (μM) | HMLERtax IC50 (μM) | selectivity for HMLERtax a |
|---|---|---|---|
| 1 | 11.20 ± 0.48 | 4.91 ± 0.86 | 2.31 |
| 2 | 14.58 ± 0.20 | 16.06 ± 4.12 | 0.91 |
| 3 | 82.80 ± 18.43 | 53.99 ± 2.45 | 1.53 |
| salinomycin | 0.49 ± 0.26 | 0.058 ± 0.01 | 8.45 |
| abamectin | 1.45 ± 0.18 | 0.64 ± 0.06 | 2.26 |
| cisplatin | 1.95 ± 0.40 | 2.06 ± 0.67 | 0.95 |
| carboplatin | 17.84 ± 0.58 | 18.19 ± 0.80 | 0.98 |
| oxaliplatin | 15.04 ± 0.41 | 26.95 ± 4.42 | 0.55 |
| satraplatin | 1.22 ± 0.06 | 2.87 ± 0.23 | 0.43 |
| Pt(IV)-C2 | 39.09 ± 9.82 | 40.64 ± 9.91 | 0.96 |
| Pt(IV)-C16 | 0.0254 ± 0.0016 | 0.1131 ± 0.0197 | 0.22 |
Selectivity = IC50 for HMLER/IC50 for HMLERtax. The values reported are an average of five independent determinations.
To determine the effect of the metal complexes on the heterogeneity of breast cancer cells, flow cytometric studies were carried out. Upon treatment of HMLERtax cells with 1 (5–20 μM for 4 days), a dose-dependent decrease in the proportion of CD44high cells was observed, indicative of CSC-specific toxicity (Figure 2B). A slight decrease in the CD44high population was also observed upon incubation of HMLER cells with increasing concentrations of 1 (5–40 μM for 4 days; Figure S13). Taken together, the results demonstrate that 1 can selectively kill CD44high CSC-like cells over bulk cancer cells. HMLERtax cells treated with clinically approved Pt(II) anticancer drugs35 cisplatin (1.5 μM for 4 days), carboplatin (15 μM for 4 days), and oxaliplatin (15 μM for 4 days) displayed little change in the fraction of cells with CD44high character compared to the untreated control (Figure S14). Moreover, upon treatment of HMLER cells with the Pt drugs under the same conditions described above, a marked increase in the relative CD44high population was detected (3.2–5.4-fold), consistent with CSC enrichment (Figure S15). These findings highlight the partiality of conventional Pt(II)-based anticancer drugs to kill bulk cancer cells over CSCs. In the same way as the Pt(II) compounds, satraplatin treatment (1.5 μM for 4 days) did not alter the CSC proportion in HMLERtax cells and propagated CSC enrichment in HMLER cells (Figures S14 and S15), consistent with non-CSC specificity.
Owing to their unlimited self-renewal ability, breast CSCs have the tendency to form de novo tumor-like structures called mammospheres in non-adherent, serum-free cell cultures.38 The tumor sphere formation assay was used to assess the ability of 1, salinomycin, paclitaxel, cisplatin, carboplatin, oxaliplatin, and satraplatin (at their respective IC30 values) to inhibit mammosphere formation from HMLER single-cell suspensions. To obtain 3D images of the mammospheres and determine the proportion of CD44-positive cells within a given mammosphere, the cells were stained with Hoechst 33258 dye (7.5 μM for 30 min) and allophycocyanin (APC)-labeled anti-CD44 antibody (15 μL, 1:133 dilution for 45 min), respectively, and imaged using a fluorescence microscope (Figure S16). The tumorsphere formation assay showed that treatment with 1 induced a 38% decrease in the number of mammospheres formed relative to the untreated control, providing strong evidence for the inhibition of CSC self-renewal (Figure 3). The microscopy studies revealed that the size of the spheroids decreased by up to 2.4-fold in the presence of 1 (Figure 3). Furthermore, upon incubation with 1, the proportion of CD44-positive cells within a given mammosphere was markedly diminished, indicative of CSC-specific toxicity (Figure S16). Collectively, these data show that 1 inhibits the clonogenic growth of HMLER mammospheres by eliminating CD44-positive, CSC-like cells. As expected, a reduction in both mammosphere number and size (up to 1.4-fold) was observed for salinomycin treatment (positive control, Figure 3). In contrast, paclitaxel, carboplatin, and satraplatin treatment led to an increase in the number of mammospheres formed (10–40%), suggestive of CSC enrichment (Figure 3). This result was corroborated by a 2–4-fold increase in the number of CD44-positive cells within the mammospheres (Figure S16). The number of mammospheres formed was marginally reduced by cisplatin and oxaliplatin treatment, but the size of the spheroids remained largely unaltered (Figure 3).
Figure 3.
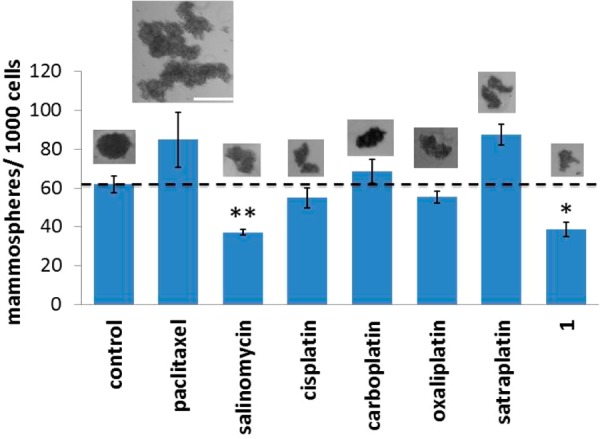
Quantification of mammosphere formation with HMLER cells untreated and treated with the investigated compounds at their respective IC30 values for 5 days. Representative bright-field images (×4) of the mammospheres formed under each condition are presented to scale. Scale bar = 0.3 mm. Student t test, p < 0.05 or p < 0.01. Error bars represent standard deviations.
To determine whether 1 could induce a durable mammosphere inhibitory response, 1-treated primary mammospheres were dissociated into single-cell suspensions, and their propensity to form secondary mammospheres was assessed (Figure S17). Control studies were also conducted with salinomycin- and paclitaxel-treated primary mammospheres. The secondary mammospheres formed from cells isolated from 1-treated primary mammospheres were 5-fold fewer than those from the untreated control. This result shows that 1 inhibits the self-renewal of HMLER mammospheres and that this effect is maintained upon serial passage. Cells extracted from salinomycin-treated primary mammospheres displayed non-clonogenic properties, similar to those observed for 1-treated cells. Single-cell suspensions of paclitaxel-treated primary mammospheres, on the other hand, produced slightly more secondary mammospheres compared to the untreated control.
To account for the CSC-specificity and mammosphere potency observed for 1, detailed mechanistic studies were conducted. The Os complex 1 was previously characterized via an RNAi signature approach39 capable of discerning drug mechanism of action.28 Although 1 did not resemble any category of drug mechanism present in the reference set, the pleiotropic mechanism of 1-induced cell death appeared to involve DNA damage. Given the nature of other Os compounds in eliciting endoplasmic reticulum (ER) stress,28 we added known ER stress inducers tunicamycin and thapsigargin to the RNAi signature training set (Figure 4A). Upon re-analysis of the RNAi signatures, we found that 1 still did not belonging to any training set category. Thus, we next examined the signatures of 1, ER stress inducers tunicamycin and thapsigargin, and representative DNA-cross-linkers cisplatin, carboplatin, and chlorambucil via principal components analysis (PCA). PCA represents the variance of a multi-dimensional data set in successive principal components where each component represents a larger portion of the data set variance than the next. By plotting the aforementioned compounds via PCA, we found that 1 lies roughly equidistant between the canonical ER stress inducers and DNA-cross-linkers (Figure 4B). This result indicated that 1 can induce cell death via both mechanisms.
Figure 4.
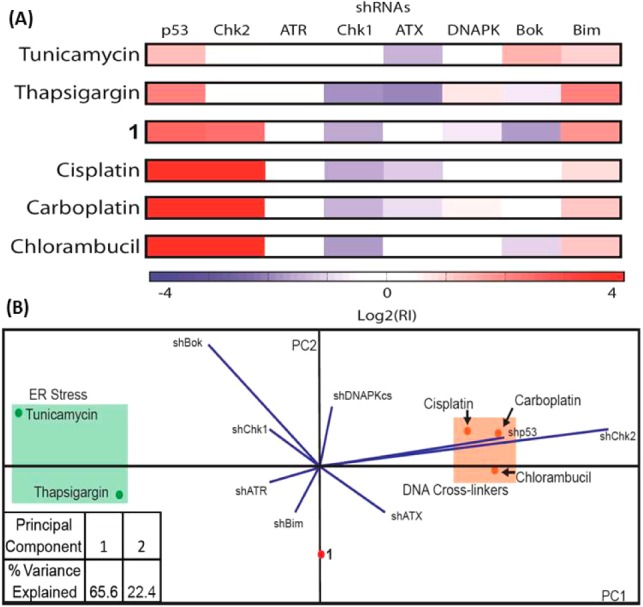
(A) RNAi signatures derived from the treatment of Eμ-Mycp19arf–/– lymphoma cells with 1, ER stress inducers tunicamycin and thapsigargin, and DNA-cross-linking agents cisplatin, carboplatin, and chlorambucil at the LD80–90 concentration for each compound. (B) Principal components analysis plot of the RNAi signatures.
Immunoblotting studies were conducted to monitor changes in expression of biomarkers related to the DNA damage and ER stress pathways. HMLER cells incubated with 1 (5–20 μM for 72 h) displayed a marked increase in the expression of the phosphorylated forms of H2AX and CHK2, indicative of DNA damage (Figure S18).40 DNA damage is usually accompanied by p53 accumulation and upregulation of downstream effectors related to cell cycle arrest, DNA repair, and apoptosis.42 Owing to the inactivation of p53 in HMLER cells, however, the expression of p21, a p53 effector, remained unchanged upon treatment with 1(5–20 μM for 72 h; Figure S18). The inability of p53 to coordinate cellular response following DNA damage in HMLER cells suggests that 1-induced DNA damage may not be a major determinant of cell death. HMLER cells dosed with 1 (5–20 μM for 72 h) exhibited an increase in the expression of proteins related to the unfolded protein response, such as phosphorylated eukaryotic initiation factor 2α (eIF2α) and C/EBP homologous protein, suggestive of ER stress (Figure S18).43 Immunofluorescence studies showed that, upon incubation of HMLER cells with 1 (25 μM for 24 h), the expression of the phosphorylated RNA-dependent protein kinase-like endoplasmic reticulum kinase (phospho-PERK) increased. This finding provided further evidence of ER stress (Figure S19).43 A similar result was also observed for HMLER cells treated with thapsigargin (0.25 μM for 24 h; Figure S19). HMLER cells treated with 1 (5–20 μM for 72 h) displayed an increase in the expression of cleaved caspase 3 and 7, and poly-ADP ribose polymerase (Figure S18). Thus, the mode of 1-induced cell death is most likely to be caspase-dependent apoptosis. Taken together, the immunoblotting and immunofluorescence data reveal that in breast cancer cells 1 initiates both DNA damage and ER stress, culminating in apoptotic cell death.
A recent study identified the vulnerability of HMLER CSC-like cells to agents that can induce ER stress through the PERK-eIF2α axis.44 In light of this report and our mechanistic data, we propose that the CSC specificity observed for 1 could be attributed to the ability of the complex to induce ER stress. To investigate this hypothesis, the toxicity of 1 against HMLER and HMLERtax cells in the absence and presence of a known ER stress inhibitor, salubrinal,45 was determined. Co-administration of HMLER and HMLERtax cells with 1 and salubrinal (10 μM) significantly reduced the cytotoxicity of 1 in CSC-enriched HMLERtax cells (t test, p < 0.05) but not in CSC-depleted HMLER cells (t test, p = 0.30; Figure S20). Thus, 1 induces ER stress-mediated cell death in CSCs more readily than in non-CSCs. Overall, our mechanistic studies show that the CSC specificity observed for 1 likely results from the ability of 1 to induce ER stress via the PERK-eIF2α pathway and the sensitivity of HMLER CSCs to ER stress inducers.
In summary, we present the anti-CSC properties of 1. To our knowledge, 1 is the first osmium-based compound to exhibit selective toxicity for breast CSC-enriched cell populations. Encouragingly, the CSC-specific potency of 1 challenges some of the most CSC-selective compounds identified to date. Additionally, 1 inhibits the formation of mammospheres by specifically targeting CD44-positive, CSC-like cells. Given our findings and the urgent medical need for CSC-specific chemotherapies to overcome cancer relapse and metastases formation in the clinic, the anti-CSC properties of 1 are pre-clinically very appealing. Overall, this study highlights the great, largely unexplored potential of metal-based complexes for CSC-directed chemotherapy and provides hints about the mechanism and targets of systemic Os toxicity.
Acknowledgments
This work is supported by the NCI under grant CA034992. K.S. is supported by a Misrock Fellowship. We thank Prof. Robert Weinberg for kindly making available the HMLER cells and members of his laboratory for useful suggestions. We are also grateful to Prof. Piyush Gupta for some experimental suggestions.
Supporting Information Available
Experimental techniques and data concerning all biophysical and cellular studies. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Gupta P. B.; Chaffer C. L.; Weinberg R. A. Nat. Med. 2009, 15, 1010. [DOI] [PubMed] [Google Scholar]; b Nguyen L. V.; Vanner R.; Dirks P.; Eaves C. J. Nat. Rev. Cancer 2012, 12, 133. [DOI] [PubMed] [Google Scholar]; c Reya T.; Morrison S. J.; Clarke M. F.; Weissman I. L. Nature 2001, 414, 105. [DOI] [PubMed] [Google Scholar]; d Visvader J. E.; Lindeman G. J. Nat. Rev. Cancer 2008, 8, 755. [DOI] [PubMed] [Google Scholar]
- a Dean M.; Fojo T.; Bates S. Nat. Rev. Cancer 2005, 5, 275. [DOI] [PubMed] [Google Scholar]; b Maugeri-Sacca M.; Vigneri P. G.; De Maria R. Clin. Cancer Res. 2011, 17, 4942. [DOI] [PubMed] [Google Scholar]; c Rich J. N. Cancer Res. 2007, 67, 8980. [DOI] [PubMed] [Google Scholar]
- a Chen K.; Huang Y. H.; Chen J. L. Acta Pharmacol. Sin. 2013, 34, 732. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lum C. T.; Wong A. S.; Lin M. C.; Che C. M.; Sun R. W. Chem. Commun. 2013, 49, 4364. [DOI] [PubMed] [Google Scholar]; c Ning X.; Shu J.; Du Y.; Ben Q.; Li Z. Cancer Biol. Ther. 2013, 14, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Al-Hajj M.; Wicha M. S.; Benito-Hernandez A.; Morrison S. J.; Clarke M. F. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3983. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Eramo A.; Lotti F.; Sette G.; Pilozzi E.; Biffoni M.; Di Virgilio A.; Conticello C.; Ruco L.; Peschle C.; De Maria R. Cell Death Differ. 2008, 15, 504. [DOI] [PubMed] [Google Scholar]; c Li C.; Heidt D. G.; Dalerba P.; Burant C. F.; Zhang L.; Adsay V.; Wicha M.; Clarke M. F.; Simeone D. M. Cancer Res. 2007, 67, 1030. [DOI] [PubMed] [Google Scholar]; d Prince M. E.; Sivanandan R.; Kaczorowski A.; Wolf G. T.; Kaplan M. J.; Dalerba P.; Weissman I. L.; Clarke M. F.; Ailles L. E. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Singh S. K.; Hawkins C.; Clarke I. D.; Squire J. A.; Bayani J.; Hide T.; Henkelman R. M.; Cusimano M. D.; Dirks P. B. Nature 2004, 432, 396. [DOI] [PubMed] [Google Scholar]
- a Janikova M.; Skarda J. Neoplasma 2012, 59, 6. [DOI] [PubMed] [Google Scholar]; b Korkaya H.; Paulson A.; Charafe-Jauffret E.; Ginestier C.; Brown M.; Dutcher J.; Clouthier S. G.; Wicha M. S. PLoS Biol. 2009, 7, e1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Takebe N.; Harris P. J.; Warren R. Q.; Ivy S. P. Nat. Rev. Clin. Oncol. 2011, 8, 97. [DOI] [PubMed] [Google Scholar]
- Gupta P. B.; Onder T. T.; Jiang G.; Tao K.; Kuperwasser C.; Weinberg R. A.; Lander E. S. Cell 2009, 138, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillmore C. M.; Kuperwasser C. Breast Cancer Res. 2008, 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder T. T.; Gupta P. B.; Mani S. A.; Yang J.; Lander E. S.; Weinberg R. A. Cancer Res. 2008, 68, 3645. [DOI] [PubMed] [Google Scholar]
- Suntharalingam K.; Johnstone T. C.; Bruno P. M.; Lin W.; Hemann M. T.; Lippard S. J. J. Am. Chem. Soc. 2013, 135, 14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Giandomenico C. M.; Abrams M. J.; Murrer B. A.; Vollano J. F.; Rheinheimer M. I.; Wyer S. B.; Bossard G. E.; Higgins J. D. Inorg. Chem. 1995, 34, 1015. [DOI] [PubMed] [Google Scholar]; b Kidani Y.; Inagaki K.; Iigo M.; Hoshi A.; Kuretani K. J. Med. Chem. 1978, 21, 1315. [DOI] [PubMed] [Google Scholar]; c Rochon F. D.; Gruia L. M. Inorg. Chim. Acta 2000, 306, 193. [Google Scholar]; d Zheng Y. R.; Suntharalingam K.; Johnstone T. C.; Yoo H.; Lin W.; Brooks J. G.; Lippard S. J. J. Am. Chem. Soc. 2014, 136, 8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B.; Spirio L.; Koerner F.; Fleming M. D.; Zimonjic D. B.; Donaher J. L.; Popescu N. C.; Hahn W. C.; Weinberg R. A. Genes Dev. 2001, 15, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louderbough J. M. V.; Schroeder J. A. Mol. Cancer Res. 2011, 9, 1573. [DOI] [PubMed] [Google Scholar]
- a Fricker S. P. Dalton Trans. 2007, 4903. [DOI] [PubMed] [Google Scholar]; b Kelland L. Nat. Rev. Cancer 2007, 7, 573. [DOI] [PubMed] [Google Scholar]
- Rose W. C.; Crosswell A. R.; Schurig J. E.; Casazza A. M. Cancer Chemother. Pharmacol. 1993, 32, 197. [DOI] [PubMed] [Google Scholar]
- Dontu G.; Abdallah W. M.; Foley J. M.; Jackson K. W.; Clarke M. F.; Kawamura M. J.; Wicha M. S. Genes Dev. 2003, 17, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Pritchard J. R.; Williams R. T.; Lauffenburger D. A.; Hemann M. T. Nat. Chem. Biol. 2011, 7, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burma S.; Chen B. P.; Murphy M.; Kurimasa A.; Chen D. J. J. Biol. Chem. 2001, 276, 42462. [DOI] [PubMed] [Google Scholar]; b Matsuoka S.; Rotman G.; Ogawa A.; Shiloh Y.; Tamai K.; Elledge S. J. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokino T.; Nakamura Y. Crit. Rev. Oncol. Hematol. 2000, 33, 1. [DOI] [PubMed] [Google Scholar]
- Kim I.; Xu W.; Reed J. C. Nat. Rev. Drug Discovery 2008, 7, 1013. [DOI] [PubMed] [Google Scholar]
- Feng Y. X.; Sokol E. S.; Del Vecchio C. A.; Sanduja S.; Claessen J. H.; Proia T. A.; Jin D. X.; Reinhardt F.; Ploegh H. L.; Wang Q.; Gupta P. B. Cancer Discovery 2014, 4, 702. [DOI] [PubMed] [Google Scholar]
- Boyce M.; Bryant K. F.; Jousse C.; Long K.; Harding H. P.; Scheuner D.; Kaufman R. J.; Ma D.; Coen D. M.; Ron D.; Yuan J. Science 2005, 307, 935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.