

Abstract
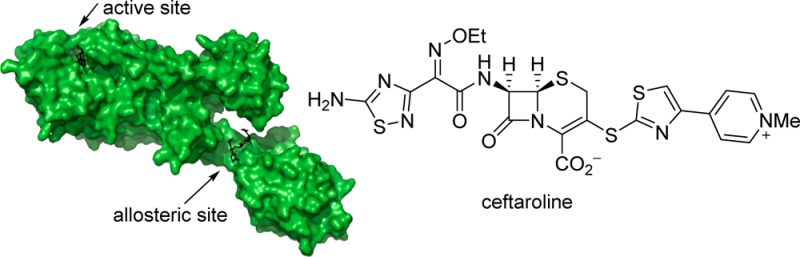
Ceftaroline, a recently approved β-lactam antibiotic for treatment of infections by methicillin-resistant Staphylococcus aureus (MRSA), is able to inhibit penicillin-binding protein 2a (PBP2a) by triggering an allosteric conformational change that leads to the opening of the active site. The opened active site is now vulnerable to inhibition by a second molecule of ceftaroline, an event that impairs cell-wall biosynthesis and leads to bacterial death. The triggering of the allosteric effect takes place by binding of the first antibiotic molecule 60 Å away from the active site of PBP2a within the core of the allosteric site. We document, by kinetic studies and by determination of three X-ray structures of the mutant variants of PBP2a that result in resistance to ceftaroline, that the effect of these clinical mutants is the disruption of the allosteric trigger in this important protein in MRSA. This is an unprecedented mechanism for antibiotic resistance.
Methicillin-resistant Staphylococcus aureus (MRSA), a problematic human pathogen, was first reported in the U.K. in 1961, but it rapidly disseminated globally.1−3 This organism is broadly resistant to antibiotics.4,5 Ceftaroline is a recently approved fifth-generation cephalosporin antibiotic with anti-MRSA activity (Figure 1).6−8 In a mechanism that has been elucidated only recently, ceftaroline manifests its anti-MRSA activity by triggering an allosteric conformational change in penicillin-binding protein 2a (PBP2a) of MRSA, which predisposes the critical enzyme to inhibition by a second molecule of antibiotic at 60 Å distance.9
Figure 1.

Chemical structures of ceftaroline, nitrocefin, ceftobiprole, and L-695,256.
Following the introduction of ceftaroline to the clinic, a recent report disclosed two sets of mutations within the PBP2a sequence that conferred resistance to this antibiotic in clinical strains.10 It was noted that the mutations were distant from the active site of PBP2a, hence their functions could not be understood at the time. Our recent structural description of allostery in PBP2a intriguingly placed these mutations within the allosteric site. One clinical variant of PBP2a is a double mutant (N146K/E150K) and the other a triple mutant (N146K/E150K/H351N). Both have the N146K and E150K mutations, which are within the allosteric domain of PBP2a. The H351N change seen in the triple mutant is outside of both the allosteric and the active sites, hence the likely residues manifesting resistance are N146K and E150K. We introduced these clinical mutations in the gene mecA from S. aureus strain ATCC 700699, which encodes PBP2a, and also cloned the single mutant variants harboring N146K and E150K individually. We report herein by functional assays and by X-ray crystallography that the clinically observed mutations that emerged in the allosteric domain of PBP2a interfere with triggering of allostery by ceftaroline. These mutations impart two traits on the mutant variants of PBP2a in manifestation of resistance to ceftaroline: one is a modest but insignificant increase in dissociation constant for ceftaroline binding to the allosteric site, and another is a disruption of the propagation of conformational change that is key to the opening of the active site. The outcome is resistance to this latest antibiotic by an unprecedented mechanism, namely interference with the fidelity of the allosteric response.
After successful introduction of the desired mutations, we next investigated interactions of ceftaroline with purified PBP2a and its mutant variants. The complication in these experiments is that the β-lactam antibiotic could interact at both the allosteric and the active sites. We chose to incubate these proteins individually in the presence of very high concentrations of oxacillin (a penicillin) to force acylation of the active sites by this antibiotic (which the high concentration achieves). The acylated PBP2a was separated from excess oxacillin by the use of size-exclusion spin columns. If binding of ceftaroline at the allosteric site would affect that of the antibiotic at the active site, then the inverse could also be true. There are a few X-ray structures that show the active site acylated by antibiotics.11,12 However, these invariably show the active site in the closed conformation, leaving the allosteric site similarly intact, with no antibiotic bound. We also note that the oxacillin-acylated PBP2a exhibits a t1/2 for deacylation of 77 h,13 hence for the purpose of our experiments, this is an irreversible modification. We used the mutants modified within the active site by oxacillin in evaluating binding of ceftaroline exclusively to the allosteric domain by quantification of the decrease in the intrinsic fluorescence of the protein. The process was saturable, giving a dissociation constant (Kd) value of 20 ± 4 μM for ceftaroline binding to the wild-type protein. The maximum concentration of ceftaroline in healthy adults given the recommended dosage of multiple 600 mg doses administered every 12 h as 1 h infusions for 14 days is 21.3 ± 4.1 μg/mL, equivalent to 35.2 ± 6.8 μM.8 This is how ceftaroline is efficacious in treatment of MRSA infections; the clinical ceftaroline dose is maintained above the Kd for the allosteric site. The clinical double and triple mutants gave modest increases in the values of the dissociation constants, but these are not significant when error in the measurements is taken into account (Table 1).
Table 1. Second-Order Rate Constants (k2/Ks) for Acylation of the Active Site of PBP2a by Nitrocefin and by Ceftaroline and Dissociation Constants for Binding of Ceftaroline to the Allosteric Site of PBP2a.
| nitrocefin | ceftaroline |
||
|---|---|---|---|
| enzymes | k2/Ks (M–1 s–1) | k2/Ks (M–1 s–1) | Kd (μM) |
| wild-type | 220 ± 25 | 4500 ± 640 | 20 ± 4 |
| N146K | 270 ± 70 | 9150 ± 1560 | 20 ± 4 |
| E150K | 65 ± 5 | 820 ± 470 | 44 ± 6 |
| N146K/E150K | 330 ± 60 | 850 ± 140 | 30 ± 7 |
| N146K/E150K/H351N | 230 ± 30 | 1400 ± 140 | 26 ± 6 |
For the assessment of the consequence of the conformational change by allostery, or the dynamical aspects, we have utilized an assay to evaluate access to the active site of PBP2a.9,13,14 As the active site is opened by the conformational change, the acylation event becomes more favorable, reflected by a larger value for the ratio k2/Ks, the second-order rate constant for active-site acylation (Table 1 and Figure S1). Unexpectedly, the N146K single mutant (not seen clinically) showed approximately a 2-fold enhancement of active-site access to ceftaroline, with essentially no change for nitrocefin (within the error for the determination). However, the E150K single mutant and both the clinical double and triple mutant variants showed significant decreases in the k2/Ks values, consistent with manifestation of resistance by these mutations. We conclude that the clinical mutant variants interfere with fidelity of the critical allosteric triggering of the PBP2a conformational change. We note that nitrocefin also binds to the allosteric site with a Kd of 120 ± 15 μM. However, it does not influence the acylation event the way ceftaroline does (Table 1). This is apparent in the k2/Ks values for nitrocefin (Table 1), which are not affected by the clinical allosteric-site mutations, in contrast to the case of ceftaroline.
We resorted next to solving the X-ray crystal structures of these mutant proteins to provide a structural context to the kinetic observations. In elucidation of the location of the allosteric site, we recently reported the X-ray structure of the complex of the wild-type PBP2a with ceftaroline bound at the allosteric domain (residues 27–326).9 In the present study, the crystal structures of the E150K, N146K, and N146K/E150K variants of PBP2a are reported (Figure 2 and Table 2). As observed for the wild-type PBP2a structure, all three mutant variants show two protein molecules in the asymmetric unit (designated as chains A and B). Chains A and B show minor differences between them, very likely due to somewhat different crystal packing contacts. We describe the structural comparisons below with the chain A molecules.
Figure 2.
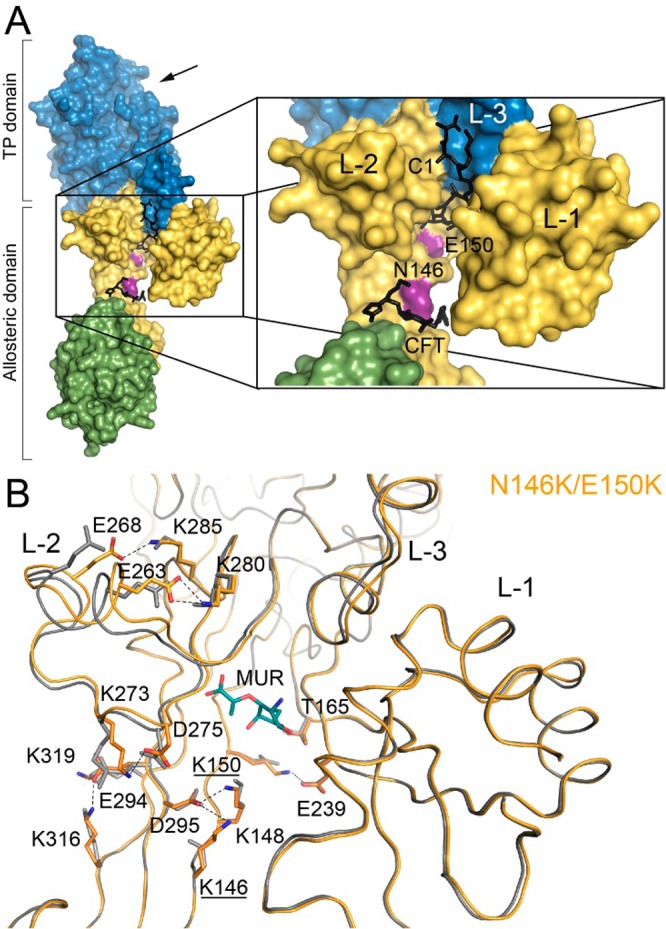
(A) Molecular surface of PBP2a. The N-terminal extension is colored in green, the remaining allosteric domain in yellow and the transpeptidase domain in blue. Active site is indicated by an arrow at 1 o’clock. The locations of the point mutations are indicated in magenta and are labeled. Lobes 1 (L-1), 2 (L-2), and 3 (L-3) of the allosteric site are labeled. The reported allosteric ligands, ceftaroline (CFT) and synthetic peptidoglycan (C1; not discussed in the manuscript) (PDB ID codes 3ZG0 and 3ZG5, respectively) are superimposed and depicted as black-capped sticks. (B) The backbone of the N146K/E150K double mutant (orange) is superimposed onto that of the wild-type PBP2a structure (PDB ID code 1VQQ colored in gray). The muramic acid (MUR) found at the allosteric site in the N146K/E150K mutant is depicted in green-capped sticks for the carbon atoms. Upon mutation, the backbone and the network of salt-bridge interactions are altered. Residues involved in these changes in L-2 are represented in capped sticks (in orange for the mutant and in gray for the wild-type PBP2a). New salt-bridges and hydrogen bonds are shown as dashed lines.
Table 2. Data Collection and Refinement Statistics.
| N146K | E150K | N146K/E150K | |
|---|---|---|---|
| Diffraction Data Statisticsa | |||
| wavelength (Å) | 1.541 79 | 1.541 79 | 0.872 90 |
| space group | P212121 | P212121 | P212121 |
| a, b, c (Å) | 81.7, 102.3, 187.1 | 81.4, 101.6,186.8 | 81.6, 101.8, 186.5 |
| α = β = γ | 90 | 90 | 90 |
| T (K) | 120 | 120 | 100 |
| X-ray source | rotating anode | rotating anode | synchrotron |
| resolution range (Å) | 69.07-(3.16–3.00) | 19.89-(2.87–2.72) | 46.64-(2.48–2.35) |
| total no. of reflections | 749983 | 602683 | 544302 |
| no. unique reflections | 30067 | 42164 | 65572 |
| Rsym | 0.10 (0.58) | 0.13 (0.40) | 0.16 (0.94) |
| ⟨I/σ(I)⟩ | 8.3 (1.9) | 16.1 (7.5) | 11.6 (2.3) |
| completeness (%) | 83.2 (75.3) | 99.2 (96.6) | 100.0 (100.0) |
| redundancy | 3.6 (3.8) | 14.1 (11.4) | 8.3 (8.4) |
| Refinement Statistics | |||
| resolution (Å) | 48.74–3.00 | 19.89–2.72 | 46.64–2.35 |
| Rwork/Rfree | 0.23/0.30 | 0.21/0.29 | 0.18/0.23 |
| No. of Atoms | |||
| protein | 10273 | 10226 | 10217 |
| MUR | 34 | – | 34 |
| ion | 11 | 11 | 15 |
| solvent | 109 | 216 | 778 |
| Average B-Factor (Å2) | |||
| protein | 76.02 | 53.28 | 47.43 |
| solvent | 29.24 | 39.76 | 43.34 |
| RMSD | |||
| bond length (Å) | 0.01 | 0.01 | 0.01 |
| bond angles (°) | 1.20 | 1.17 | 1.15 |
| Ramachandran | |||
| favored/outliers (%) | 93.9/0.9 | 96.4/0.6 | 95.5/0.3 |
| PDB code | 4BL3 | 4BL2 | 4CPK |
Value for the highest resolution shell is shown in parentheses.
The crystal structures of the E150K, N146K, and N146K/E150K variants present overall the same structure seen for the wild-type PBP2a (PDB ID code 1VQQ) (Figure 2B). No changes were observed in their active sites, and all of them gave a closed active-site conformation (Figure S2). Interestingly, these mutations produce changes in the backbone of Lobe-2 and also, to a lesser degree, of Lobe-3 (Figure 2B), both major structural components of the allosteric site. Changes in Lobe-2 result in displacements of up to 1.8 Å in its backbone. We recently proposed that the recognition of ceftaroline at the allosteric site propagates a conformational change involving swapping of ion-paired side chains of a series of acidic and basic residues, akin to dominoes falling.9 The mutations in the allosteric site alter these interactions, as we will elaborate below.
We also note that in vitro selection of resistance in MRSA strains to ceftobiprole (Figure 1)15 or to L-695,256 (Figure 1),16 both β-lactam antibiotics with anti-MRSA activities,17,18 identified E150K (as indicated, selected clinically against ceftaroline), in addition to E237K, and E239K, all of which are located at the core of the allosteric site. Our focus in the present study was on the clinically identified mutants that emerged from challenge by ceftaroline. The crystal structure of the N146K/E150K double mutant (Figure 2B) provides an interpretation for our findings. These mutations alter the aforementioned network of salt-bridge interactions within the allosteric site (Figure 2B), which are involved in the allosteric trigger. There are multiple points of entry into the conformational change within Lobes 1–3 of the allosteric site, which converge to the active site. The K146 side chain in the allosteric site interacts in the mutant protein with the side chain of D295, which in turn interacts with that of K148. On the other hand, the K150 side chain establishes a new salt-bridge with E239 in Lobe-1 (identified by in vitro selection, as described above). This alteration of the interaction network for the allosteric effect occurs by creating new salt-bridges (D275-K273; E294-K319; E294-K316; E268-K285; and E263-K280) that are not present in the wild-type PBP2a (Figure 2B, Figure S3 and Table S1). This altered connectivity within the allosteric site seen in the X-ray structures is consistent with the effects of the kinetic measurements that we report in Table 1. Thus, the salt-bridge observed between E378 and K382 (both in Lobe-3) in the wild-type protein is no longer observed in the clinical mutant. On the contrary, a new salt-bridge is found connecting Lobe-1 (K219) with Lobe-3 (D367) (Table S1). Therefore, the clinical double mutations at positions 146 and 150 not only alter the pattern of interactions around the mutated positions but also, more importantly, tamper with the salt-bridge network among many other residues within Lobe-2 and Lobe-3, as far away as 35 Å from the mutated residues.
A more intuitive observation is the change in the electrostatic potential within the allosteric site (Figure 3). This difference extends beyond the immediate location of the mutated amino acids (Figure 3B) and would be expected to influence the initial complexation with the allosteric trigger (ceftaroline) and the ensuing conformational change that gives access to the active site to the antibiotic.
Figure 3.
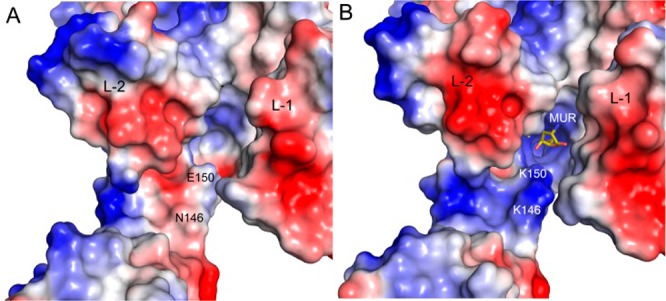
Comparison between the electrostatic potential on the molecular surface of (A) wild-type PBP2a and (B) the clinical N146K/E150K double mutant. Acidic regions are colored in red and basic in blue. The locations of the point mutations are labeled. Lobes 1 (L-1) and 2 (L-2) are labeled. The mutations at positions 146 and 150 provoke a strong change in the electrostatic potential in the entire allosteric site. The new allosteric site presenting a marked basic character extending from the intact ceftaroline binding-site (see Figure 2A) to the MUR binding-site. The muramic acid (MUR) found at the allosteric site in the N146K/E150K mutant is depicted in yellow-capped sticks for the carbon atoms.
As discerned from the X-ray structures, the single mutations (N146K or E150K) result in an attenuated effect on the salt-bridge interactions network. Structural changes in the single mutants are concentrated around the mutated position and do not extend beyond (Figures S4 and S5). Conformations of residues K146 or K150 are not the same in the single and double mutants, providing different interaction networks in each case, even locally within the site of mutation. In other words, the structural effects observed in the clinical double mutant (N146K/E150K) are not merely the sum of the structural effects seen for the single mutants at the same positions.
Resistance to antibiotics typically emerges by the loss of affinity in the active site of the target protein for antibiotic, by enzymatic modification of the antibiotic itself, or by efflux mechanisms.19−22 The resistance to ceftaroline by MRSA that we have described is unprecedented in the considerable literature of antibiotic resistance. The findings also argue for the critical mechanistic role that allostery plays in these events. In light of the importance of allostery to many biological processes,23,24 it is likely that disruption of allostery in other systems in manifestation of antibiotic resistance awaits discovery.
Acknowledgments
This work was supported by a grant from the U.S. National Institutes of Health (AI104987) and by grants BFU2011-25326 (the Spanish Ministry of Economy and Competitiveness) and S2010/BMD-2457 (the Government of Community of Madrid).
Supporting Information Available
Experimental procedures for structural determination, cloning, purification, and kinetic analysis of PBP2a mutants. The crystallographic coordinates are deposited in the Protein Data Bank (PDB codes 4BL2, 4BL3, and 4CPK for E150K, N146K, and N146K/E150K mutants, respectively). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chambers H. F. Clin. Microbiol. Rev. 1997, 10, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright M. C.; Robinson D. A.; Randle G.; Feil E. J.; Grundmann H.; Spratt B. G. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevons M. P. Br. Med. J. 1961, 1, 124. [Google Scholar]
- Wu S. W.; de Lencastre H.; Tomasz A. Microbial. Drug Resist. 2005, 11, 215. [DOI] [PubMed] [Google Scholar]
- Fuda C. C.; Fisher J. F.; Mobashery S. Cell. Mol. Life Sci. 2005, 62, 2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez P. O.; Lema S.; Tyring S. K.; Mendoza N. Infect. Drug Resist. 2012, 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell D. J.; Castanheira M.; Mendes R. E.; Sader H. S.; Jones R. N. Clin. Infect. Dis. 2012, 55, S206. [DOI] [PubMed] [Google Scholar]
- Laudano J. B. J. Antimicrob. Chemother. 2011, 66, iii11. [DOI] [PubMed] [Google Scholar]
- Otero L. H.; Rojas-Altuve A.; Llarrull L. I.; Carrasco-Lopez C.; Kumarasiri M.; Lastochkin E.; Fishovitz J.; Dawley M.; Hesek D.; Lee M.; Johnson J. W.; Fisher J. F.; Chang M.; Mobashery S.; Hermoso J. A. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes R. E.; Tsakris A.; Sader H. S.; Jones R. N.; Biek D.; McGhee P.; Appelbaum P. C.; Kosowska-Shick K. J. Antimicrob. Chemother. 2012, 67, 1321. [DOI] [PubMed] [Google Scholar]
- Lim D.; Strynadka N. C. Nat. Struct. Biol. 2002, 9, 870. [DOI] [PubMed] [Google Scholar]
- Lovering A. L.; Gretes M. C.; Safadi S. S.; Danel F.; de Castro L.; Page M. G.; Strynadka N. C. J. Biol. Chem. 2012, 287, 32096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda C.; Suvorov M.; Vakulenko S. B.; Mobashery S. J. Biol. Chem. 2004, 279, 40802. [DOI] [PubMed] [Google Scholar]
- Graves-Woodward K.; Pratt R. F. Biochem. J. 1998, 332, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R.; Gretes M.; Basuino L.; Strynadka N.; Chambers H. F. Antimicrob. Agents Chemother. 2008, 52, 2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y.; Zhang H.-Z.; Chambers H. F. Antimicrob. Agents Chemother. 2004, 48, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidaillac C.; Rybak M. J. Pharmacotherapy 2009, 29, 511. [DOI] [PubMed] [Google Scholar]
- Rylander M.; Rollof J.; Jacobsson K.; Norrby S. R. Antimicrob. Agents Chemother. 1995, 39, 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox G.; Wright G. D. Int. J. Med. Microbiol. 2013, 303, 287. [DOI] [PubMed] [Google Scholar]
- Fisher J. F.; Mobashery S.. 8.13 - Enzymology of Bacterial Resistance. In Comprehensive Natural Products II; Liu H.-W.; Mander L., Eds.; Elsevier: Oxford, 2010; pp 443–487. [Google Scholar]
- Hawkey P. M. Br. Med. J. 1998, 317, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G. D. Curr. Opin. Chem. Biol. 2003, 7, 563. [DOI] [PubMed] [Google Scholar]
- Goodey N. M.; Benkovic S. J. Nat. Chem. Biol. 2008, 4, 474. [DOI] [PubMed] [Google Scholar]
- Changeux J.-P. Annu. Rev. Biophys. 2012, 41, 103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.