

Abstract
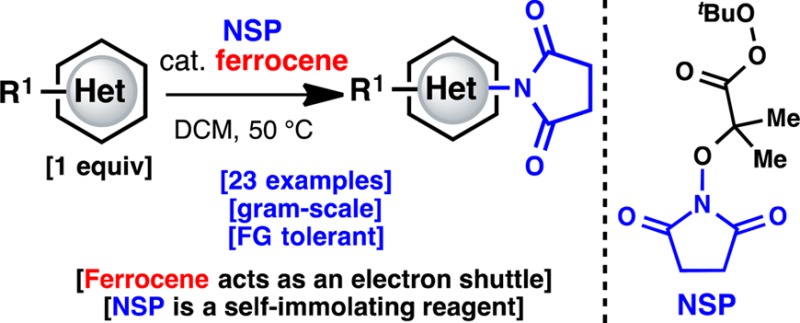
A simple method for direct C–H imidation is reported using a new perester-based self-immolating reagent and a base-metal catalyst. The succinimide products obtained can be easily deprotected in situ (if desired) to reveal the corresponding anilines directly. The scope of the reaction is broad, the conditions are extremely mild, and the reaction is tolerant of oxidizable and acid-labile functionality, multiple heteroatoms, and aryl iodides. Mechanistic studies indicate that ferrocene (Cp2Fe) plays the role of an electron shuttle in the decomposition of the perester reagent, delivering a succinimidyl radical ready to add to an aromatic system.
Direct, intermolecular C–H amination of arenes is a highly sought-after transformation in organic chemistry. The ubiquity of amines in medicine, agrochemicals, and materials demands efficiency in their synthesis.1 Great strides have been made toward these goals using both guided and innate C–H functionalization logic.2 Guided approaches (Figure 1A) can be extremely useful if common functional groups can serve the dual role of directing groups, as has been expertly demonstrated by the groups of Yu, Nakamura, Daugulis, Sanford, and others.2b−2j The area of innate arene amination is just beginning to blossom. In 2011, Cho and Chang described a metal-free oxidative C–H phthalimidation of benzene using PhI(OAc)2 as oxidant (Figure 1A).2k Although the reaction was conducted at 140 °C with solvent quantities of substrate, it was an important proof of principle. Hartwig discovered that adding a Pd catalyst to the Cho–Chang system enables a lower operating temperature and higher selectivity.2m In the same year, Zhang and co-workers reported a guided imidation of arenes using Pd(OAc)2 and NFSI as the amine source.2e Ritter demonstrated that the directing group was not needed in the Zhang system by utilizing a novel Pd system.2n The Ritter conditions also broadened the substrate scope by including pyrroles and thiophenes. Other significant reports in this area involve the Cu-catalyzed C–H amination of azoles.2o−2q This Communication describes a fundamentally different approach to C–H imidation enabled by an imide-radical precursor and an unusual catalyst system employing ferrocene (Cp2Fe). The reaction is scalable and mild and, most importantly, functions on both electron-rich and -poor (hetero)arenes.
Figure 1.
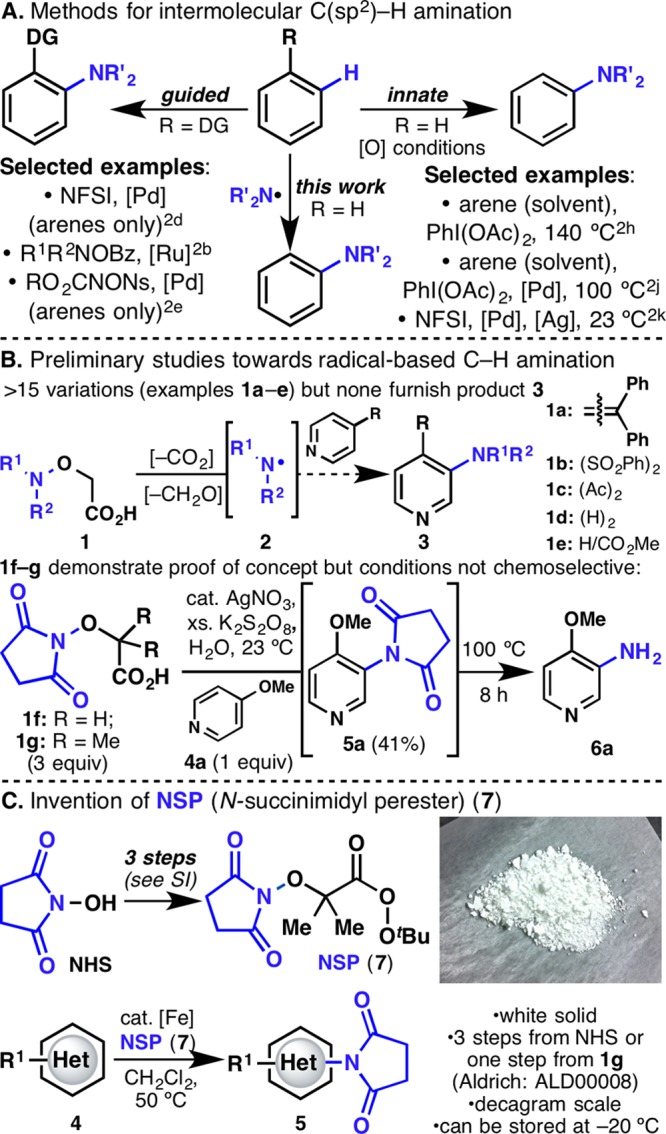
(A) Examples of intermolecular oxidative amination of arenes. (B) Preliminary studies toward a radical-based C–H amination reagent. (C) Invention of NSP (7).
Our laboratory has a longstanding interest in radical-based C–H functionalization of heteroaromatic systems.3 The present study was aimed at forging C–N bonds via N-centered radicals (N-radicals). However, intermolecular radical amination is a notoriously difficult reaction, due to the propensity of N-radicals to undergo hydrogen abstraction or to engage in other degradation pathways.4 These challenges are evident from prior studies where a large excess (solvent quantities) of simple arenes was needed in the context of aromatic C–H imidation.2s−2v
Drawing inspiration from Forrester’s work on iminyl radicals,5 we synthesized a library of over 15 closely related precursors 1 by modifying the substituents on the nitrogen atom (Figure 1B). The guiding principle in this design was to generate N-radicals 2 upon radical decarboxylation/deformylation. Preliminary studies were performed on 4-methoxypyridine (4a), and gratifyingly one of the reagents, 1f, was met with success. This result was surprising, considering (1) Minisci’s prediction that such reactions on pyridines were bound to fail6 and (2) numerous reports pointing to succinimidyl radicals as being inherently unstable, undergoing immediate ring-opening.7 Subsequent succinimide deprotection was achieved by heating the crude mixture to 100 °C (8 h) to deliver amine 6a directly, resulting in a one-pot aniline synthesis. It was also found that radical precursor 1g gave identical yields, except that the rate of decarboxylation was faster.8 Unfortunately, attempts to expand the scope of this reaction by including more electron-rich substrates led only to decomposition of starting material due to the strongly oxidizing nature of the reaction media (excess K2S2O8).
Having achieved a proof of concept for the basic reactivity, we turned our attention to developing a more chemoselective system. Thus, perester 7 (N-succinimidyl perester, NSP) was targeted, as we envisaged that homolysis of the weak O–O bond would lead to the same radical under neutral conditions (Figure 1C). Perester 7 can be easily synthesized (decagram scale) in three steps from N-hydroxysuccinimide. This compound is a white solid, can be stored without degradation at −20 °C, and is currently in the process of commercialization (precursor 1g is available through Aldrich: cat. no. ALD00008). Gratifyingly, even in the absence of catalyst, NSP (7) (3 equiv) reacted directly with 4a in CH2Cl2 at 50 °C to furnish 5a in 7% yield (Table 1). Since Cu(I) or Fe(II) salts are commonly used to promote the decomposition of peresters or peroxides, an extensive search for the optimal catalyst was undertaken.9 Surprisingly, and after significant exploration (Table 1 summarizes >100 experiments), it was found that Cp2Fe (5 mol%) was ideal (see Supporting Information). It is also noteworthy that the more redox-active Cp*2Fe10 gave a lower yield.11a The mechanistic implications of this finding are discussed below and, to the best of our knowledge, represent an uncommon use of Cp2Fe in a preparative setting.9e,9f,11
Table 1. Catalyst Optimization Summary for Reaction between NSP (7) and 4a.

5–10 mol% of catalyst was used.
Yield is based on NMR comparison.
This new reagent and catalyst system could be employed on a variety of electron-rich and -poor (hetero)aromatic systems (Table 2). The imidation process occurs on a wide variety of pyridines and phenyl rings with good yields in most cases. The observed regioselectivity of the imidation is similar to that of electrophilic aromatic substitution reactions: electron-donating groups such as alkoxy/alkyl are ortho/para directing (5a–c and 5g–k). This procedure is tolerant of sensitive groups such as esters, halogens, and silyl protecting groups, which is particularly important if subsequent functionalization is required. Interestingly, highly electron-rich heterocycles (such as 5l–r) and electron-poor substrates (such as 5s–w) can be imidated successfully. Alternate approaches to aromatic amine synthesis (e.g., nitration/reduction), in many cases, would not result in a comparable yield. For instance, the nitration of pyrroles is a classic problem, with little in the way of progress with respect to the yield. Many of the compounds, which demonstrate efficient reactivity under these conditions, are useful to the pharmaceutical industry, where the synthesis of aromatic amines is a key area of need. Although the reaction could be conducted under air, an inert atmosphere generally gave more consistent results. One limitation of the process was the occasional observation of ipso-substitution in 1,4-disubstituted benzenes (5i (15:2)) as a side reaction.
Table 2. Substrate Scope of Ferrocene-Catalyzed C–H Imidation of (Hetero)Arenes 4.
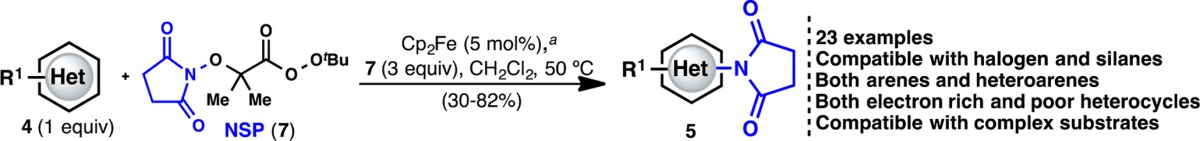
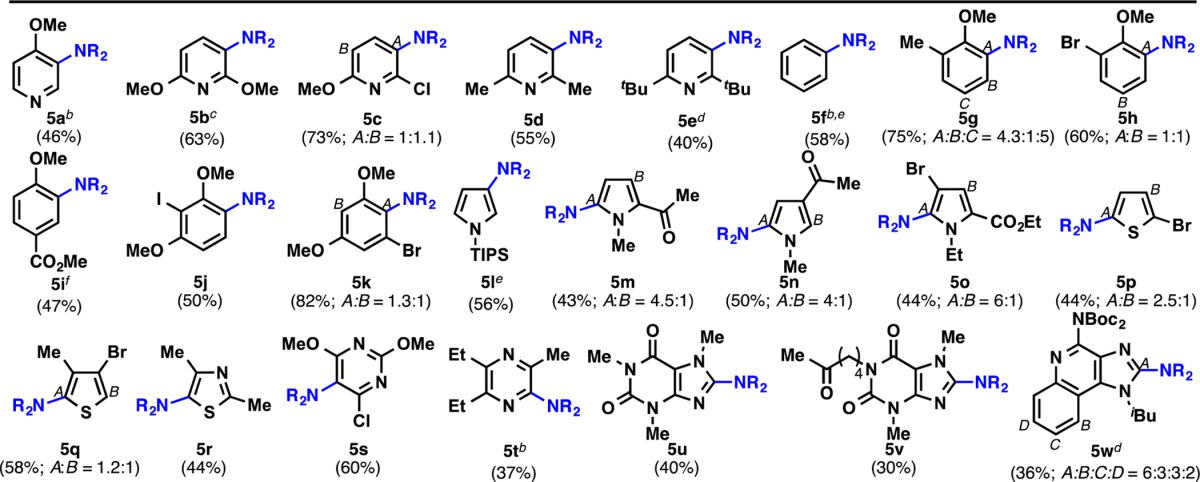
Cp2Fe (5 mol%), (hetero)arene 4 (0.2 mmol), NSP (7) (3 equiv), CH2Cl2 (0.05 M), 50 °C, 2–7 h; isolated yields reported.
NSP (7) (4 equiv) was used.
NSP (7) (2.75 equiv) was used.
NSP (7) (5 equiv) was used.
Cp2Fe (10 mol%) was used.
Ipso-substitution observed.
A plausible mechanistic proposal for this reaction is described in Figure 2. Outer-sphere single-electron transfer from Cp2Fe to NSP (7) initiates O–O bond cleavage, resulting in concerted fragmentation, generating the imidyl radical, CO2, acetone, and ferrocenium tert-butoxide. C–N bond formation between the imidyl radical and 4 leads to radical 8. One-electron oxidation of 8, followed by deprotonation, yields 5 and regenerates Cp2Fe in the process. Although there are a few examples of Cp2Fe acting either as a Fenton-like reagent9e,9f or to mediate aryl radical formation from diazonium salts,11 Cu(I) salts are normally employed for perester activation.9a,9b Figure 2B depicts a 1H NMR study that supports the role of Cp2Fe in the reaction. The spectra overlay shows clearly that the Cp2Fe signal (I) diminishes as 7 is added to the mixture (II) and then completely disappears upon heating to 50 °C (III). Increasing the signal peak intensity reveals a broad signal (δ 5.14 ppm) (IV), characteristic of a paramagnetic species. This observation is consistent with a ferrocenium species, which are known to be paramagnetic.10 A control experiment with a mixture of Cp2Fe and 4a was conducted in an identical fashion, and the Cp2Fe signal remained unchanged. Additional evidence of ferrocenium ions is the green color (VI) achieved upon heating of the yellow Cp2Fe mixture with NSP (7) (V). The use of Cp2Fe as an electron shuttle forms the essence of this interesting catalytic cycle.
Figure 2.
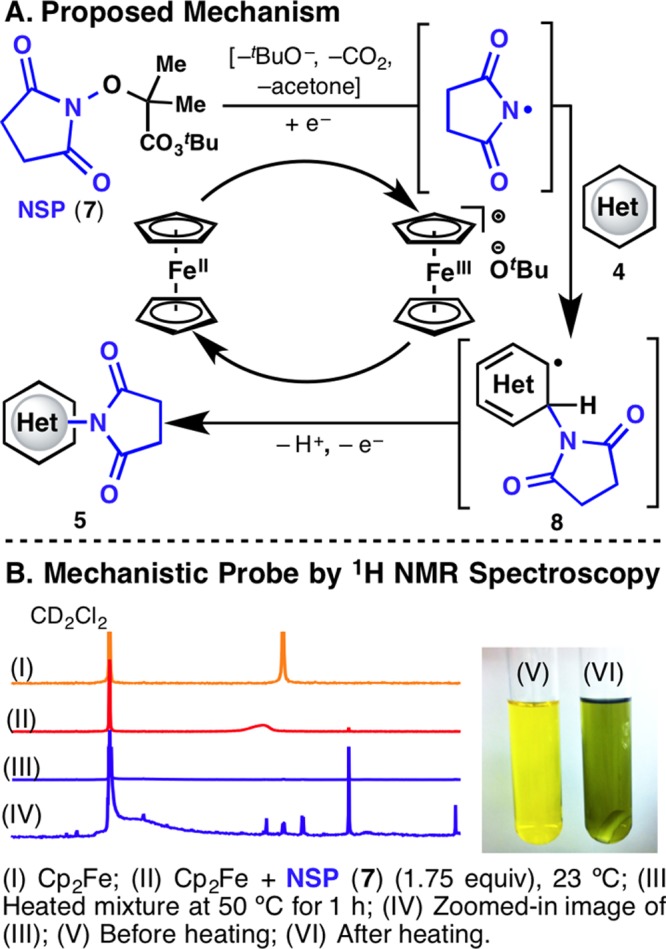
(A) Proposed catalytic cycle. (B) Mechanistic probe by 1H NMR spectroscopy.
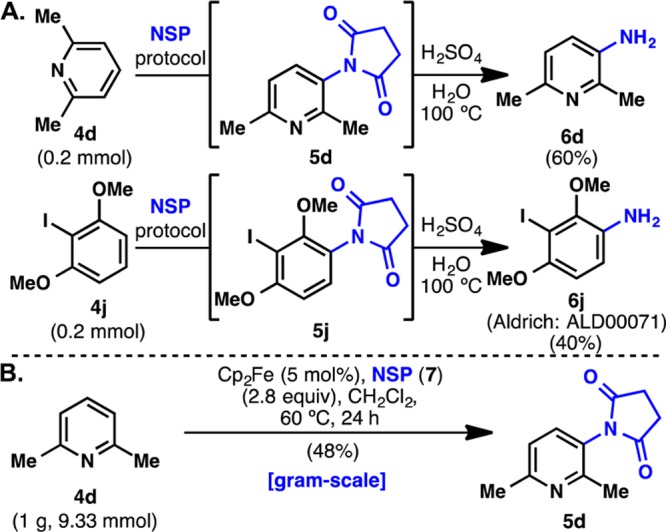
To demonstrate the utility of NSP (7) as a potential direct amination reagent, a one-pot imidation/deprotection procedure was developed (Scheme 1A). Upon completion of the imidation step, the reaction was quenched with MeOH, concentrated, and heated to reflux in a dilute solution of H2SO4. This procedure was effective for the one-pot amination of 4d to give 6d in 60% yield. Iodoarene 4j was directly aminated using a similar procedure to give amine 6j in 40% yield (6j has been commercialized by Aldrich using this method: cat. no. ALD00071). It was found that the hydrolysis of imide 5j was significantly slower, and more acid was added to accelerate the deprotection. Finally, the C–H imidation was successfully carried out on gram scale with 4d, giving the desired imide 5d in 48% yield. This is comparable to the result obtained on 0.2 mmol scale as shown in Table 2.
Scheme 1. (A) One-Pot C–H Imidation/Deprotection of 4d and 4j and (B) Gram-Scale C–H Imidation of 4d.
The mild and chemoselective nature of this imidation reaction (and subsequent deprotection to an amine) bodes well for its use at both the early and late stages of a synthesis program. The ability to aminate in the presence of oxidizable functionality and halogen atoms enables sequential aryl functionalizations. NSP (7) represents a unique self-immolating12 reagent that might inspire the design of other synthetically useful reactive species. Finally, the use of Cp2Fe as a mild electron shuttle in other reaction types is a topic worthy of further investigation.
Acknowledgments
Financial support was provided by Bristol-Myers Squibb, NSS (PhD) A*STAR (predoctoral fellowship to K.F.), G. E. Hewitt Foundation (postdoctoral fellowship to E.S.), AvH Foundation (postdoctoral fellowship to I.T.), and the NIH/NIGMS (GM-106210). We thank Prof. A. L. Rheingold, Dr. C. E. Moore, and Dr. J. A. Golen for X-ray crystallographic analysis.
Supporting Information Available
Experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For intramolecular (hetero)arene C–H amination, see:; a Tsang W. C. P.; Zheng N.; Buchwald S. L. J. Am. Chem. Soc. 2005, 127, 14560. [DOI] [PubMed] [Google Scholar]; b Tan Y.; Hartwig J. F. J. Am. Chem. Soc. 2010, 132, 3676. [DOI] [PubMed] [Google Scholar]; c Engle K. M.; Mei T.-S.; Wang X.; Yu J.-Q. Angew. Chem., Int. Ed. 2011, 50, 1478. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jordan-Hore J. A.; Johansson C. C. C.; Gulia M.; Beck E. M.; Gaunt M. J. J. Am. Chem. Soc. 2008, 130, 16184. [DOI] [PubMed] [Google Scholar]; e Wang H.; Wang Y.; Peng C.; Zhang J.; Zhu Q. J. Am. Chem. Soc. 2010, 132, 13217. [DOI] [PubMed] [Google Scholar]; f Antonchick A. P.; Samanta R.; Kulikov K.; Lategahn J. Angew. Chem., Int. Ed. 2011, 50, 8605. [DOI] [PubMed] [Google Scholar]; g King A. E.; Huffman L. M.; Casitas A.; Costas M.; Ribas X.; Stahl S. S. J. Am. Chem. Soc. 2010, 132, 12068. [DOI] [PubMed] [Google Scholar]; h Brasche G.; Buchwald S. L. Angew. Chem., Int. Ed. 2008, 47, 1932. [DOI] [PubMed] [Google Scholar]; For reviews on (hetero)arene C–H aminations, see:; i Hartwig J. F. Acc. Chem. Res. 2012, 45, 864. [DOI] [PubMed] [Google Scholar]; j Mueller P.; Fruit C. Chem. Rev. 2003, 103, 2905. [DOI] [PubMed] [Google Scholar]; k Dick A. R.; Sanford M. S. Tetrahedron 2006, 62, 2439. [Google Scholar]; l Davies H. M. L.; Long M. S. Angew. Chem., Int. Ed. 2005, 44, 3518. [DOI] [PubMed] [Google Scholar]; m Davies H. M. L.; Manning J. R. Nature 2008, 451, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Cho S. H.; Kim J. Y.; Kwak J. Y.; Kwak J.; Chang S. Chem. Soc. Rev. 2011, 40, 5068. [DOI] [PubMed] [Google Scholar]
- a Brückl T.; Baxter R. D.; Ishihara Y.; Baran P. S. Acc. Chem. Res. 2012, 45, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]; For guided (hetero)arene C–H amination, see:; b Shang M.; Zeng S.-H.; Sun S.-Z.; Dai H.-X.; Yu J.-Q. Org. Lett. 2013, 15, 5286. [DOI] [PubMed] [Google Scholar]; c Yoo E. J.; Ma S.; Mei T.-S.; Chan K. S. L.; Yu J.-Q. J. Am. Chem. Soc. 2011, 133, 7652. [DOI] [PubMed] [Google Scholar]; d Shang M.; Sun S.-Z.; Dai H.-X.; Yu J.-Q. J. Am. Chem. Soc. 2014, 136, 3354. [DOI] [PubMed] [Google Scholar]; e Sun K.; Li Y.; Xiong T.; Zhang J.; Zhang Q. J. Am. Chem. Soc. 2011, 133, 1694. [DOI] [PubMed] [Google Scholar]; f Thu H.-Y.; Yu W.-Y.; Che C.-M. J. Am. Chem. Soc. 2006, 128, 9048. [DOI] [PubMed] [Google Scholar]; g Ng K.-H.; Chan A. S. C.; Yu W.-Y. J. Am. Chem. Soc. 2010, 132, 12862. [DOI] [PubMed] [Google Scholar]; h Dick A. R.; Remy M. S.; Kampf J. W.; Sanford M. S. Organometallics 2007, 26, 1365. [Google Scholar]; i Matsubara T.; Asako S.; Ilies L.; Nakamura E. J. Am. Chem. Soc. 2014, 136, 646. [DOI] [PubMed] [Google Scholar]; j Tran L. D.; Roane J.; Daugulis O. Angew. Chem., Int. Ed. 2013, 52, 6043. [DOI] [PMC free article] [PubMed] [Google Scholar]; For innate (hetero)arene intermolecular C–H amination, see:; k Kim H. J.; Kim J.; Cho S. H.; Chang S. J. Am. Chem. Soc. 2011, 133, 16382. [DOI] [PubMed] [Google Scholar]; l Kantak A. A.; Potavathri S.; Barham R. A.; Romano K. M.; DeBoef B. J. Am. Chem. Soc. 2011, 133, 19960. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Shrestha R.; Mukherjee P.; Tan Y.; Litman Z. C.; Hartwig J. F. J. Am. Chem. Soc. 2013, 135, 8480. [DOI] [PubMed] [Google Scholar]; n Boursalian G. B.; Ngai M.-Y.; Hojczyk K. N.; Ritter T. J. Am. Chem. Soc. 2013, 135, 13278. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Kawano T.; Hirano K.; Satoh T.; Miura M. J. Am. Chem. Soc. 2010, 132, 6900. [DOI] [PubMed] [Google Scholar]; p Wang Q.; Schreiber S. L. Org. Lett. 2009, 11, 5178. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Monguchi D.; Fujiwara T.; Furukawa H.; Mori A. Org. Lett. 2009, 11, 1607. [DOI] [PubMed] [Google Scholar]; r Citterio A.; Gentile A.; Minisci F.; Navarrini V.; Serravalle M.; Ventura S. J. Org. Chem. 1984, 49, 4479. [Google Scholar]; s Lidgett R. A.; Lynch E. R.; McCall E. B. J. Chem. Soc. 1965, 3754. [Google Scholar]; t Cadogan J. I. G.; Rowley A. G. J. Chem. Soc., Perkin Trans. 1 1975, 1069. [Google Scholar]; u Day J. C.; Katsaros M. G.; Kocher W. D.; Scott A. E.; Skell P. S. J. Am. Chem. Soc. 1978, 100, 1950. [Google Scholar]; v Lu F.-L.; Naguib Y. M. A.; Kitadani M.; Chow Y. L. Can. J. Chem. 1979, 57, 1967. [Google Scholar]; w Togo H.; Hoshina Y.; Muraki T.; Nakayama H.; Yokoyama M. J. Org. Chem. 1998, 63, 5193. [Google Scholar]
- Fujiwara Y.; Dixon J. A.; O’Hara F.; Daa Funder E.; Dixon D. D.; Rodriguez R. A.; Baxter R. D.; Herlé B.; Sach N.; Collins M. R.; Ishihara Y.; Baran P. S. Nature 2012, 492, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zard S. Z. Chem. Soc. Rev. 2008, 37, 1603. [DOI] [PubMed] [Google Scholar]; b Danen W. C.; Neugebauer F. A. Angew. Chem., Int. Ed. 1975, 14, 783. [Google Scholar]; c Stella L. Angew. Chem., Int. Ed. 1983, 22, 337. [Google Scholar]
- a Forrester A. R.; Gill M.; Meyer C. J.; Sadd J. S.; Thomson R. H. J. Chem. Soc., Chem. Commun. 1975, 291. [Google Scholar]; b Boivin J.; Fouquet E.; Zard S. Z. Tetrahedron 1994, 50, 1769. [Google Scholar]
- Minisci F.Synthetic and Mechanistic Organic Chemistry, Topics in Current Chemistry 62; Springer: Berlin/Heidelberg, 1976; p 1. [DOI] [PubMed] [Google Scholar]
- a Lüning U.; Seshadri S.; Skell P. S. J. Org. Chem. 1986, 51, 2071. [Google Scholar]; b Day J. C.; Lindstrom M. J.; Skell P. S. J. Am. Chem. Soc. 1974, 96, 5616. [Google Scholar]; c Hedaya E.; Hinman R. L.; Schomaker V.; Theodoropulos S.; Kyle L. M. J. Am. Chem. Soc. 1967, 89, 4875. [Google Scholar]; d O’Reilly R. J.; Karton A.; Radom L. J. Phys. Chem. A 2013, 117, 460. [DOI] [PubMed] [Google Scholar]
- Hilborn J. W.; Pincock J. A. J. Am. Chem. Soc. 1991, 113, 2683. [Google Scholar]
- For Cu(I)-catalyzed O–O cleavage, see:; a Kharasch M. S.; Sosnovsky G. J. Am. Chem. Soc. 1958, 80, 756. [Google Scholar]; b Beckwith A. L. J.; Zavitsas A. A. J. Am. Chem. Soc. 1986, 108, 8230. [Google Scholar]; For Fe(II)-catalyzed O–O cleavage, see:; c Fenton H. J. H. J. Chem. Soc., Trans. 1894, 65, 899. [Google Scholar]; d Yang H.; Yan H.; Sun P.; Zhu Y.; Lu L.; Liu D.; Rong G.; Mao J. Green Chem. 2013, 15, 976. [Google Scholar]; e Wertz S.; Leifer D.; Studer A. Org. Lett. 2013, 15, 928. [DOI] [PubMed] [Google Scholar]; f Ohtsuka Y.; Yamakawa T. Tetrahedron 2011, 67, 2323. [Google Scholar]
- Yang E. S.; Chan M.-S.; Wahl A. C. J. Phys. Chem. 1980, 84, 3094. [Google Scholar]
- a Marciasini L. D.; Richy N.; Vaultier M.; Pucheault M. Adv. Synth. Catal. 2013, 355, 1083. [Google Scholar]; b Chernyak N.; Buchwald S. L. J. Am. Chem. Soc. 2012, 134, 12466. [DOI] [PubMed] [Google Scholar]
- Sagi A.; Weinstein R.; Karton N.; Shabat D. J. Am. Chem. Soc. 2008, 130, 5434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.